

Abstract
Elucidation of the interaction of proteins with different molecules is of significance in the understanding of cellular processes. Computational methods have been developed for the prediction of protein–protein interactions. But insufficient attention has been paid to the prediction of protein–RNA interactions, which play central roles in regulating gene expression and certain RNA-mediated enzymatic processes. This work explored the use of a machine learning method, support vector machines (SVM), for the prediction of RNA-binding proteins directly from their primary sequence. Based on the knowledge of known RNA-binding and non-RNA-binding proteins, an SVM system was trained to recognize RNA-binding proteins. A total of 4011 RNA-binding and 9781 non-RNA-binding proteins was used to train and test the SVM classification system, and an independent set of 447 RNA-binding and 4881 non-RNA-binding proteins was used to evaluate the classification accuracy. Testing results using this independent evaluation set show a prediction accuracy of 94.1%, 79.3%, and 94.1% for rRNA-, mRNA-, and tRNA-binding proteins, and 98.7%, 96.5%, and 99.9% for non-rRNA-, non-mRNA-, and non-tRNA-binding proteins, respectively. The SVM classification system was further tested on a small class of snRNA-binding proteins with only 60 available sequences. The prediction accuracy is 40.0% and 99.9% for snRNA-binding and non-snRNA-binding proteins, indicating a need for a sufficient number of proteins to train SVM. The SVM classification systems trained in this work were added to our Web-based protein functional classification software SVMProt, at http://jing.cz3.nus.edu.sg/cgi-bin/svmprot.cgi. Our study suggests the potential of SVM as a useful tool for facilitating the prediction of protein–RNA interactions.
Keywords: RNA-binding proteins, RNA–protein interactions, rRNA, mRNA, tRNA, snRNA, support vector machine
INTRODUCTION
Knowledge regarding how proteins interact with each other and with other molecules is essential in the understanding of cellular processes (Siomi and Dreyfuss 1997; Draper 1999; Lengeler 2000; Downward 2001). With the accumulation of sequence information, attention has been paid to the development of methods for the prediction of protein function (Fetrow and Skolnick 1998) and interactions (Dandekar et al. 1998; Overbeek et al. 1999; Bock and Gough 2001) from sequence. Several computational methods have been developed for the prediction of protein–protein interactions using support vector machines (SVM; Bock and Gough 2001) and for the prediction of protein–protein interaction maps by Rosetta/gene fusion (Enright et al. 1999; Marcotte et al. 1999), phylogenetic profile (Pellegrini et al. 1999), gene neighbor (Dandekar et al. 1998; Overbeek et al. 1999), and interacting domain profile pair (Eisen et al. 1998) methods.
Although progress has been made in the development of predictive methods for protein–protein interactions, insufficient attention has been paid to the development of predictive methods for protein–RNA interactions. Most cellular RNAs work in concert with protein partners, and protein–RNA interactions are critically important in regulation of different steps of gene expression (Siomi and Dreyfuss 1997). Moreover, binding of proteins to some catalytic RNA molecules is known to activate or enhance the activity of these molecules (Frank and Pace 1998). Therefore, prediction of protein–RNA interactions is of significance in a more comprehensive understanding of how cellular processes and networks work.
RNA recognition by proteins is primarily mediated by certain classes of RNA binding domains and motifs (Draper 1999; Fierro-Monti and Mathews 2000; Peculis 2000; Perez-Canadillas and Varani 2001). Hence, as in the case of protein–protein interactions (Casari et al. 1995; Pawson 1995; Elcock and McCammon 2001), correlated patterns of sequence and substructure in RNA-binding proteins can be recognized to bind to specific RNA sequences and folds. The SVM approach, successfully used for the prediction of protein–protein interactions from primary sequences (Bock and Gough 2001), is therefore expected to be applicable for recognizing this pattern and thus predicting RNA-binding proteins from protein primary sequence.
In the present study, we explored the use of SVM for the prediction of RNA-binding proteins from protein primary sequence. The SVM method was used for the prediction of individual classes of rRNA-, mRNA-, and tRNA-binding proteins, as well as all RNA-binding proteins. There are other groups of RNA-binding proteins, such as snRNA-binding and snoRNA-binding proteins, with small numbers of proteins and fewer available sequences (Tomasevic and Peculis 1999; Singh 2002). A search of protein family and sequence databases revealed a total of 60 sequences of snRNA-binding proteins and 21 sequences of snoRNA-binding proteins, which is fewer than the 80–100 sequences typically needed to properly train an SVM protein classification system (Cai et al. 2003a). Nevertheless, to evaluate its performance on classification of a small protein class, SVM was used for the prediction of snRNA-binding proteins. Proteins of small RNA-binding classes as well as other RNA-binding proteins were included in training and testing the SVM classification of all RNA-binding proteins.
SVM is a relatively new and promising algorithm for binary classification by means of supervised learning which was originally developed by Vapnik and his coworkers (Vapnik 1995; Burges 1998) and applied to a wide range of problems including text categorization (Drucker et al. 1999; Kim et al. 2001; de Vel et al. 2001), hand-written digit recognition (Vapnik 1995), tone recognition (Thubthong and Kijsirikul 2001), image classification and object detection (Ben-Yacoub et al. 1999; Karlsen et al. 2000; Papageorgiou and Poggio 2000; Huang et al. 2002), flood stage forecasting (Liong and Sivapragasam 2002), cancer diagnosis (Furey et al. 2000; Ramaswamy et al. 2001; Fritsche 2002), microarray gene expression data analysis (Brown et al. 2000), inhibitor classification (Burbidge et al. 2001), prediction of protein solvent accessibility (Yuan et al. 2002), protein fold recognition (Ding and Dubchak 2001), protein secondary structure prediction (Hua and Sun 2001), prediction of protein–protein interaction (Bock and Gough 2001) and protein functional class classification (Karchin et al. 2002; Cai et al. 2003a). These studies have demonstrated that SVM is consistently superior to other supervised learning methods including classification methods (Brown et al. 2000; Burbidge et al. 2001; Cai et al. 2002b). In the present study, SVM was further tested regarding its capability to predict protein–RNA interactions.
RESULTS AND DISCUSSION
Overall prediction accuracy
The numbers and prediction results of specific classes of RNA-binding proteins and non-class members are given in Table 1 ▶. In the able, TP stands for true positive (correctly predicted RNA-binding proteins of a specific class), FN for false negative (specific class of RNA-binding proteins incorrectly predicted as non-class members), TN for true negative (correctly predicted non-class members), and FP for false positive (non-class members incorrectly predicted as a specific class of RNA-binding proteins). The predicted sensitivity (SE) for rRNA-, mRNA-, tRNA-, and snRNA-binding proteins and all RNA-binding proteins, which measures the overall prediction accuracy for each class of RNA-binding proteins, is 94.1%, 79.3%, 94.1%, 41.0%, and 97.8%, respectively. The predicted specificity (SP) for non-rRNA-, non-mRNA-, non-tRNA-, and non-snRNA-binding proteins and all non-RNA-binding proteins, which measures prediction accuracy for each group of non-RNA-binding proteins, is 98.7%, 96.5%, 99.9%, 99.7%, and 96.0%, respectively.
TABLE 1.
Prediction accuracies and number of positive and negative samples in the training, testing, and independent evaluation set of rRNA-, mRNA-, tRNA-, and snRNA-binding proteins and of all RNA-binding proteins
| Training set | Testing set | Independent evaluation set | |||||||||||
| positive | negative | positive | negative | ||||||||||
| Protein family | positive | negative | TP | FN | TN | FP | TP | FN | SE (%) | TN | FP | SP (%) | Q (%) |
| RNA-binding | 2161 | 2965 | 1844 | 6 | 6802 | 14 | 437 | 10 | 97.8 | 4685 | 196 | 96.0 | 96.1 |
| rRNA-binding | 708 | 972 | 1243 | 2 | 9031 | 13 | 95 | 6 | 94.1 | 4931 | 66 | 98.7 | 98.6 |
| mRNA-binding | 277 | 2106 | 129 | 0 | 10164 | 0 | 130 | 34 | 79.3 | 5833 | 213 | 96.5 | 96.0 |
| tRNA-binding | 94 | 792 | 114 | 0 | 9295 | 2 | 48 | 3 | 94.1 | 5028 | 5 | 99.9 | 99.8 |
| snRNA-binding | 33 | 1988 | 7 | 0 | 10373 | 1 | 9 | 11 | 41.0 | 6133 | 18 | 99.7 | 99.5 |
Predicted results are given in TP (true positive), FN (false negative), TN (true negative), FP (false positive), sensitivity SE = TP/(TP + FN), specificity SP = TN/(TN + FP), and Q (overall accuracy, Q = (TN + TP)/(TP + FN + TN + FP)). Number of positive or negative samples in the testing and independent evaluation sets is TP + FN or TN + FP, respectively.
