

Abstract
Post-transcriptional processes such as alternative splicing and RNA editing have a huge impact on the diversity of the proteome. Detecting alternatively spliced transcripts is difficult when they are rare. In addition, edited transcripts often differ from the genomic sequence by only a few nucleotides. Denaturing high performance liquid chromatography (DHPLC) is routinely used for single nucleotide polymorphism detection and we used this method to detect alternatively spliced or edited transcripts. As the sites of RNA editing appear to be conserved within gene families, we investigated whether editing sites are conserved in the murine homologue of the Drosophila cacophony transcript encoding the α1 subunit of a voltage-gated calcium channel that is edited at 10 independent positions. Although DHPLC analysis detects RNA editing at as low as 3% in control transcripts, no evidence of RNA editing was found in the analysed murine transcript. However an alternative exon was identified at the 3′ end of the mouse Cacna1α transcript and an alternative micro-exon encoding only two amino acids (NP) was found in the extracellular loop before the IVS4 helix in the same transcript. In the homologous Drosophila transcript a micro-exon also encoding two amino acids was found at the same position before the IVS4 helix.
INTRODUCTION
With the completion of sequencing of many genomes attention is now focused on the proteome. Post-transcriptional processes such as alternative splicing play an important role in creating a complex proteome and recent estimates suggest that 35–59% of human genes undergo alternative splicing (1–3). Since alternative splicing of a transcript can vary during development and between different tissues and considering that in several cases human genes can produce hundreds or thousands of different mRNAs (4,5), the probability remains that some alternative splicing has gone undetected (4). Therefore methods are necessary for detecting the full range of different transcripts that a gene can encode. At present there are various techniques for detecting alternatively spliced products, of which the alignment of multiple expressed sequence tags (ESTs) is perhaps the most accurate (6). In the last few years, bioinformatics studies have identified more alternative splicing events than were previously found (6), but since most of the ESTs correspond to the 5′ or 3′ ends of transcripts many alternatively spliced exons remain undetected. In fact, the greatest disadvantage of the bioinformatics method is the risk of a high rate of false negatives (4).
Another post-transcriptional event that can increase the complexity of the proteome is RNA editing (7,8). The difficulty of detecting edited transcripts is similar to the problem of detecting micro-exons as editing creates very subtle changes in the sequence. The most abundant type of RNA editing found in higher eukaryotes is the conversion of adenosine to inosine by the ADAR (adenosine deaminases that act on RNA) family of enzymes (for reviews see 7,9). The abundance of inosine in poly(A)+ mRNA has been estimated at one base in 17 000 in rat brain and one in 33 000 in rat heart (10). Since very few edited transcripts have been identified (7), this suggests that many edited transcripts remain to be found. Inosine is read as if it were guanosine by the translation machinery and editing can change the encoded protein (11). This can have dramatic effects on the function of the protein, as has been observed both in the glutamate-gated ion channel and serotonin receptors (for a review see 8). With one exception, editing at a specific position is not 100% efficient so RT–PCR from an edited transcript produces a heterogeneous pool of cDNAs with adenosine/guanosine differences at the edited site. To confirm that this difference between cDNAs is truly RNA editing and not a single nucleotide polymorphism (SNP) a comparison of cDNAs and the corresponding genomic sequence must verify an adenosine at the edited position in the genomic sequence. A screen for editing events based on chemical modification of inosine in RNA molecules has been developed by Bass and colleagues but this is not a rapid method and many cDNAs and genomic DNA have to be sequenced to eliminate false positives (12).
Editing sites in vertebrate glutamate-gated ion channel receptor transcripts are conserved at the same positions in different genes of the glutamate receptor family (13). Screening homologues of known edited transcripts thus offers one possible approach to find new targets of RNA editing. The Dmca1A (cacophony) transcript that encodes the α1 subunit of a voltage-gated calcium channel in Drosophila melanogaster is edited at 10 positions causing 10 amino acid changes (Fig. 1) (14,15). The amino acid sequence of the edited regions is well conserved in homologous vertebrate calcium channel transcripts as are the intron–exon boundaries (15), suggesting that RNA editing sites might also be conserved. Voltage-gated ion channel receptors play a significant role in human diseases and as drug targets (16). Therefore it is important to determine how much transcript diversity is introduced by RNA processing events such as alternative splicing and RNA editing.
Figure 1.

Schematic representation of the α1 subunit of the voltage-gated calcium channel. The pore-forming α1 subunit consists of a single polypeptide chain with four (I–IV) repeated domains of six membrane spanning α-helical segments, represented as rectangles (S1–S6), in each of the four domains. The α-helix S4 segments are positively charged in all four repeats. In this representation the dots represent the edited amino acid changes in the Drosophila channel. The positions of the new alternatively spliced exons found in the mouse Cacna1α transcript are indicated by black rectangles. The mini NP insertion is located in the IVS3/IVS4 extracellular loop, like the HD exon in the fly, and the mutually exclusive exon is close to the C-terminus.
In this report we describe the application of a heteroduplex detection method, which is extensively used for genomic SNP and mutation detection, to search for RNA editing and alternative splicing within an ion channel transcript in the mouse brain. Denaturing high performance liquid chromatography (DHPLC) detects heteroduplexes in PCR products by subjecting the crude PCR products to ion pair reverse-phase liquid chromatography under partially denaturing conditions (17,18). We show that this method is sufficiently sensitive to detect RNA editing at a specific position at a frequency as low as 3%, in control experiments, as well as detecting subtle splicing events. A micro-exon of two amino acids was found in the mouse Cacna1α transcript as well as an alternative exon in the C-terminus that is the same length as the exon it replaces.