A direct comparison with results from previous protein studies is inappropriate, because of the differences in the specific aspects of proteins classified, data set, descriptors, and classification methods. Nonetheless, a tentative comparison may provide some crude estimate regarding the level of accuracy of our method with respect to those achieved by other studies of proteins. With the exception of snRNA-binding proteins, the range of accuracy for the prediction of each class of RNA-binding proteins from our study is from 79.3% to 97.8%, which is comparable to or better than the level of accuracy obtained from other SVM studies of proteins (Bock and Gough 2001; Ding and Dubchak 2001; Cai et al. 2002a,b, 2003a).
As a statistical learning method, a sufficient number of samples is needed in order to properly train and test an SVM classification system. Our analysis of SVM classification of a number of protein families (Cai et al. 2003a) suggested that protein classification accuracy is significantly reduced if the number of protein sequences in the positive training set is substantially less than 80–100. Fewer samples in a positive training set tend to be less adequate in representing all types of proteins in a class. As described below, this imbalance also helps to compromise the ability of SVM classification by increasing the imbalance between the number of samples in the positive and negative training sets (for protein classification there are typically hundreds or more samples in the negative training set due to the large number of protein families). The total number of available snRNA-binding protein sequences is only 60, from which a very small training set of 33 sequences was generated in the present study. It is thus not surprising to find that the prediction accuracy for this RNA-binding class is at a very low level of 40%, in contrast to the level of 79.3%–97.8% for other RNA-binding classes.
The prediction accuracy for each group of non-RNA-binding proteins appears to be better than that for the corresponding group of RNA-binding proteins. The higher prediction accuracy for non-RNA-binding proteins likely results from the availability of a sufficiently diverse set of non-RNA-binding proteins compared to that of RNA-binding proteins, which enables SVM to perform better statistical learning for recognition of non-RNA-binding proteins. Based on the statistics provided on the Web page of the Pfam database (Bateman et al. 2002), there are more than 5000 families of proteins, from which one can generate a diverse set of non-RNA-binding proteins.
Examples of the predicted true positive, false negative, true negative, and false positive protein sequences and their host species for each class are provided in Table 2 ▶. The host species of some protein sequences are not given in Table 2 ▶, because the relevant information is not yet available in the protein sequence database. There is no statistically significant number of incorrectly predicted proteins in one species.
TABLE 2.
Examples of the predicted true positive (TP), true negative (TN), false positive (FP), false negative (FN) protein sequences and host species of different RNA-binding classes
| Protein class | Prediction category | Example of predicted protein (host species) |
| RNA-binding | TP | 30S ribosomal protein S3 (Anaeroplasma abactoclasticum) |
| 30S ribosomal protein S4 (Chlorobium tepidum) | ||
| 30S ribosomal protein S5 (Shewanella oneidensis) | ||
| 30S ribosomal protein S11 (Mycoplasma penetrans) | ||
| 30S ribosomal protein S12 (Leptospira interrogans) | ||
| Matrix protein M1 (Influenza A virus [strain A/Bangkok/1/79], Influenza A virus [strain A/Wilson-Smith/33]) | ||
| Methionyl-tRNA synthetase | ||
| Nonstructural RNA-binding protein 53 (Simian 11 rotavirus [serotype 3/strain SA11-Patton]) | ||
| Ribonuclease P protein component (Bifidobacterium longum) | ||
| Transactivating regulatory protein (Bovine immunodeficiency virus [isolate 106]) | ||
| TN | DNA-binding 11 kDa phosphoprotein (Vaccinia virus [strain Copenhagen]) | |
| DNA polymerase V (Schizosaccharomyces pombe) | ||
| Hypothetical AL4 protein (Indian cassava mosaic virus) | ||
| Mating type protein mtA-1 (Sordaria fimicola) | ||
| NGFI-A binding protein 1 (Mus musculus) | ||
| Nonstructural protein 2 (Human coronavirus [strain OC43]) | ||
| Nucleolar phosphoprotein p130 (Homo sapiens) | ||
| Virulence-associated V antigen (Yersinia pestis) | ||
| XAP-5 protein (Homo sapiens) | ||
| FP | Arsenate reductase (Acidiphilium multivorum) | |
| Delta-atracotoxin-Hv1b (Hadronyche versuta) | ||
| DNA-binding protein HU 2 (Bacillus subtilis) | ||
| Cytochrome c oxidase polypeptide VIII, mitochondrial precursor (Candida albicans) | ||
| Elongation factor 1-beta (Methanobacterium thermoautotrophicum) | ||
| Hypothetical protein AF1917 (Archaeoglobus fulgidus) | ||
| Hypothetical protein HP0309 | ||
| Hypothetical protein PH0461 (Pyrococcus horikoshii) | ||
| Hypothetical protein yhbY (Escherichia coli) | ||
| Insecticidal toxin fragment | ||
| Nerve growth factor fragment | ||
| Nitrogenase GLNBA subunit | ||
| Prefoldin subunit 6 (Homo sapiens) | ||
| Putative cell surface protein homolog | ||
| Putative nucleolar protein K01G5.5 (Caenorhabditis elegans) | ||
| Putative MUDRA-like RETROTRANSPOSON-associated protein | ||
| Ribosomal protein L13A fragment | ||
| Zinc finger protein 263 (Homo sapiens) | ||
| FN | 2′,5′-oligoadenylate synthetase-like 11 | |
| 30S ribosomal protein S7P fragment (Methanosarcina thermophila) | ||
| 30S ribosomal protein S6, chloroplast precursor fragment | ||
| Bicoid protein fragment | ||
| Coat protein (Bacteriophage Q-beta) | ||
| Hypothetical 56.7 kD protein | ||
| Matrix protein M1 fragment (Influenza A virus [strain A/Camel/Mongolia/82]) | ||
| Putative heterogeneous nuclear ribonucleoprotein X fragment | ||
| RNA helicase DbpA | ||
| U2 small nuclear ribonucleoprotein 40K | ||
| rRNA-binding | TP | 30S ribosomal protein S1 (Escherichia coli, Helicobacter pylori J99, Mycobacterium tuberculosis) |
| 30S ribosomal protein S3 (Anaeroplasma abactoclasticum) | ||
| 30S ribosomal protein S4 (Chlorobium tepidum, Shigella flexneri) | ||
| 30S ribosomal protein S5 (Shewanella oneidensis) | ||
| 30S ribosomal protein S7 (Rhodobacter capsulatus) | ||
| 30S ribosomal protein S20 fragment | ||
| 30S ribosomal protein S12 fragment | ||
| 50S ribosomal protein L2 (Aquifex pyrophilus) | ||
| 50S ribosomal protein L3 (Aquifex pyrophilus) | ||
| TN | Apolipoprotein B mRNA editing enzyme (Rattus norvegicus) | |
| Aspartoacylase (Bos taurus) | ||
| DNA-directed RNA polymerase III 80 kD polypeptide (Mus musculus) | ||
| DNA polymerase V (Schizosaccharomyces pombe) | ||
| Hypothetical protein MG248 homolog (Mycoplasma pneumoniae) | ||
| Membrane protein C21orf4 (Homo sapiens) | ||
| Mitochondrial 24 kD protein (Zea mays) | ||
| Probable RNA 3′-terminal phosphate cyclase (Methanobacterium thermoautotrophicum) | ||
| RNA-binding protein VP2 (Bovine rotavirus [strain RF]) | ||
| RNA polymerase transcriptional regulation mediator, subunit 6 homolog (Homo sapiens) | ||
| FP | Bcn92 protein (Drosophila melanogaster) | |
| Cell division topological specificity factor (Escherichia coli) | ||
| DNA-directed RNA polymerases I, II, and III 7.0 kD polypeptide (Homo sapiens) | ||
| DNA repair protein radC homolog (Aquifex aeolicus) | ||
| GyrA fragment | ||
| Hypothetical protein AQ_1922 (Aquifex aeolicus) | ||
| Hypothetical protein C24H6.02c in chromosome I (Schizosaccharomyces pombe) | ||
| Hypothetical protein Rv2842c (Mycobacterium tuberculosis) | ||
| Imidazoleglycerol-phosphate dehydratase [Archaeoglobus fulgidus] | ||