MATERIALS AND METHODS
Isolation of genomic DNA
DNA was extracted by standard procedures from adult BALB-C mouse tails (19). To obtain DNA from Drosophila, 100 flies (deprived of yeast for 2–3 h) were ground in liquid nitrogen with a mortar and pestle. The powder was mixed with homogenisation buffer (10 mM Tris–HCl, pH 8, 60 mM NaCl, 10 mM EDTA, 0.15 mM spermidine, 0.5% Triton X-100) and spun at 6000 g for 7 min at 4°C in a Sorval centrifuge. Supernatant was collected and respun at the same speed for 15 min. The pellet was resuspended in the same homogenisation buffer plus 20% Sarkosyl and 0.1 mg/ml proteinase K at 50°C for 2–3 h. Phenol/chloroform extraction and precipitation of the genomic DNA was performed. The DNA was quantitated on a UV spectrophotometer. For PCRs 200–300 ng of genomic DNA was used (20).
Preparation of RNA
Between 10 and 20 frozen flies were ground in 3–4 ml of Tri Reagent (Sigma) with a homogeniser (IKA Labortechnik). The RNA was phenol/chloroform extracted and ethanol precipitated. The RNA was quantitated on a UV spectrophotometer and 5 µg was used directly for RT–PCR experiments. Drosophila melanogaster adult poly(A)+ RNA purchased from Clontech was also used in some experiments. Total RNA was also prepared from frozen adult mouse brain using the standard guanidinium thiocyanate lysis buffer and centrifuged through a caesium chloride cushion (21).
Preparation of cDNA
RT–PCR was performed with gene-specific primers (Table 1) on 5 µg of total RNA or on 2 µg of poly(A)+ RNA using 200 U of Superscript II reverse transcriptase (Gibco BRL) at 42°C for 40 min in 20 µl final volume. The final volume of the PCRs was 50 µl and contained 1× Gold buffer (Perkin Elmer), 1.5 mM MgCl2, 20 pmol of each primer, 1 U of Pfu (Promega) + 1 U of Gold (Perkin Elmer) and 2 µl of specific first strand cDNA. The conditions for PCR amplification varied depending on the different substrates; the reactions were performed in a Perkin Elmer 9600 Thermal Cycler. The PCR products were electrophoresed on agarose gels and directly subcloned in the T-easy vector (Promega) or denatured and gradually reannealed for DHPLC analysis. If more than one PCR product was present after electrophoresis on an agarose gel we processed only the RT–PCR product that corresponded to the size of the predicted product as we were not interested in analysing alternative exons that can be distinguished by size.
Table 1. Ion channel transcripts analysed in this study.
 |
Dmca1A (U55776) (14); GluR-5 (U31443) (27); Cacna1α (U76716) (30). All the RT–PCR products were electrophoresed on agarose gels before DHPLC analysis. Products amplified from the Dmca1A exon 15 were used in the mixing experiments. RT–PCR of Dmca1A exon 24 was performed to determine the presence of the micro-exon in the fly.
DHPLC analysis
The search for hypothetical editing events or alternative splicing was performed by DHPLC scanning on an automated HPLC instrument (Transgenomic WAVE™ DNA Fragment Analysis System). To generate heteroduplex molecules, PCR products were subjected to denaturation for 3 min at 95°C followed by a gradual reannealing for 30 min in the thermal cycler. Aliquots of 5–10 µl of reannealed PCR product was loaded on a DNASep column (Transgenomic, Santa Clara, CA). PCR products were eluted from the column using an acetonitrile gradient in a 0.1 M triethylamine acetate (TEAA) buffer, pH 7, at a constant flow rate of 0.9 ml/min. The gradient was created by mixing buffers A and B (buffer A, 0.1 M TEAA, 0.1 mM Na4EDTA; buffer B, 25% acetonitrile in 0.1 M TEAA). The start and end points of the gradient depend on the size of the PCR fragments and on the composition of the DNA fragments (22). The temperature required for successful resolution of heteroduplex molecules was determined by computer program; the web site for the melting algorithm is http://insertion.stanford.edu/melt.html.
Sequencing
After subcloning in T-easy vector (Promega) the PCR products were sequenced in both orientations with T7- or SP6-specific primers on an ABI 373 automated sequencer using a big dye-terminator cycle sequencing reaction kit (Perkin Elmer).
RESULTS
Detection of RNA editing using DHPLC
DHPLC is a mutation detection method that is widely used to detect sequence polymorphisms. PCR products, containing heterogeneity, are denatured and allowed to reanneal slowly so that some heteroduplex molecules are formed. This DNA mixture is chromatographed over a DNA affinity resin under partially denaturing conditions of high temperature within a linear acetonitrile gradient. Heteroduplexes having internal sequence variation display reduced column retention times (x-axis) relative to their homoduplex counterparts; the eluted DNA fragments were detected by an UV detector at 254 nm and converted to millivolts (y-axis) (23).
The DHPLC method has been used successfully for detecting point mutations at very low percentages of heterogeneity, as low as 0.5–5%, in mitochondrial DNA and genomic DNA samples (24,25). To verify that this method can be used to study RNA editing (A→G conversion), we did control experiments using wild-type and edited clones amplified from the Drosophila calcium channel Dmca1A (cacophony) transcript (exon 15) (Table 1). Equal amounts of PCR products of 500 nt amplified from edited and unedited cDNA clones were mixed, denatured and reannealed to allow some heteroduplex formation. This PCR product mix was chromatographed over a DHPLC column and the heteroduplex was readily detected. Figure 2 shows merged DHPLC profiles of the unedited (green line) and edited (red line) clones and the blue line represents the 50% mixture of the two products. To determine if PCR product length affects detection of editing, experiments were also performed with different primers producing PCR products of different lengths. Edited and unedited PCR products of 200, 500 or 800 nt or 1 kb (Table 1) were mixed at different ratios of edited to total fragments (3, 6, 10, 25, 40 and 50% edited) and analysed by DHPLC using experimental conditions suggested by the software provided by Transgenomic. An editing event could be detected when the edited product comprised as little as 3% of the total transcripts (Fig. 3B). Mixing experiments using different percentages of unedited and edited PCR products of 200 nt are shown in Figure 3. The retention time of the heteroduplex peaks is written over the peaks; this value is only given when a true peak is recognised above background. The resolution of the 3 and 6% experimental points can be increased using different running conditions and temperatures, but even the simple conditions suggested by the Transgenomic program are sufficient to detect heterogeneous products.
Figure 2.
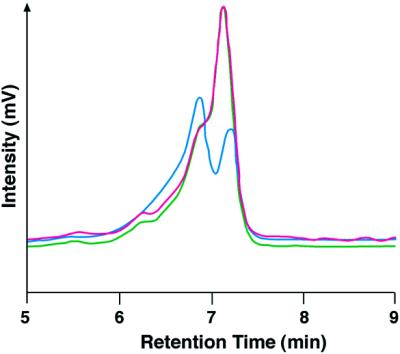
DHPLC analyses of PCR products encompassing the edited position in exon 15 of Drosophila Dmca1A. PCR products of a region of exon 15 (500 nt) amplified from either unedited or edited clones or mixtures of both were analysed by DHPLC. The red DHPLC profile represents the completely unedited version of exon 15 (homogeneous PCR population), the green DHPLC profile represents the completely edited version (A→G at position 2092) of the same product (homogeneous population). The blue DHPLC profile represents a mixture of 50% of both unedited and edited PCR products (heterogeneous population). The x-axis represents the retention time in minutes, which is dependent on the length of the product analysed; the y-axis is a measure of absorbency (converted to mV). The temperature recommended by the Trangenomic program for the resolution of the heteroduplex molecules was between 59 and 61°C. The resolution depicted above was at 59°C and the gradient used was eluent buffer B (25% acetonitrile) increasing from 50 to 58.9% over 30 s and from 58.9 to 64.3% over 3 min.
Figure 3.

Mixing experiments and DHPLC analysis to determine the sensitivity of the DHPLC to find sites of RNA editing. PCR products of a region of Drosophila Dmca1A exon 15 (200 nt) were amplified from both unedited and edited clones and mixed at different ratios (3, 6, 10, 25, 40 and 50%) and analysed by DHPLC: (A) wild-type; (B) 3%; (C) 6%; (D) 10%; (E) 25%; (F) 40%; (G) 50%. The gradient used was eluent buffer B (25% acetonitrile) increasing from 50 to 58.9% over 30 s and from 58.9 to 64.3% over 3 min. The temperatures suggested for the resolution of the heteroduplex are 59–61°C. (B) and (C) show DHPLC analysis of the 3 and 6% mixed products resolved at 61°C; all the other DHPLC profiles were resolved at 60°C. The retention times written on the tops of the peaks are automatically given by the Trangenomic program only when a real heterogeneous product is recognised above the background.
The multiple peaks of the 10–50% mix population, under the same running conditions but at a different temperature, reflect the different nature of the duplex molecules that have been formed, such as two distinguishable heteroduplex and homoduplex peaks (edi:edi, wt:wt, edi:wt and wt:edi) (23,26). The different heights of the peaks in the DHPLC profiles reflect the difference in the amount of PCR product loaded over the DNAsep column in the different experiments (see Materials and Methods). The retention time, between 4 and 5 min, is equivalent for all the samples in Figure 3, reflecting the same length of PCR product analysed (200 nt).
These mixing experiments were performed on cDNA. A potential problem with DHPLC analysis of RT–PCR products is that a true representation of the RNA population may not be obtained. To address this question nested primers were designed so that 6 nt of random sequence were incorporated close to the 3′ end of the product. Six separate RT–PCRs were performed with these primers and the products were subcloned and sequenced. In five independent experiments each cDNA had a unique hexamer tag sequence and the number of clones sequenced was 26, 24, 27, 26 and 29. The only occasion when clones were not independent was when more than 35 PCR cycles were required to obtain a good product yield from a rare transcript. Therefore RT–PCR products generated in less than 30 PCR cycles represent the mRNA population with an accuracy more than sufficient for DHPLC analysis. On average 25 PCR cycles were used in this study as all PCR products gave a good yield.
To test this method on RT–PCR products made from transcripts that are edited in vivo, we extracted total mRNA from the brain of inbred BALB/C mice. Transcripts encoding the GluR-5 subunit of the kainate receptor are edited at the Q/R site to 40% (27). Primers flanking the Q/R editing site in GluR-5 were used to generate RT–PCR products (261 nt) that were denatured, reannealed and analysed by DHPLC or directly sequenced. After 7 min of DHPLC analysis the RT–PCR product of GluR-5 displays a double peak indicating the presence of a heterogeneous population (Fig. 4A), as predicted. However, when the same RT–PCR product is analysed directly by sequencing the heterogeneity at this edited position is not observed due to low resolution of the A/G mix bases and cannot be distinguished over background (Fig. 4B). This problem of detecting editing events by sequencing RT–PCR products has previously been reported (28). The 40% editing of the GluR-5 transcript, in our sample, was confirmed by sequencing independent clones.