| Photosystem I reaction center subunit IV, chloroplast precursor (Chlamydomonas reinhardtii) | ||
| Putative RNA binding protein KOC | ||
| RlpA-like lipoprotien precursor (Aquifex aeolicus) | ||
| FN | 30S ribosomal protein S6, chloroplast precursor fragment | |
| 50S ribosomal protein L23 (Aquifex pyrophilus) | ||
| 50S ribosomal protein L4 (Aquifex pyrophilus) | ||
| Chloroplast 50S ribosomal protein L1 fragment | ||
| Chloroplast 50S ribosomal protein L29 fragment | ||
| mRNA-binding | TP | 30S ribosomal protein S1 (Escherichia coli, Helicobacter pylori J99, Mycobacterium tuberculosis) |
| 30S ribosomal protein S3 (Acholeplasma axanthum, Acholeplasma sp. [strain ATCC J233], Alder yellows phytoplasma, Aquifex aeolicus, Bacillus halodurans) | ||
| 30S ribosomal protein S4 (Chlorobium tepidum, Shigella flexneri) | ||
| Cap specific mRNA (nucleoside-2′-O)-methyltransferase (Variola virus, Swinepox virus [strain Kasza]) | ||
| Eukaryotic translation initiation factor 3 subunit 4 (Homo sapiens, Mus musculus, Arabidopsis thaliana, Medicago truncatula) | ||
| Fertility inhibition protein | ||
| Fragile X mental retardation protein 1 homolog | ||
| Heterogeneous nuclear ribonucleoprotein D0 (Homo sapiens) | ||
| Heterogeneous nuclear ribonucleoprotein F (Homo sapiens) | ||
| Interleukin enhancer-binding factor 3 (Mus musculus) | ||
| Iron-responsive element binding protein 2 (Homo sapiens) | ||
| Maternal exuperantia protein | ||
| Polyadenylate-binding protein 1 (Homo sapiens) | ||
| Pyrimidine operon regulatory protein | ||
| PyrR bifunctional protein (Bacillus caldolyticus, Clostridium acetobutylicum, Listeria monocytogenes, Thermoanaerobacter tengcongensis) | ||
| Pre-mRNA splicing factor PRP9 (Candida albicans) | ||
| Splicing factor 3A subunit 3 (Drosophila melanogaster) | ||
| Splicing factor SC35 | ||
| Transcription termination factor rho | ||
| U1 small nuclear ribonucleoprotein A (Drosophila melanogaster) | ||
| TN | 3-deoxy-D-manno-octulosonic acid kinase (Pasteurella multocida) | |
| Adenosylhomocysteinase (Sulfolobus tokodaii) | ||
| Decarboxylase DEC1 (Cochliobolus heterostrophus) | ||
| DNA replication terminus site-binding protein | ||
| DNA terminal protein | ||
| Holliday junction DNA helicase ruvB (Staphylococcus aureus [strain Mu50/ATCC 700699]) | ||
| rRNA processing protein EBP2 (Candida albicans) | ||
| Transcription factor-like protein MORF4 (Homo sapiens) | ||
| Virion membrane protein FPV182 (Fowlpox virus) | ||
| FP | 40S ribosomal protein S25 (Arabidopsis thaliana) | |
| 50S ribosomal protein L22 (Mycoplasma gallisepticum) | ||
| Cell division protein ftsB homolog (Xanthomonas axonopodis [pv. citri]) | ||
| DNA mismatch repair protein MutS fragment | ||
| Hypothetical protein TC0713 (Chlamydia muridarum) | ||
| Hypothetical protein yjiX (Escherichia coli) | ||
| Hypothetical protein in LEU2 3′ region fragment (Pichia angusta) | ||
| Opioid growth factor receptor (Homo sapiens) | ||
| Putative metal-dependent hydrolase | ||
| Putative transition state regulator abh (Bacillus subtilis) | ||
| Squamosa-promoter binding protein 1 (Antirrhinum majus) | ||
| Trimethylamine methyltransferase mttB (Methanosarcina barkeri) | ||
| FN | 30S ribosomal protein S1 (Rickettsia prowazekii) | |
| Cap specific mRNA (Capripoxvirus [strain KS-1]) | ||
| Double-stranded RNA-binding protein Staufen homolog (Homo sapiens) | ||
| Eukaryotic translation initiation factor 3 RNA-binding subunit (Candida albicans) | ||
| Heterogenous nuclear ribonucleoprotein U (Homo sapiens) | ||
| Polyadenylate-binding protein 5 (Homo sapiens) | ||
| Putative eukaryotic translation initiation factor 3 subunit 7 (Caenorhabditis elegans) | ||
| U1 small nuclear ribonucleoprotein A (Candida albicans) | ||
| U6 snRNA-associated Sm-like protein LSm4 (Candida albicans) | ||
| tRNA-binding | TP | 30S ribosomal protein S7 (Haemophilus ducreyi, Chlamydia pneumoniae, Vibrio cholerae, Campylobacter jejuni) |
| 30S ribosomal protein S12 (Spirulina platensis, Mycobacterium gordonae, Leptospira interrogans, Streptococcus mutans, Bartonella henselae) | ||
| 60S ribosomal protein L35a | ||
| Methionyl-tRNA synthetase (Xanthomonas campestris, [pv. campestris], Pyrococcus furiosus) | ||
| Multisynthetase complex auxiliary component p43 | ||
| Phenylalanyl-tRNA synthetase beta chain (Chlamydia pneumoniae, Rickettsia prowazekii) | ||
| Zipcode-binding protein | ||
| TN | 4-hydroxythreonine-4-phosphate dehydrogenase (Sphingomonas aromaticivorans) | |
| Capsid protein VP26 | ||
| DNA repair protein RAD9 (Schizosaccharomyces octosporus) | ||
| Histone deacetylase HST1 (Candida albicans) | ||
| Putative RNA-directed RNA polymerase (Avian infectious bursal disease virus [strain Australian 002-73]) | ||
| Single-stranded DNA-binding protein 2 (Homo sapiens) | ||
| TAP42 protein (Candida albicans) | ||
| Transport protein particle 20 kD subunit (Candida albicans) | ||
| Zinc finger protein Rp-8 (Mus musculus) | ||
| FP | 60S ribosomal protein L18 fragment (Cicer arietinum) | |
| 40S ribosomal protein S26 (Schizophyllum commune) | ||
| SsrA-binding protein (Bacillus subtilis) | ||
| Cytochrome c oxidase polypeptide VIc-1 (Rattus norvegicus) | ||
| Thiamine biosynthesis protein, putative | ||
| FN | Phenylalanyl-tRNA synthetase beta chain (Ureaplasma parvum, Deinococcus radiodurans) | |
| Probable methionyl-tRNA synthetase (Oryza sativa) | ||
| snRNA-binding | TP | Octamer-binding transcription factor I (Homo sapiens, Sus scrofa) |
| U1 small nuclear ribonucleoprotein A (Homo sapiens) | ||
| U1 small nuclear ribonucleoprotein 70 kD (Drosophila melanogaster, Xenopus laevis) | ||
| U2 small nuclear ribonucleoprotein A′ (Mus musculus) | ||
| U6 snRNA-associated Sm-like protein LSm4 (Fagus sylvatica, Oryza sativa, Candida albicans) | ||
| TN | Acetylglutamate kinase | |
| DNA binding protein S1FA (Arabidopsis thaliana) | ||
| DNA mismatch repair protein mutS (Vibrio vulnificus) | ||
| DNA-directed RNA polymerase beta′ chain (Porphyra purpurea) | ||
| Guanine nucleotide exchange factor MSS4 homolog (Drosophila melanogaster) | ||
| Glutaminyl-tRNA synthetase (Lupinus luteus) | ||
| Heme/hemopexin-binding protein precursor (Haemophilus influenzae) | ||
| Nonstructural protein NS2 | ||
| Probable arginyl-tRNA—protein transferase (Xylella fastidiosa) | ||
| RNA polymerase sigma-54 factor (Salmonella typhimurium) | ||
| FP | CG1622 protein | |
| CG17446 protein | ||
| F46B6.3b protein | ||
| Heparan sulfate 2-sulfotransferase fragment | ||
| Homoserine dehydrogenase fragment | ||
| Hypothetical 44.5 kD protein | ||
| Hypothetical protein BH2667 (Bacillus halodurans) | ||
| Hypothetical protein PYRAB10580 (Pyrococcus abyssi) | ||
| Hypothetical protein spyM18_1551 | ||
| MiaE fragment | ||
| ORF FPV166 Molluscum contagiosum virus MC105L homolog | ||
| US3ii/US3iv protein | ||
| FN | 80 kD nuclear cap binding protein NCBP 80 kD subunit (Homo sapiens) | |
| Hypothetical 28.9 kD protein (Caenorhabditis elegans) | ||
| NHP2-like protein 1 (Homo sapiens) | ||
| Probable U6 snRNA-associated Sm-like protein LSm3 (Schizosaccaromyces pombe) | ||
| Small nuclear ribonucleoprotein E (Candida albicans) | ||
| Small nuclear ribonucleoprotein D1 homolog (Candida albicans) | ||
| U6 snRNA-associated Sm-like protein LSm2 (Homo sapiens) | ||
| U6 snRNA-associated Sm-like protein LSm3 (Candida albicans) | ||
| U6 snRNA-associated Sm-like protein LSm6 (Homo sapiens) | ||
| Zinc finger protein 143 (SPH-binding factor) (Homo sapiens) |
Only proteins in the independent evaluation sets are included. Host species of some protein sequences are not provided because the relevant information is not yet available in the protein sequence database.