Figure 4.

Chromatograms representing DHPLC analysis of the Q/R edited site present in the transcript encoding the GluR-5 subunit of the kainate receptor. (A) Total RNA from mouse brain was used to generate the RT–PCR products (261 nt) for DHPLC analysis. The pre-mRNA encoding the GluR-5 receptor is edited to 40% (A→G, nt 1352) in vivo. The DHPLC resolution temperature was 60°C and the gradient was eluent buffer B (25% acetonitrile) increasing from 46 to 51% over 60 s and from 51 to 65% over 7 min. (B) The sequence profile of the same RT–PCR product analysed directly by sequencing. The arrow indicates the edited position.
Detection of micro-processing events by DHPLC analysis
The hypothetical secondary structure of the pore-forming α1 subunit of a voltage-gated calcium channels is diagrammed in Figure 1. These large proteins contain four repeats of the same domain (I–IV) and a large intracellular domain at the C-terminus. Each repeated domain contains six hydrophobic transmembrane segments; in particular, the S4 helix contains positively charged residues critical for voltage gating (16). Most of the RNA editing in the Drosophila cacophony transcript that is preferentially expressed in the CNS change the amino acids close to or within the transmembrane domains III and IV (Fig. 1) (14). In addition, micro-exons have been identified before the IVS4 helix in this fly channel (14). The amino acid sequence is highly conserved among vertebrate and invertebrate voltage-gated calcium channels (29), therefore we used DHPLC to analyse the homologous vertebrate ion channel transcript from mouse brain for subtle evidence of RNA processing events such as RNA editing or the presence of micro-exons. The mouse Cacna1α, which encodes the α1A subunit of the P/Q type calcium channel (GenBank accession no. U76716), is the vertebrate homologue of the Drosophila Dmca1A gene so we choose its transcript to investigate if it undergoes micro-processing events such as RNA editing and splicing. Like cacophony, this is a CNS-specific transcript and the tottering mutation in the mouse Cacna1α gene causes defects in movement (30), as do mutations in Drosophila Dmca1A (31).
Regions of the Cacna1α transcript corresponding to the edited regions in the cacophony transcript were amplified from RNA from whole mouse brain to generate a set of seven RT–PCR products (Table 1, fragments tot1–tot7). Each product was separately denatured, reannealed and analysed by DHPLC using the conditions of temperature and buffer gradients that were calculated based on the sequence of each fragment. For five out of seven PCR products only a single homogeneous peak was obtained at different temperatures (one example is shown in Fig. 5A), indicating that there was no evidence of micro-processing in these products. However, two of the PCR products (tot3 and tot6) did show heterogeneity (Fig. 5B and C). To confirm the DHPLC data, 25–40 independent clones of each of the seven RT–PCRs were sequenced confirming the presence of heterogeneous products only in the tot3 and tot6 products.
Figure 5.

Chromatograms representing DHPLC analysis of three different RT–PCR products encoding different regions of the mouse α1A subunit of the P/Q type calcium channel (Cacna1α). (A) An example of a DHPLC profile of a homogeneous population. This profile is of the RT–PCR of the fragment tot5 (300 nt). (B) A heterogeneous population (fragments tot3, 223 nt). (C) A heterogeneous population (fragments tot6, 229 nt). The temperature used for successful resolution of the heteroduplex molecules of the RT–PCR products was 60°C. The conditions used for the experiments shown were: tot3, eluent buffer B (25% acetonitrile) increasing from 50 to 52.8% over 30 s and from 52.8 to 58.2% over 3 min at 60°C; tot6, eluent buffer B (25% acetonitrile) increasing from 50 to 52.2% over 30 s and from 52.2 to 57.6% over 3 min at 60°C; tot5, eluent buffer B (25% acetonitrile) increasing from 50 to 50.7% over 30 s and from 50.7 to 56.1% over 3 min at 60°C.
The sequence analysis of tot3 revealed the presence of two different products that differ by the inclusion or exclusion of a micro-exon of 6 nt, encoding just two amino acids (Fig. 6A). We found that 40% of the sequences analysed contained the insertion of the 6 nt resulting in a modification of the extracellular loop between transmembrane segments IVS3 and IVS4 of the mouse Cacna1α (Fig. 1), changing the amino acid sequence GNNF to GNPNNF (Fig. 6A).
Figure 6.

Alternatively spliced exons detected in the α1A subunit of the mouse P/Q type calcium channel. (A) Alternatively spliced isoforms of the α1A subunit of the P/Q type calcium channel present in mouse brain (Cacna1α). The sequence analysis of the tot3 RT–PCR products reveals an insertion of 6 nt that results in a modification of the extracellular loop between the transmembrane IVS3 and IVS4 of the mouse α1A subunit changing the amino acid sequence GNNF to GNPNNF. (B) Alternative splicing in the tot6 RT–PCR products results in the insertion of an exon of the same size (96 nt) as the one it replaces; arrows denote the mutually exclusive exons. Many of the amino acids are identical (black box) but different at the nucleotide level (dots below the sequence); the result is that only 10 amino acids are different between the two isoforms.
The second heterogeneous RT–PCR product tot6 spans a region within the intracellular C-terminal domain of Cacna1α (Fig. 1). Sequencing individual clones of this PCR product revealed the presence of an alternative exon of identical length to the exon that is replaced (96 nt) in 30% of the sequences analysed (Fig. 6B). What is interesting about these mutually exclusive alternative exons is that they are very homologous at the amino acid level with only 10 out of 32 amino acids being different.
RT–PCR was performed to generate PCR products that included the IVS3 transmembrane segment and the C-terminus of the mouse brain Cacna1α transcript encompassing both tot3 and tot6. Sequencing of these PCR products revealed that the C-terminus alternative exon and NP micro-exon were present together in ∼25% of the analysed sequences but also both exons were found on their own at a very low frequency.
Micro-exons and editing events in Drosophila are developmentally regulated
It has been reported that the Drosophila calcium channel α1 subunit gene, Dmca1A, produces different transcripts by alternative splicing and RNA editing (14). In particular, the extracellular IVS3–IVS4 loop can be modified by the inclusion of micro-exons encoding one, two or three amino acids (14). In addition, there is an editing event (N→S) in the same extracellular loop one amino acid after the micro-splicing insertion (Fig. 7B). Thus, in this small region there is the potential for significant transcript variability.
Figure 7.

Micro-exons present in transcripts encoding the α1 subunit of the mouse and Drosophila voltage-gated calcium channels are positioned in front of the positively charged voltage sensor helix IVS4. (A) A micro-exon present in exon 24 in the D.cacophony transcript. Either one (H), two (HD) or three (HDD) amino acids can be inserted. The developmental analysis shows a higher frequency of insertion of the two amino acid micro-exon at all stages of development in the fly. (B) The micro-exons of two amino acids in the mouse and Drosophila transcripts of the α1 subunit encode different amino acids but occur at the same frequency in adults in vertebrates and invertebrates. Both micro-exons are present in the same position just in front of the IVS4 positively charged helix, increasing the length of the extracellular loop IVS3–IVS4. RNA editing at a nearby position (N→S) is present in the Drosophila channel. The amino acid (S) that is the result of RNA editing is above the line.
Introns 22 and 23 covering the region of the micro-exon were sequenced in Drosophila and an exon of 6 nt encoding histidine and aspartate (the HD micro-exon) was found 2 kb downstream of the 5′ splice site, as previously reported (14). An additional new exon of 3 nt encoding aspartate (the D micro-exon) was found a further 3.14 kb downstream. These micro-exons have canonical consensus splice sites. Two independent RT–PCRs were performed on poly(A)+ RNA from embryo, larva and adult and 46–70 independent clones were sequenced to investigate which micro-exon was preferentially included during a particular stage of development. We found that the micro-exons are either included or excluded apparently independently of one another so that mRNA may encode none, two, one or three extra amino acids (Fig. 7). The HD micro-exon was included more frequently than it was excluded during all stages of development and the percentage of inclusion in adult fly is 47.9%. In addition, we found that the percentage of editing at position 4106, one amino acid after the micro-exon, increases from embryo to adult with 36.1% editing in adult fly (L.P.Keegan and A.Gallo, unpublished results) (Fig. 7).
It is intriguing that from fly to mouse in the homologous calcium channel transcript we found a micro-exon, of only two amino acids, present at ∼40%, in the same position in the adult stage. Surprisingly we did not find any RNA editing in the mouse channel transcript close to the micro-exon as occurs in adult fly (Fig. 7), considering that this position is highly edited in adult flies.
DISCUSSION
DHPLC as a method for detecting micro-processing events in transcripts
Alternative splicing and RNA editing are essential biological processes that generate multiple different transcripts from the same pre-mRNA (4,7). Aberrant splicing as well as failed editing of precursor transcripts has been associated with human diseases and pathological status (32–34). In the future, analysis of all transcripts that a gene can encode in different cells or tissues will provide important insights into normal cellular physiology as well as into some disease processes.
DHPLC has been used successfully for SNP analysis but has not previously been used to search for sequence variation in transcripts. The single base pair changes caused by RNA editing are among the most subtle sequence alterations that can occur during RNA processing. Detection of editing clearly benefits from using a very sensitive method like DHPLC that can detect editing occurring at a very low percentage (3%) (Fig. 3) as well as subtle splicing events (Fig. 5).
Alternatively spliced exons can be detected by different methods, one of the most commonly used being the alignment of multiple ESTs to the genomic sequence using specific databases such as ISIS, TAP or HASDB (35–37). The limitation of this method is the misdetection of rare splicing events that are amplified less efficiently or splicing events localised far from the 3′ and 5′ ends of the mRNA, considering that most of the ESTs correspond to the ends of the transcripts. In addition, the risks of chimeric ESTs and sequencing artefacts have to be considered. To determine how sensitive bioinformatics is to detect micro-exons, we looked for a micro-exon (NP) in the mouse genomic sequence of Cacna1α between exons 27 and 31. Bioinformatics programs did not detect the micro-exon, although it is present in brain cDNAs. In addition, there are still gaps in the mouse genomic sequence. This emphasises the importance of alternative methods to detect micro-exons.
However, despite these disadvantages, bioinformatics does provide a large source of ‘hypothetical’ new spliced variants, but fast and cheap methods are necessary to verify all this potential. One method, routinely used at present, is based on RT–PCR of hypothetical spliced products and gel electrophoresis, but this method fails in detecting mutually exclusive exons of the same length or micro-exons, as identified in this study (Fig. 6). In addition, direct sequencing of RT–PCR products fails to detect RNA editing (Fig. 4B).