Inspection of individual misclassified protein sequences of different RNA-binding and non-RNA-binding classes, including those false negatives and false positives in Table 2 ▶, shows that a significant portion of these sequences are either a protein fragment or described as hypothetical, probable, or putative. Sequence incompleteness likely contributes to some of the prediction errors in this work. Many of the hypothetical, probable, and putative proteins are so described primarily based on some form of distant sequence similarity relationship with existing proteins of known functions. Our earlier study of SVM classification of protein families suggested that prediction accuracy for distantly related proteins is substantially lower than those of closely related proteins (Cai et al. 2003a). It is thus possible that the prediction error for some of the sequences in this work may be partly due to their low sequence similarity to other protein sequences in the same class.
A substantial number of incorrectly predicted protein sequences in each non-RNA-binding class, some of which are shown in Table 2 ▶, are DNA-binding proteins and proteins of other RNA-binding classes. Because of the certain degree of common structural features among different classes of ssRNAs and between dsRNAs and dsDNAs, some RNA-binding proteins and DNA-binding proteins might share a certain degree of common structural features that makes it more difficult for a statistical classification system such as SVM to unambiguously distinguish the features between these proteins, which likely contributes to a higher prediction error for some of these sequences.
Because of the differences in the number of RNA-binding proteins and that of non-RNA-binding proteins in each class, there is an imbalance between each data set. SVM based on an unbalanced data set tends to produce feature vectors that push the hyperplane towards the side with a smaller number of data (Veropoulos et al. 1999), which can lead to a reduced accuracy for the set either with a smaller number of samples or of less diversity. This might partly explain why the prediction accuracy for RNA-binding proteins is lower than that for non-RNA-binding proteins. It is however inappropriate to simply reduce the size of non-RNA-binding proteins to artificially match that of RNA-binding proteins, because this compromises the diversity needed to fully represent all non-RNA-binding proteins. Computational methods for re-adjusting a biased shift of hyperplane have been introduced (Brown et al. 2000). Application of these methods may help improving SVM prediction accuracy in this and other cases involving unbalanced data.
Classification of proteins with specific characteristics
A number of RNA-binding proteins have a modular structure and contain RNA-binding domains of 70–150 amino acids that mediate RNA recognition (Mattaj 1993; Perez-Canadillas and Varani 2001). Three classes of RNA-binding domains have been documented to bind RNA in a sequence-independent manner: These domains are RNA-recognition motif (RRM), double-stranded RNA-binding motif (dsRM), and K-homology (KH) domain (Perez-Canadillas and Varani 2001). A fourth class of RNA-binding domain, S1 RNA-binding domain, has also been found in a number of RNA-associated proteins (Bycroft et al. 1997). These domains have distinguished structural features responsible for RNA recognition and binding. Thus the performance of SVM classification of RNA-binding proteins can be evaluated by examining whether or not proteins containing one of these domains can be correctly classified as RNA-binding proteins.
A search of protein family and sequence databases shows that there are a total of 260, 74, 190, and 41 RNA-binding protein sequences known to contain the RRM, dsRM, KH, and S1 RNA-binding domain, respectively. The majority of these sequences are included in the training and testing set of all RNA-binding proteins. In the corresponding independent evaluation set, there are 35, 16, 93, and 10 sequences containing the RRM, dsRM, KH, and S1 RNA-binding domain, respectively. The prediction status and examples of these protein sequences are given in Table 3 ▶. All but one protein sequence are correctly classified as RNA-binding by SVM, which shows the capability of our trained SVM classification system. The only incorrectly predicted protein sequence is HnRNP-E2 protein fragment in the group that contains KH domain. The incompleteness of this sequence might partially contribute to its incorrect prediction by SVM.
TABLE 3.
Predication statistics, examples, and host species of RNA-binding protein sequences known to contain one of the RNA-recognition motif (RRM), double-stranded RNA-binding motif (dsRM), K-homology (KH), and S1 RNA-binding domain
| RNA-binding proteins known to contain domain | ||||
| RNA-binding domain | Number of RNA-binding proteins with domain | Number of proteins correctly predicted as RNA-binding | Example of correctly predicted protein (host species) | Prediction accuracy (%) |
| RRM | 35 | 35 | CUG triplet repeat RNA-binding protein 1 (Homo sapiens) | 100% |
| ELAV-like protein (Mus musculus) | ||||
| ELAV-like protein 4 (Homo sapiens, Rattus norvegicus) | ||||
| Heterogeneous nuclear ribonucleoprotein A1 (Mus musculus) | ||||
| Heterogeneous nuclear ribonucleoprotein A3 (Homo sapiens, Xenopus laevis) | ||||
| Heterogeneous nuclear ribonucleoprotein H (Homo sapiens) | ||||
| Matrin 3 (Rattus norvegicus) | ||||
| Nuclear polyadenylated RNA-binding protein NAB4 (Candida albicans) | ||||
| Polypyrimidine tract-binding protein 1 (Rattus norvegicus) | ||||
| RNA-binding protein FUS (Mus musculus) | ||||
| RNA-binding region containing protein 2 (Mus musculus) | ||||
| Splicing factor, arginine/serine-rich 4 (Mus musculus) | ||||
| Splicing factor, arginine/serine-rich 5 (Homo sapiens) | ||||
| Splicing factor U2AF 65 kD subunit (Mus musculus, Caenorhabditis elegans) | ||||
| dsRM | 16 | 16 | ATP-dependent RNA helicase A (Bos taurus) | 100% |
| Interleukin enhancer-binding factor 3 (Mus musculus, Rattus norvegicus) | ||||
| Ribonuclease III (Escherichia coli, Ralstonia solanacearum, Brucella melitensis, Salmonella typhi, Yersinia pestis, Rhizobium meliloti, Staphylococcus aureus [strain N315], Neisseria meningitidis [serogroup A], Neisseria meningitidis [serogroup B], Chlamydia muridarum, Helicobacter pylori J99) | ||||
| SON protein (Mus musculus) | ||||
| KH | 94 | 93 | 30S ribosomal protein S3 (Mycobacterium bovis, Escherichia coli, Mycoplasma pneumoniae, Buchnera aphidicola [subsp. Acyrthosiphon kondoi], Acholeplasma florum, Buchnera aphidicola [subsp. Acyrthosiphon pisum], Synechocystis sp. [strain PCC 6803], Thermus thermophilus, Phytoplasma sp. [strain STRAWB2], Mycoplasma capricolum, Acholeplasma sp. [strain ATCC J233], Fusobacterium nucleatum [subsp. nucleatum], etc.) | 98.9% |
| A kinase anchor protein 1 (Homo sapiens, Mus musculus) | ||||
| GTP-binding protein era homolog (Streptococcus pyogenes [serotype M3], Streptococcus pneumoniae, Fusobacterium nucleatum [subsp. nucleatum], Clostridium perfringens, Anabaena sp. [strain PCC 7120], Mycoplasma pulmonis, Staphylococcus aureus [strain Mu50/ATCC 700699], Neisseria meningitidis [serogroup A], Neisseria meningitidis [serogroup B], Bacillus halodurans, Lactococcus lactis [subsp. lactis], Helicobacter pylori J99) | ||||