Microarray technology takes advantage of bioinformatics to detect alternative splicing using oligo probes that span specific exon–exon junctions. However, if bioinformatics fails to detect splice sites, microarray techniques reflect these limitations (6,38). Microarrays cannot be used to screen all the post-transcriptional modifications that a single gene can produce and cannot be used to detect unknown splicing events.
DHPLC is a very sensitive method that can detect one base change directly in a crude RT–PCR. The major advantages of this method are that single nucleotide substitutions, deletions and insertions are detected successfully within 3–7 min in unpurified RT–PCR products (Figs 2 and 3), the sample loading and analysis are essentially automated and a few nanograms of RT–PCR products are required for the analysis. These features and its low cost make DHPLC one of the most powerful tools for the analysis of different transcripts. Using DHPLC to detect subtle RNA processing events will be most useful in situations where the primer sets and DHPLC conditions are established for the same transcript that can be analysed in different tissues or during different stages of development or to compare the same transcript between normal and diseased tissue.
Identifying rare post-transcriptional events that happen in a subset of cells in a particular tissue is becoming very important. The combination of single cell RT–PCR with DHPLC analysis could be used instead of sequencing a large number of cDNAs considering that DHPLC is a fast method and at least 10 times less expensive than sequencing reactions due to savings in reagent and labour costs (23,39).
In this study we have been able to take advantage of inbred mouse strains to reduce the background due to genomic SNPs. One problem with using DHPLC to analyse human transcripts is that the heterogeneity observed for the A→G substitution could be due to SNPs. Our use of inbred mice in this study largely eliminates this problem. Now that both genomic and cDNA sequences are becoming available from the same species, DHPLC could become a powerful tool in analysing mouse transcripts to develop a full catalogue of alternatively spliced and modified transcripts. Genes with shared functions in two distantly related organisms should maintain a level of conservation within the coding sequence and splicing variants during evolution, so that the analysis of mouse transcripts by DHPLC could define a prototype for transcript complexity in mammals.
Alternative splicing events detected in Cacna1α transcripts by DHPLC
The PCR product tot3 when analysed by DHPLC showed an altered peak (Fig. 5B) due to a mixture of PCR products that either did or did not contain the 6 nt insertion (two amino acids, NP) (Fig. 6A). This micro-exon is present in 40% of the sequences analysed and it consists of an insertion in the extracellular loop between transmembrane segments IVS3 and IVS4 of the Cacna1α transcript (Fig. 1). The NP micro-exon was previously identified in exactly the same position in the rat orthologue of this ion channel (40,41). In particular, it was found that +NP and –NP transcripts are expressed in different regions of the rat nervous system and the –NP variant activates a calcium channel at a rate 1.7-fold faster compared with the +NP variant (41). A micro-exon of two amino acids (HD) is also found in 47.9% of the Drosophila α1 subunit of the voltage-gated calcium channel Dmca1A at the same position (Fig. 7A).
The region just in front of the helix IVS4 in the voltage-gated calcium channel is very important for the physiology of the channel (40). It is interesting that this region of the transcript is the target for subtle RNA processing events such as RNA editing in Drosophila or the inclusion of a micro-exon in both vertebrates and invertebrates. The high frequency of inclusion of these micro-exons of only two amino acids in vertebrates and invertebrates suggests that perhaps the length of the extracellular loop between IVS3 and IVS4 is critical.
Another PCR product (tot6) of the Cacna1α ion channel also displayed an altered DHPLC profile. This PCR product gave a single homogeneous band by gel electrophoresis but analysis by DHPLC revealed that it was a mixed PCR population. Sequencing the PCR products revealed the presence of two exons of the same length but different in nucleotide composition. Of the sequences analysed, 30% contained this mutually exclusive exon, located in the C-terminus immediately after the fourth repeated domain (Fig. 1). What is surprising is that it is very homologous at the amino acid level to the exon that it replaces with only 10 amino acids different out of a total 32 residues (Fig. 6B). The sequence is different at the nucleotide level, suggesting that there is high selective pressure to retain these amino acids. This alternative exon is in a region of the ion channel identified by homology as a ‘calcium binding region’ and could be involved in binding of a specific protein/modulator with different affinities for the two different protein isoforms. This hypothesis is supported by the observation that a similar alternative exon, with 96% amino acid identity, is also present at the same position in the homologous human channel (GenBank accession no. O00555).
In 25% of the sequences analysed both the NP micro-exon and the alternative C-terminus exon were present. The NP micro-exon in rat brain transcript has been found in specific areas of the brain (41). Considering we found a correlation between NP micro-exon splicing and the C-terminus exon, it is possible that both exons are preferentially expressed in mouse Cacna1α in specific regions or cell subsets of the mouse brain.
Non-conservation of RNA editing targets between vertebrates and invertebrates?
RNA editing of the Drosophila transcript encoding the α1 subunit of the voltage-gated calcium channel is targeted mainly to the transmembrane regions that come together to form the ion pore and voltage sensor (Fig. 1). The transmembrane regions of the ion channels are strongly conserved throughout the gene family and only a few amino acid changes can be tolerated without affecting the function of the channel. We searched for evidence of RNA editing in the same regions of the homologous mouse Cacna1α transcript within seven RT–PCR products using DHPLC. No editing was found in the homologous mouse transcript in regions corresponding to portions of the Drosophila ion channel where editing produces amino acid changes. However, we cannot exclude the possibility that the Cacna1α mouse channel could show additional post-transcriptional modifications to the ones that we found in this study, considering that we analysed RT–PCRs from RNA from whole mouse brain and not specific RNA products from a selected subset of cells of the mouse brain.
Why is the edited form of the Drosophila channel not conserved in the mouse transcript at the same positions? One explanation could be that these channels have evolved by sequential duplication events in chordate genomes (29); in this situation the genome can produce a large number of different transcripts by alternative splicing and not require RNA editing to generate variability. This hypothesis is intriguing if we think that in Drosophila there are a small number of genes encoding voltage-gated calcium channels and the editing could be a method to create diverse transcripts in combination with alternative splicing (14). As of yet we do not understand why the fly edits transcripts encoding the voltage-gated calcium channel and what biological role the isoforms have. It is possible that their function is very specialised and unique to flies. Therefore if this function is not required in vertebrates, the ability to edit voltage-gated calcium transcript has been lost.
Acknowledgments
ACKNOWLEDGEMENTS
This work has been supported by the MRC and a grant from the British Heart Foundation (PG/98086).
REFERENCES
- 1.Brett D., Hanke,J., Lehmann,G., Haase,S., Delbruck,S., Krueger,S., Reich,J. and Bork,P. (2000) EST comparison indicates 38% of human mRNAs contain possible alternative splice forms. FEBS Lett., 474, 83–86. [DOI] [PubMed] [Google Scholar]
- 2.Hanke J., Brett,D., Zastrow,I., Aydin,A., Delbruck,S., Lehmann,G., Luft,F., Reich,J. and Bork,P. (1999) Alternative splicing of human genes: more the rule than the exception? Trends Genet., 15, 389–390. [DOI] [PubMed] [Google Scholar]
- 3.Mironov A.A., Fickett,J.W. and Gelfand,M.S. (1999) Frequent alternative splicing of human genes. Genome Res., 9, 1288–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graveley B.R. (2001) Alternative splicing: increasing diversity in the proteomic world. Trends Genet., 17, 100–107. [DOI] [PubMed] [Google Scholar]
- 5.Roberts G.C. and Smith,C.W. (2002) Alternative splicing: combinatorial output from the genome. Curr. Opin. Chem. Biol., 6, 375–383. [DOI] [PubMed] [Google Scholar]
- 6.Modrek B. and Lee,C. (2002) A genomic view of alternative splicing. Nature Genet., 30, 13–19. [DOI] [PubMed] [Google Scholar]
- 7.Keegan L.P., Gallo,A. and O’Connell,M.A. (2001) The many roles of an RNA editor. Nature Rev. Genet., 2, 869–878. [DOI] [PubMed] [Google Scholar]
- 8.Emeson R.B. and Singh,M. (2000) Adenosine-to-inosine RNA editing: substrates and consequences. In Bass,B.L. (ed.), RNA Editing. Oxford University Press, Oxford, UK, pp. 109–138.
- 9.Hough R.F. and Bass,B.L. (2000) Adenosine deaminases that act on RNA. In Bass,B.L. (ed.), RNA Editing. Oxford University Press, Oxford, UK, pp. 77–108.
- 10.Paul M. and Bass,B.L. (1998) Inosine exists in mRNA at tissue-specific levels and is most abundant in brain mRNA. EMBO J., 17, 1120–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basilio C., Wahba,A.J., Lengyel,P., Speyer,J.F. and Ochoa,S. (1962) Synthetic polynucleotides and the amino acid code. Proc. Natl Acad. Sci. USA, 48, 613–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morse D.P. and Bass,B.L. (1997) Detection of inosine in messenger RNA by inosine-specific cleavage. Biochemistry, 36, 8429–8434. [DOI] [PubMed] [Google Scholar]
- 13.Seeburg P.H. (1996) The role of RNA editing in controlling glutamate receptor channel properties. J. Neurochem., 66, 1–5. [DOI] [PubMed] [Google Scholar]
- 14.Smith L.A., Wang,X.J., Peixoto,A.A., Neumann,E.K., Hall,L.M. and Hall,J.C. (1996) A Drosophila calcium channel α1 subunit gene maps to a genetic locus associated with behavioural and visual defects. J. Neurosci., 16, 7868–7879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peixoto A.A., Smith,L.A. and Hall,J.C. (1997) Genomic organization and evolution of alternative exons in a Drosophila calcium channel gene. Genetics, 145, 1003–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Catterall W.A. (2000) Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell. Dev. Biol., 16, 521–555. [DOI] [PubMed] [Google Scholar]
- 17.Underhill P.A., Jin,L., Lin,A.A., Mehdi,S.Q., Jenkins,T., Vollrath,D., Davis,R.W., Cavalli-Sforza,L.L. and Oefner,P.J. (1997) Detection of numerous Y chromosome biallelic polymorphisms by denaturing high-performance liquid chromatography. Genome Res., 7, 996–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu W., Smith,D.I., Rechtzigel,K.J., Thibodeau,S.N. and James,C.D. (1998) Denaturing high performance liquid chromatography (DHPLC) used in the detection of germline and somatic mutations. Nucleic Acids Res., 26, 1396–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laird P.W., Zijderveld,A., Linders,K., Rudnicki,M.A., Jaenisch,R. and Berns,A. (1991) Simplified mammalian DNA isolation procedure. Nucleic Acids Res., 19, 4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bingham P.M., Levis,R. and Rubin,G.M. (1981) Cloning of DNA sequences from the white locus of D. melanogaster by a novel and general method. Cell, 25, 693–704. [DOI] [PubMed] [Google Scholar]