| Hypothetical UPF0109 protein TC0030 (Chlamydia muridarum) | ||||
| N utilization substance protein A homolog (Bacillus halodurans, Rickettsia conorii) | ||||
| Poly(rC)-binding protein 1 (Oryctolagus cuniculus) | ||||
| Poly(rC)-binding protein 2 (Homo sapiens) | ||||
| Poly(rC)-binding protein 3 (Mus musculus) | ||||
| Poly(rC)-binding protein 4 (Mus musculus) | ||||
| Polyribonucleotide nucleotydyltransferase (Bacillus subtilis, Buchnera aphidicola [subsp. Schizaphis graminum]) | ||||
| Probable exosome complex RNA-binding protein 1 (Methanosarcina mazei, Thermoplasma acidophilum, Pyrococcus abyssi) | ||||
| Heterogeneous nuclear ribonucleoprotein K (Oryctolagus cuniculus) | ||||
| Vigilin (Gallus gallus) | ||||
| Zipcode-binding protein 2 (Gallus gallus) | ||||
| S1 RNA-binding domain | 10 | 10 | 30S ribosomal protein S1 (Chlamydia trachomatis, Chlamydia pneumoniae) | 100% |
| Eukaryotic translation initiation factor 2 (Rattus norvegicus) | ||||
| N utilization substance protein A homolog (Buchnera aphidicola [subsp. Schizaphis graminum]) | ||||
| Probable translation initiation factor 2 alpha subunit (Methanopyrus kandleri, Pyrococcus furiosus, Sulfolobus tokodaii, Pyrococcus abyssi) | ||||
| Ribonuclease E (Buchnera aphidicola [subsp. Schizaphis graminum]) | ||||
Only those RNA-binding proteins in the independent evaluation sets are included. Host species of some protein sequences are not provided because the relevant information is not yet available in the protein sequence database. The only incorrectly predicted protein sequence with KH domain is HnRNP-E2 protein fragment.
Some proteins bind to RNA in a primarily sequence-specific manner. Typical examples are ribosomal proteins (Draper and Reynaldo 1999) and a U8 snoRNA-specific binding protein (Tomasevic and Peculis 1999). The majority of the ribosomal protein entries are correctly predicted as rRNA-binding proteins. Inspection of the ribosomal protein entries that are incorrectly predicted as a non-rRNA-binding protein shows that some of these entries are protein fragment and some are described as hypothetical, probable, or putative. It is possible that the prediction error for some of these sequences may be partly due to sequence incompleteness or low sequence similarity to those of other protein sequences in each class. Some ribosomal proteins are known to bind to mRNA and tRNA as well as rRNA; examples of these proteins are 30S ribosomal proteins S1, S3, S4. The multiple binding nature of these proteins likely makes it more difficult for a statistical classification system such as SVM to unambiguously distinguish the features between rRNA-binding, mRNA-binding, and tRNA-binding, which is another possible reason for the inaccurate classification of these sequences.
Some proteins, such as dihydrofolate reductase and thymidylate synthase, are known to bind to their own mRNA (Zhang and Rathod 2002). Not all of these proteins are listed as RNA-binding proteins in protein sequence databases. As a result, these mRNA-binding proteins may not be included in the right protein group, which probably affects prediction accuracy for these proteins. Hence, additional work is needed to search for these proteins and include them in the group of mRNA-binding proteins.
Contribution of feature properties to the classification of RNA-binding proteins
In this work, a total of nine feature properties was used to describe physicochemical characteristics of each protein, which have been routinely used in previous studies of proteins (Bock and Gough 2001; Ding and Dubchak 2001; Cai et al. 2002a,b, 2003a). It has been reported that not all feature vectors contribute equally to the classification of proteins; some have been found to play a relatively more prominent role than others in specific aspects of proteins (Ding and Dubchak 2001). It is therefore of interest to examine which feature properties play more prominent roles in the classification of RNA-binding proteins.
In an earlier study, the contribution of individual feature properties to protein classification was investigated by conducting classifications using each feature property separately (Ding and Dubchak 2001). The same method was employed here. An analysis of the classification of the group of all RNA-binding proteins seemed to suggest that, in order of prominence, the amino acid composition, charge, polarity, and hydrophobicity play more prominent roles than the other feature properties examined. Amino acid composition and hydrophobicity are important factors for the interaction of a protein with other biomolecules, as well as for structural folding. On the other hand, charge and polarity are important for electrostatic interactions and hydrogen-bonding to RNA. As the backbone of RNA is charged, charge and polarity are expected to be particularly important feature properties for the binding of a protein with its RNA-substrate. A study of the dynamics of protein–RNA interfaces showed that cations condensed around RNA affect the binding of protein to RNA (Hermann and Westhof 1999), which is indicative of the strong effect of charges and polarity.
Conclusion
SVM appears to be a potentially useful tool for the prediction of various RNA-binding proteins. The prediction accuracy may be further enhanced with the improvement of SVM algorithms, particularly for unbalanced data sets and with expanded knowledge about RNA-binding proteins. The SVM RNA-binding protein classification systems developed in this work have been added to our Web-based protein functional classification software SVMProt (Cai et al. 2003a) which is accessible at http://jing.cz3.nus.edu.sg/cgi-bin/svmprot.cgi. Thus, SVMProt may be used as one of the Web-based tools in facilitating the prediction of RNA-binding proteins as well as proteins of other functional classes.
MATERIALS AND METHODS
Support vector machine
The theory of SVM has been extensively described in the literature (Vapnik 1995; Burges 1998; Evgeniou and Pontil 2001). Thus only a brief description is given here. SVM is based on the structural risk minimization (SRM) principle from statistical learning theory (Vapnik 1995). In linearly separable cases, SVM constructs a hyperplane that separates two different classes of feature vectors. A feature vector represents the structural and physicochemical properties of a protein. There are a number of hyperplanes for an identical group of training data. The classification objective of SVM is to separate the training data with a maximum margin while maintaining reasonable computing efficiency. This is done by finding another vector w and a parameter b that minimizes ∥w∥2 and satisfies the following conditions:
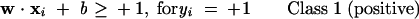 |
(1) |
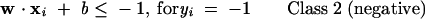 |
(2) |
In this study, a feature vector corresponds to a protein, and this vector is represented by xi with protein descriptors as its components, yi is the class index, w is a vector normal to the hyperplane, |b|/∥w∥ is the perpendicular distance from the hyperplane to the origin, and ∥w∥2 is the Euclidean norm of w. After the determination of w and b, a given vector xi can be classified by:
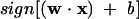 |
(3) |
The hyperplane determined by w0 and b0 is called optimal separating hyperplane (OSH).
In nonlinearly separable cases, SVM maps the input variable into a high-dimensional feature space using a kernel function K(xi, xj) followed by the construction of OSH in the feature space. An example of a kernel function is the Gaussian kernel, which is frequently used by others (Burbidge et al. 2001; Czeminski et al. 2001):
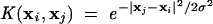 |
(4) |
Earlier studies have indicated that the Gaussian kernel consistently gives better results than other kernel functions (Ding and Dubchak 2001; Cai et al. 2002b). Hence the Gaussian kernel function was used in the present work. Linear SVM is applied to this feature space, and then the decision function is given by:
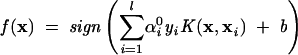 |
(5) |
where the coefficients αi0 and b are determined by maximizing the following Langrangian expression:
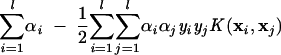 |
(6) |
under the following conditions:
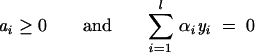 |
(7) |
Positive or negative value from Eq. 3 or Eq. 5 indicates that the vector x belongs to the positive or negative class, respectively. To further reduce the complexity of parameter selection, hard-margin SVM with a threshold instead of soft-margin SVM with a threshold was used in our own SVM program SVM★ (Cai et al. 2003b). A soft margin is introduced by adding a constraint on αi to simultaneously reduce the training error and maximize the margin (Vapnik 1995). A hard margin is under the condition that 0≤αi∞.
As in the case of all discriminative methods (Baldi et al. 2000; Roulston 2002), the performance of SVM classification can be measured by the quantity of true positives (TP), true negatives (TN), false positives (FP), false negatives (FN), sensitivity, SE = TP/(TP + FN), specificity, SP = TN/(TN + FP), and the overall accuracy (Q) given below:
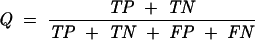 |
(8) |
Selection of RNA-binding proteins and non-RNA-binding proteins
All RNA-binding proteins used in this study are from a comprehensive search of the Swissprot database at http://www.expasy.ch/sprot (Bairoch and Apweiler 2000). A total of 4458 RNA-binding protein sequences were obtained, which include 2054 rRNA-, 570 mRNA-, 259 tRNA-, 60 snRNA-, and 21 snoRNA-binding proteins. The distribution of RNA-binding proteins in different kingdoms and in the top 10 host species is given in Table 4 ▶, and that of each class of RNA-binding proteins is given in Table 5 ▶. From these two tables one finds that these proteins are from a diverse range of species, and all species appear to be fairly adequately represented.
TABLE 4.
Distribution of RNA-binding proteins in different kingdoms and in top 10 host species of each kingdom
| Kingdom | Eucaryote | Eubacteria | Archaea |
| Number of proteins in kingdom | 986 | 1854 | 294 |
| List of top 10 species and number of proteins in each species | Homo sapiens (168) | Escherichia coli (75) | Methanococcus jannaschii (22) |
| Mus musculus (78) | Bacillus subtilis (64) | Methanobacterium thermoautotrophicum (21) | |
| Candida albicans (77) | Haemophilus influenzae (60) | Archaeoglobus fulgidus (20) | |
| Schizosaccharomyces pombe (52) | Buchnera aphidicola (subsp. Acyrthosiphon pisum) (50) | Halobacterium sp. (19) | |
| Drosophila melanogaster (45) | Helicobacter pylori (49) | Pyrococcus horikoshii (19) | |
| Arabidopsis thaliana (42) | Buchnera aphidicola (subsp. Schizaphis graminum) (47) | Pyrococcus abyssi (18) | |
| Xenopus laevis (30) | Aquifex aeolicus (45) | Sulfolobus solfataricus (18) | |
| Rattus norvegicus (28) | Mycobacterium tuberculosis (45) | Aeropyrum pernix (18) | |
| Caenorhabiditis elegans (26) | Rickettsia prowazekii (44) | Methanopyrus kandleri (15) | |
| Porphyra purpurea (19) | Mycoplasma pneumoniae (43) | Thermoplasma volcanium (14) |
Not all protein sequences studied in this work are included because the host species information of some protein sequences is not yet available in the protein sequence database. Moreover, there are 108 viral RNA-binding proteins used in this work.
TABLE 5.
Distribution of rRNA-, mRNA-, tRNA- and snRNA-binding proteins in different kingdoms and in top 10 host species
| rRNA-binding | mRNA-binding | tRNA-binding | snRNA-binding | |||||
| kingdom or species | no. of proteins | kingdom or species | no. of proteins | kingdom or species | no. of proteins | kingdom or species | no. of proteins | |
| Protein distribution in kingdom | Eucaryote | 493 | Eucaryote | 310 | Eucaryote | 19 | Eucaryote | 50 |
| Eubacteria | 1330 | Eubacteria | 235 | Eubacteria | 230 | Eubacteria | – | |
| Archaea | 181 | Archaea | – | Archaea | 10 | Archaea | – | |
| Protein distribution in top 10 species | Thermus thermophilus | 32 | Homo sapiens | 77 | Thermus thermophilus | 6 | Homo sapiens | 18 |
| Aquifex aeolicus | 29 | Candida albicans | 41 | Homo sapiens | 5 | Candida albicans | 15 | |
| Mycobacterium leprae | 28 | Mus musculus | 36 | Bacillus subtilis | 5 | Mus musculus | 5 | |
| Chlamydia pneumoniae | 28 | Schizosaccharomyces pombe | 21 | Escherichia coli | 5 | Xenopus laevis | 3 | |
| Helicobacter pylori | 28 | Escherichia coli | 21 | Pasteurella multocida | 4 | Drosophila melanogaster | 3 | |
| Rickettsia prowazekii | 28 | Arabidopsis thaliana | 19 | Mycoplasma genitalium | 4 | Schizosaccharomyces pombe | 3 | |
| Thermotoga maritima | 28 | Caenorabditis elegans | 18 | Deinococcus radiodurans | 4 | Caenorhabditis elegans | 2 | |
| Chlamydia trachomatis | 28 | Drosophila melanogaster | 15 | Neisseria meningitidis (serogroup A) | 4 | Rattus norvegicus | 2 | |
| Borrelia burgdorferi | 28 | Rattus norvegicus | 14 | Helicobacter pylori | 4 | Arabidopsis thaliana | 2 | |
| Buchnera aphidicola | 28 | Nicotiana tabacum | 11 | Campylobacter jejuni | 4 | Macropus eugenii | 1 | |
Not all protein sequences studied in this work are included because the host species information of some protein sequences is not yet available in the protein sequence database.
Not all of the protein sequences in each of the above-described five RNA-binding classes are specified as such in the protein sequence database. An effort was made to manually check all of the selected RNA-binding protein sequences to determine whether or not some of them belong to each of the five classes. It is expected that some of these proteins may not be selected and thus not included in each class. However, these proteins were included in the all RNA-binding protein class. The number of known snRNA- and snoRNA-binding proteins is significantly smaller than those in the other groups (Tomasevic and Peculis 1999; Singh 2002), and it is substantially below the number of 80–100 sequences needed to properly train an SVM protein classification system (Cai et al. 2003a). Hence, at present, SVM is expected to be useful only for classification of rRNA-, mRNA-, and tRNA-binding proteins, respectively, as well as for all RNA-binding proteins as a single group. Nevertheless, to evaluate its performance on classification of a small protein class, SVM was applied to the prediction of snRNA-binding proteins.
All distinct members in each group were used to construct positive samples for training, testing, and independent evaluation of the SVM classification system. The negative samples for training and testing were selected from seed proteins of the curated protein families in the Pfam database (Bateman et al. 2002) excluding those that belong to the group of RNA-binding proteins under study. For each group of non-rRNA-, non-mRNA-, non-tRNA-, and non-snRNA-binding proteins, distinct members in the other three groups were added to the negative samples of each of the training, testing, and independent evaluation sets. For instance, distinct members of mRNA-, tRNA-, and snRNA-binding proteins were added to the negative samples of the non-rRNA-binding proteins. It is expected that the number of negative samples in each of these three groups may be higher than that in the group of negative samples for all RNA-binding proteins.
Training sets of both positive and negative samples were further screened so that only essential proteins that optimally represent each family were retained. The SVM training system for each group was optimized and tested by using separate testing sets of both positive and negative samples composed of all of the remaining distinct proteins of a group and those outside the group, respectively. The performance of SVM classification was further evaluated by using independent sets of both positive and negative samples composed of all of the remaining proteins of a group and those outside the group, respectively. No duplicate protein was used in the training, testing, or independent evaluation set for each group. For those with a sufficient number of distinct members, multiple entries were assigned to each set. For those with less than three distinct members, the proteins were assigned in the order of priority of training, testing, and independent evaluation set.