- 21.Chirgwin J.M., Przybyla,A.E., MacDonald,R.J. and Rutter,W.J. (1979) Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry, 18, 5294–5299. [DOI] [PubMed] [Google Scholar]
- 22.Huber C.G., Oefner,P.J., Preuss,E. and Bonn,G.K. (1993) High-resolution liquid chromatography of DNA fragments on non-porous poly(styrene-divinylbenzene) particles. Nucleic Acids Res., 21, 1061–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao W. and Oefner,P.J. (2001) Denaturing high-performance liquid chromatography: a review. Hum. Mutat., 17, 439–474. [DOI] [PubMed] [Google Scholar]
- 24.van Den Bosch B.J., de Coo,R.F., Scholte,H.R., Nijland,J.G., van Den Bogaard,R., de Visser,M., de Die-Smulders,C.E. and Smeets,H.J. (2000) Mutation analysis of the entire mitochondrial genome using denaturing high performance liquid chromatography. Nucleic Acids Res., 28, e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolford J.K., Blunt,D., Ballecer,C. and Prochazka,M. (2000) High-throughput SNP detection by using DNA pooling and denaturing high performance liquid chromatography (DHPLC). Hum. Genet., 107, 483–487. [DOI] [PubMed] [Google Scholar]
- 26.Ke S.H. and Wartell,R.M. (1993) Influence of nearest neighbor sequence on the stability of base pair mismatches in long DNA; determination by temperature-gradient gel electrophoresis. Nucleic Acids Res., 21, 5137–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herb A., Higuchi,M., Sprengel,R. and Seeburg,P.H. (1996) Q/R site editing in kainate receptor GluR5 and GluR6 pre-mRNAs requires distant intronic sequences. Proc. Natl Acad. Sci. USA, 93, 1875–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith L.A., Peixoto,A.A. and Hall,J.C. (1998) RNA editing in the Drosophila DMCA1A calcium-channel alpha 1 subunit transcript. J. Neurogenet., 12, 227–240. [DOI] [PubMed] [Google Scholar]
- 29.Strong M., Chandy,K.G. and Gutman,G.A. (1993) Molecular evolution of voltage-sensitive ion channel genes: on the origins of electrical excitability. Mol. Biol. Evol., 10, 221–242. [DOI] [PubMed] [Google Scholar]
- 30.Fletcher C.F., Lutz,C.M., O’Sullivan,T.N., Shaughnessy,J.D.,Jr, Hawkes,R., Frankel,W.N., Copeland,N.G. and Jenkins,N.A. (1996) Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell, 87, 607–617. [DOI] [PubMed] [Google Scholar]
- 31.Kawasaki F., Felling,R. and Ordway,R.W. (2000) A temperature-sensitive paralytic mutant defines a primary synaptic calcium channel in Drosophila. J. Neurosci., 20, 4885–4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang Z.H. and Wu,J.Y. (1999) Alternative splicing and programmed cell death. Proc. Soc. Exp. Biol. Med., 220, 64–72. [DOI] [PubMed] [Google Scholar]
- 33.Crook R., Verkkoniemi,A., Perez-Tur,J., Mehta,N., Baker,M., Houlden,H., Farrer,M., Hutton,M., Lincoln,S., Hardy,J., Gwinn,K., Somer,M., Paetau,A., Kalimo,H., Ylikoski,R., Poyhonen,M., Kucera,S. and Haltia,M. (1998) A variant of Alzheimer’s disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nature Med., 4, 452–455. [DOI] [PubMed] [Google Scholar]
- 34.Higuchi M., Maas,S., Single,F.N., Hartner,J., Rozov,A., Burnashev,N., Feldmeyer,D., Sprengel,R. and Seeburg,P.H. (2000) Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature, 406, 78–81. [DOI] [PubMed] [Google Scholar]
- 35.Croft L., Schandorff,S., Clark,F., Burrage,K., Arctander,P. and Mattick,J.S. (2000) ISIS, the intron information system, reveals the high frequency of alternative splicing in the human genome. Nature Genet., 24, 340–341. [DOI] [PubMed] [Google Scholar]
- 36.Kan Z., Rouchka,E.C., Gish,W.R. and States,D.J. (2001) Gene structure prediction and alternative splicing analysis using genomically aligned ESTs. Genome Res., 11, 889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Modrek B., Resch,A., Grasso,C. and Lee,C. (2001) Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res., 29, 2850–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yeakley J.M., Fan,J.B., Doucet,D., Luo,L., Wickham,E., Ye,Z., Chee,M.S. and Fu,X.D. (2002) Profiling alternative splicing on fiber-optic arrays. Nat. Biotechnol., 20, 353–358. [DOI] [PubMed] [Google Scholar]
- 39.Wagner T., Stoppa-Lyonnet,D., Fleischmann,E., Muhr,D., Pages,S., Sandberg,T., Caux,V., Moeslinger,R., Langbauer,G., Borg,A. and Oefner,P. (1999) Denaturing high-performance liquid chromatography detects reliably BRCA1 and BRCA2 mutations. Genomics, 62, 369–376. [DOI] [PubMed] [Google Scholar]
- 40.Hans M., Urrutia,A., Deal,C., Brust,P.F., Stauderman,K., Ellis,S.B., Harpold,M.M., Johnson,E.C. and Williams,M.E. (1999) Structural elements in domain IV that influence biophysical and pharmacological properties of human alpha1A-containing high-voltage-activated calcium channels. Biophys. J., 76, 1384–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin Z., Lin,Y., Schorge,S., Pan,J.Q., Beierlein,M. and Lipscombe,D. (1999) Alternative splicing of a short cassette exon in alpha1B generates functionally distinct N-type calcium channels in central and peripheral neurons. J. Neurosci., 19, 5322–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]