The number of positive and negative samples for each of the training, testing, and independent evaluation sets for each group of RNA-binding proteins is given in Table 1 ▶. The training set was composed of 708 rRNA-binding and 972 non-rRNA-binding proteins, 277 mRNA-binding and 2106 non-mRNA-binding proteins, 94 tRNA-binding and 792 non-tRNA-binding proteins, 33 snRNA-binding proteins and 1988 non-snRNA-binding proteins, and 2161 RNA-binding proteins and 2965 non-RNA-binding proteins. The testing set was comprised of 1245 rRNA-binding and 9044 non-rRNA-binding proteins, 129 mRNA-binding and 10164 non-mRNA-binding proteins, 114 tRNA-binding and 9297 non-tRNA-binding proteins, and 1850 RNA-binding proteins and 6816 non-RNA-binding proteins. The independent evaluation set was made of 101 rRNA-binding and 4997 non-rRNA-binding proteins, 164 mRNA-binding and 6046 non-mRNA-binding proteins, 51 tRNA-binding and 5033 non-tRNA-binding proteins, 20 snRNA-binding and 6151 non-snRNA-binding proteins, and 447 RNA-binding proteins and 4881 non-RNA-binding proteins.
Feature vector construction
Construction of the feature vector for each RNA-binding or non-RNA-binding protein was based on the formula used in the prediction of protein–protein interaction (Bock and Gough 2001), protein fold recognition (Ding and Dubchak 2001), and protein family classification (Cai et al. 2003a). Details of the formula can be found in the respective publications and references therein. Each feature vector was constructed from encoded representations of tabulated residue properties including amino acid composition, hydrophobicity, normalized Van der Waals volume, polarity, polarizability, charge, surface tension, secondary structure, and solvent accessibility.
Three descriptors—composition (C), transition (T), and distribution (D)—were used to describe the global composition of each of these properties (Dubchak et al. 1995). C is the number of amino acids of a particular property (such as hydrophobicity) divided by the total number of amino acids in a protein sequence. T characterizes the percent frequency with which amino acids of a particular property is followed by amino acids of a different property. D measures the chain length within which the first, 25%, 50%, 75%, and 100% of the amino acids of a particular property are located, respectively.
A hypothetical protein sequence AEAAAEAEEAAAAAEAEEE AAEEAEEEAAE, as shown in Figure 1 ▶, has 16 alanines (n1 = 16) and 14 glutamic acids (n2 = 14). The composition for these two amino acids is n1×100.00/(n1 + n2) = 53.33 and n2×100.00/(n1 + n2) = 46.67, respectively. There are 15 transitions from A to E or from E to A in this sequence, and the percent frequency of these transitions is (15/29)×100.00 = 51.72. The first, 25%, 50%, 75%, and 100% of As are located within the first 1, 5, 12, 20, and 29 residues, respectively. The D descriptor for As is thus 1/30 ×100.00 = 3.33, 5/30×100.00 = 16.67, 12/30×100.00 = 40.0, 20/30×100.00 = 66.67, 29/30×100.00 = 96.67. Likewise, the D descriptor for Es is 6.67, 26.67, 60.0, 76.67, 100.0. Overall, the amino acid composition descriptors for this sequence are C = (53.33, 46.67), T = (51.72), and D = (3.33, 16.67, 40.0, 66.67, 96.67, 6.67, 26.67, 60.0, 76.67, 100.0).
FIGURE 1.

The sequence of a hypothetic protein for illustration of derivation of the feature vector of a protein. Sequence index indicates the position of an amino acid in the sequence. The index for each type of amino acids in the sequence (A or E) indicates the position of the first, second, third, ... of that type of amino acid (e.g., the position of the first, second, third, ..., A is at 1, 3, 4, ...). A/E transition indicates the position of AE or EA pairs in the sequence.
Descriptors for other properties can be computed by a similar procedure, and all of the descriptors are combined to form the feature vector. In most studies, amino acids are divided into three classes for each property, and thus the three descriptors for each property consist of 21 elements: three for C, three for T, and 15 for D (Bock and Gough 2001; Karchin et al. 2002; Yuan et al. 2002).
There is some level of overlap in the descriptors for hydrophobicity, polarity, and surface tension. Thus the dimensionality of the feature vectors may be reduced by principle component analysis (PCA). Our own study suggests that the use of PCA-reduced feature vectors only moderately improves the accuracy for some of the families. It is thus unclear to what extent this overlap affects the accuracy of SVM classification. We note that reasonably accurate results have been obtained using these overlapping descriptors in various protein classification studies (Bock and Gough 2001; Ding and Dubchak 2001; Cai et al. 2002a,b, 2003a).
Acknowledgments
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.5890304.
REFERENCES
- Bairoch, A. and Apweiler, R. 2000. The SWISS-PROT protein sequence database and its supplement tremble in 2000. Nucleic Acids Res. 28: 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldi, P., Brunak, S., Chauvin, Y., Andersen, C.A.F., and Nielsen, H. 2000. Assessing the accuracy of prediction algorithms for classification: An overview. Bioinformatics 16: 412–424. [DOI] [PubMed] [Google Scholar]
- Bateman, A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L., Eddy, S.R., Griffiths-Jones, S., Howe, K.L., Marshall, M., and Sonnhammer, E.L. 2002. The Pfam protein families database. Nucleic Acids Res. 30: 276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Yacoub, S., Abdeljaoued, Y., and Mayoraz, E. 1999. Fusion face and speech data for person identity verification. IEEE Trans. Neural Netw. 10: 1065–1074. [DOI] [PubMed] [Google Scholar]
- Bock, J.R. and Gough, D.A. 2001. Predicting protein–protein interactions from primary structure. Bioinformatics 17: 455–460. [DOI] [PubMed] [Google Scholar]
- Brown, M.P.S., Grundy, W.N., Lin, D., Cristianini, N., Sugnet, C.W., Furey, T.S., Ares, M., and Haussler, D. 2000. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc. Natl. Acad. Sci. 97: 262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbidge, R., Trotter, M., Buxton, B., and Holden, S. 2001. Drug design by machine learning: Support vector machines for pharmaceutical data analysis. Comput. Chem. 26: 5–14. [DOI] [PubMed] [Google Scholar]
- Burges, C.J.C. 1998. A tutorial on support vector machine for pattern recognition. Data Min. Knowl. Disc. 2: 121–167. [Google Scholar]
- Bycroft M., Hubbard, T.J.P., Proctor, M., Freund, S.M.V., and Murzin, A.G. 1997. The solution structure of the S1 RNA binding domain: A number of an ancient nucleic acid-binding fold. Cell 88: 235–242. [DOI] [PubMed] [Google Scholar]
- Cai, Y.D., Liu, X.J., Xu, X.B., and Chou, K.C. 2002a. Prediction of protein structural classes by support vector machines. Comput. Chem. 26: 293–296. [DOI] [PubMed] [Google Scholar]
- ———. 2002b. Support vector machines for predicting HIV protease cleavage sites in protein. J. Comput. Chem. 23: 267–274. [DOI] [PubMed] [Google Scholar]
- Cai, C.Z., Han, L.Y., Ji, Z.L., Chen, X., and Chen, Y.Z. 2003a. SVM-Prot: Web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res. 31: 3692–3697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, C.Z., Wang, W.L., and Chen, Y.Z. 2003b. Support vector machine classification of physical and biological datasets. Inter. J. Mod. Phys. C. 14: 575–585. [Google Scholar]
- Casari, G., Sander, C., and Valencia, A. 1995. A method to predict functional residues in proteins. Nat. Struct. Biol. 2: 171–178. [DOI] [PubMed] [Google Scholar]
- Czerminski, R., Yasri, A., and Hartsough, D. 2001. Use of support vector machine in pattern classification: Application to QSAR studies. Quant. Struct.-Act. Relat. 20: 227–240. [Google Scholar]
- Dandekar, T., Snel, B., Huynen, M., and Bork, P. 1998. Conservation of gene order: A fingerprint of proteins that physically interact. Trends Biochem. Sci. 23: 324–328. [DOI] [PubMed] [Google Scholar]
- de Vel, O., Anderson, A., Corney, M., and Mohay, G. 2001. Mining e-mail content for author identification forensics. SIGMOD Record 30: 55–64. [Google Scholar]
- Ding, C.H.Q. and Dubchak, I. 2001. Multi-class protein fold recognition using support vector machines and neural networks. Bioinformatics 17: 349–358. [DOI] [PubMed] [Google Scholar]
- Downward, J. 2001. The ins and outs of signalling. Nature 411: 759–762. [DOI] [PubMed] [Google Scholar]
- Draper, D.E. 1999. Themes in RNA-protein recognition. J. Mol. Biol. 293: 255–270. [DOI] [PubMed] [Google Scholar]
- Draper, D.E. and Reynaldo, L.P. 1999. RNA binding strategies of ribosomal proteins. Nucleic Acids Res. 27: 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drucker, H., Wu, D.H., and Vapnik, V.N. 1999. Support vector machines for spam categorization. IEEE Trans. Neural Netw. 10: 1048–1054. [DOI] [PubMed] [Google Scholar]
- Dubchak, I., Muchnik, I., Holbrook, S.R., and Kim, S.H. 1995. Prediction of protein folding class using global description of amino acid sequence. Proc. Natl. Acad. Sci. 92: 8700–8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen, M.B., Spellman, P.T., Brown, P.O., and Botstein, D. 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. 95: 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elcock, A.H. and McCammon, J.A. 2001. Calculation of weak protein–protein interactions: The pH dependence of the second virial coefficient. Biophysical 80: 613–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright, A.J., Iliopoulos, I., Kyrpides, N.C., and Ouzounis, C.A. 1999. Protein interaction maps for complete genomes based on gene fusion events. Nature 402: 86–90. [DOI] [PubMed] [Google Scholar]
- Evgeniou, T. and Pontil, M. 2001. Support vector machines: Theory and applications. In Machine learning and its applications. Advanced lectures (eds. G. Paliouras et al.), pp.249–257. Springer, New York.
- Fetrow, J.S. and Skolnick, J. 1998. Method for prediction of protein function from sequence using sequence-to-structure-to-function paradigm with application to glutaredoxins/thioredoxins and T1 ribonucleases. J. Mol. Biol. 281: 949–968. [DOI] [PubMed] [Google Scholar]
- Fierro-Monti, I. and Mathews, M.B. 2000. Proteins binding to duplexed RNA: One motif, multiple functions. Trends Biochem. Sci. 25: 241–246. [DOI] [PubMed] [Google Scholar]
- Frank, D.N. and Pace, N.R. 1998. Ribonuclease P: Unity and diversity in a tRNA processing ribozyme. Annu. Rev. Biochem. 67: 153–180. [DOI] [PubMed] [Google Scholar]
- Fritsche, H.A. 2002. Tumor markers and pattern recognition analysis: A new diagnostic tool for cancer. J. Clin. Ligand Assay 25: 11–15. [Google Scholar]
- Furey, T.S., Cristianini, N., Duffy, N., Bednarski, D.W., Schummer, M., and Haussler, D. 2000. Support vector machine classification and validition of cancer tissue samples using microarray expression data. Bioinformatics 16: 906–914. [DOI] [PubMed] [Google Scholar]
- Hermann, T. and Westhof, E. 1999. Simulations of the dynamics at an RNA-protein interface. Nat. Struct. Biol. 6: 540–544. [DOI] [PubMed] [Google Scholar]
- Hua, S.J. and Sun, Z.R. 2001. A novel method of protein secondary structure prediction with high segment overlap measure: Support vector machine approach. J. Mol. Biol. 308: 397–407. [DOI] [PubMed] [Google Scholar]
- Huang, C., Davis, L.S., and Townshend, J.R.G. 2002. An assessment of support vector machines for land cover classification. Int. J. Remote Sens. 23: 725–749. [Google Scholar]
- Karchin, R., Karplus, K., and Haussler, D. 2002. Classifying G-protein coupled receptors with support vector machines. Bioinformatics 18: 147–159. [DOI] [PubMed] [Google Scholar]
- Karlsen, R.E., Gorsich, D.J., and Gerhart, G.R. 2000. Target classification via support vector machines. Opt. Eng. 39: 704–711. [Google Scholar]
- Kim, K.I., Jung, K., Park, S.H., and Kim, H.J. 2001. Support vector machine-based text detection in digital video. Pattern Recognition 34: 527–529. [Google Scholar]
- Lengeler, J.W. 2000. Metabolic networks: A signal-oriented approach to cellular models. Biol. Chem. 381: 911–920. [DOI] [PubMed] [Google Scholar]
- Liong, S.Y. and Sivapragasam, C. 2002. Flood stage forecasting with support vector machines. J. Am. Water Resour. As. 38: 173–186. [Google Scholar]
- Marcotte, E.M., Pellegrini, M., Thompson, M.J., Yeates, T.O., and Eisenberg, D. 1999. A combined algorithm for genome-wide prediction of protein function. Nature 402: 83–86. [DOI] [PubMed] [Google Scholar]
- Mattaj, I.W. 1993. RNA recognition: A family matter? Cell 73: 837–840. [DOI] [PubMed] [Google Scholar]
- Overbeek, R., Fonstein, M.D., D’Souza, M., Pusch, G.D., and Maltsev, N. 1999. The use of gene clusters to infer functional coupling. Proc. Natl. Acad. Sci. 96: 2896–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papageorgiou, C. and Poggio, T. 2000. A trainable system for object detection. Inter. J. Comput. Vision. 38: 15–33. [Google Scholar]
- Pawson, T. 1995. Protein modules and signaling networks. Nature 373: 573–580. [DOI] [PubMed] [Google Scholar]
- Peculis, B.A. 2000. RNA-binding proteins: If it looks like a sn(o)RNA. Curr. Biol. 10: R916–R918. [DOI] [PubMed] [Google Scholar]
- Pellegrini, M., Marcotte, E.M., Thompdon, M.J., Eisenberg, D., and Yeates, T.O. 1999. Assigning protein functions by comparative genome analysis: Protein phylogenetic profiles. Proc. Natl. Acad. Sci. 96: 4285–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Canadillas, J.-M. and Varani, G. 2001. Recent advances in RNA-protein recognition. Curr. Opin. Struct. Biol. 11: 53–58. [DOI] [PubMed] [Google Scholar]
- Ramaswamy, S., Tamayo, P., Rifkin, R., Mukherjee, S., Yeang, C.H., Angelo, M., Ladd, C., Reich, M., Latulippe, E., Mesirov, J.P., et al. 2001. Multiclass cancer diagnosis using tumor gene expression signatures. Proc. Natl. Acad. Sci. 98: 15149–15154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulston, J.E. 2002. Screening with tumor markers. Mol. Biotechnol. 20: 153–162. [DOI] [PubMed] [Google Scholar]
- Singh, R. 2002. RNA-protein interactions that regulate pre-mRNA splicing. Gene Expr. 10: 79–92. [PMC free article] [PubMed] [Google Scholar]
- Siomi, H. and Dreyfuss, G. 1997. RNA-binding proteins as regulators of gene expression. Curr. Opin. Genetics Dev. 7: 345–353. [DOI] [PubMed] [Google Scholar]
- Thubthong, N. and Kijsirikul, B. 2001. Support vector machines for Thai phoneme recognition. Inter. J. Uncertain. Fuzz. 9: 803–813. [Google Scholar]
- Tomasevic, N. and Peculis, B. 1999. Identification of a U8 snoRNA-specific binding protein. J. Biol. Chem. 274: 35914–35920. [DOI] [PubMed] [Google Scholar]
- Vapnik, V. 1995. The Nature of statistical learning theory. Springer, New York.
- Veropoulos, K., Campbell, C., and Cristianini, N. 1999. Controlling the sensitivity of support vector machines. In Proceedings of the International Joint Conference on Artificial Intelligence (ed. T. Dean), pp.55–60. Morgan Kaufmann, Stockholm, Sweden.
- Yuan, Z., Burrage, K., and Mattick, J.S. 2002. Prediction of protein solvent accessibility using support vector machines. Proteins 48: 566–570. [DOI] [PubMed] [Google Scholar]
- Zhang, K. and Rathod, P.K. 2002. Divergent regulation of dihydrofolate reductase between malaria parasite and human host. Science 296: 545–547. [DOI] [PMC free article] [PubMed] [Google Scholar]