

Abstract
Epithin is a type II transmembrane serine protease that exists in a soluble and membrane-bound form. Shedding is thought to be important in regulating its action, but little is known regarding the intracellular events that trigger such shedding. Here, we show that phorbol myristate acetate (PMA) causes the release of epithin. It also causes accumulation of the protein at the site of cell–cell contacts, and this accumulation is dependent on the formation of cortical actin. In addition, we have identified the actin-binding protein, filamin, as the linker between epithin and the actin cytoskeleton. The interaction of epithin and filamin was enhanced by PMA, and epithin was not released from filamin-deficient M2 cells. We also show that the release of epithin does not require its own activity and is blocked by a metalloprotease inhibitor, GM6001. These results show that filamin has an essential role in shedding by linking epithin to the as yet unidentified metalloprotease-shedding enzyme(s).
Keywords: ectodomain shedding, epithin, filamin, actin filaments, PMA
Introduction
Several transmembrane proteins containing carboxy-terminal extracellular serine protease domains have been identified and grouped as type II transmembrane serine proteases. These proteases have common structural features, such as a proteolytic domain, a stem region containing modular structural signatures, a transmembrane domain and a short cytoplasmic domain (Hooper et al, 2001). Epithin (also known as membrane typeserine protease 1 (MT-SP1)/matriptase, in humans) is a member of the group that has all the above features (Fig 1A; Kim et al, 1999). Similarly to some other members of the group, epithin exists in a soluble and membrane-bound form (Lin et al, 1997; Cho et al, 2001). It has been implicated in tumour cell invasion and metastasis by proteolytic activation of a urokinase-type plasminogen activator (Takeuchi et al, 2000) and hepatocyte growth factor/scatter factor (Lee et al, 2000).
Figure 1.
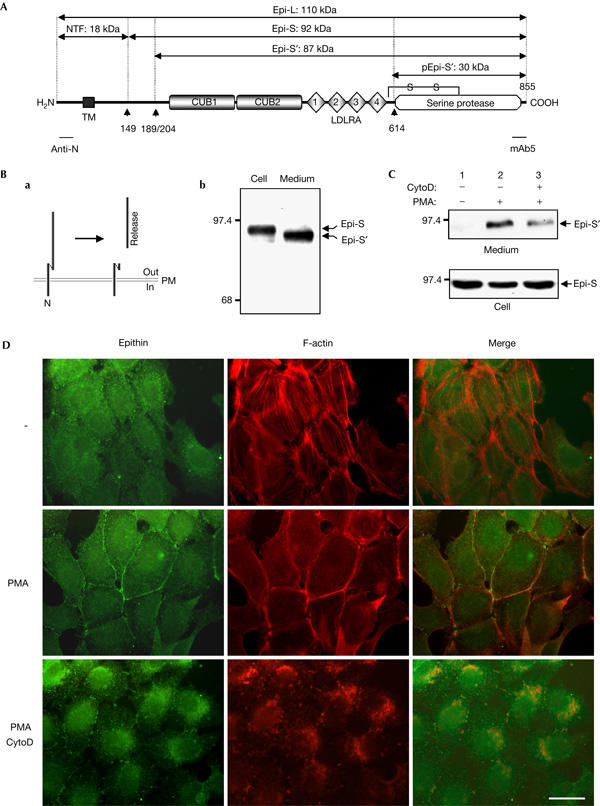
Phorbol myristate acetate induces local accumulation and release of epithin with concurrent actin reorganization. (A) Domain structure of epithin. CUB, CUB domain; LDLRA, low-density lipoprotein receptor ligand-binding repeats; TM, transmembrane region. Possible cleavage sites for amino-terminal processing (▴149), release (▴189/204; Benaud et al, 2001) and activation (▴614) are indicated, and the estimated molecular masses (in kDa) of the resulting fragments are designated (full-length epithin; Epi-L, 1–149; NTF, 149–855; Epis, 189/204–855; Epi-S′, 614–855; pEpi-S′). Regions that are recognized by the anti-N antibody and mAb5 are marked. (B) (a) Schematic diagram showing that epithin exists as a membrane-bound form linked to the N-terminal fragment or as a released form. (b) Comparison of the molecular mass of membrane-bound and soluble forms of epithin. Epithin was detected by western blotting with mAb5. (C) 427.1.86 cells were treated with 0.5 μM cytochalasin D (CytoD) and/or 1 μM phorbol myristate acetate (PMA) in serum-free media, as indicated. Proteins in the media were precipitated with trichloroacetic acid (TCA) and analysed by western blotting with mAb5. Amounts of epithin in the cell lysates are shown in the lower panel. (D) The localization of epithin and F-actin was analysed by fluorescence microscopy after staining with anti-epithin antiserum and rhodamine-conjugated phalloidin, respectively. Scale bar, 20 μm.
We have reported previously that epithin is a 110 kDa type II transmembrane serine protease that is converted to a 92 kDa form linked to its own amino-terminal fragment after processing at Gly 149 (Fig 1A). We had suggested that epithin can undergo further cleavage or structural change as a consequence of an unknown triggering event and be released as a soluble protein (Cho et al, 2001). However, the signalling events that trigger this release are still largely unknown.
In many transmembrane proteins, such as transmembrane growth factors, membrane receptors and cell adhesion molecules, the extracellular region can be released into the medium by proteolysis at the cell surface. This is essential for regulating the cellular function of these transmembrane proteins (Arribas & Borroto, 2002). In many instances, the phorbol ester phorbol myristate acetate (PMA), which activates protein kinase C, stimulates the release of a transmembrane protein, such as transforming growth factor, tumour necrosis factor and Lselectin (Arribas et al, 1996). Type II transmembrane serine proteases, including epithin, have a short cytoplasmic domain. The potentials of these domains in interactions with cytoskeletal components and signalling molecules—and in the targeting of the proteases to a particular cell surface—have been suggested (Hooper et al, 2001). The cytoplasmic domain of epithin is composed of 55 amino acids and contains a consensus phosphorylation site for protein kinase C. Therefore, it is possible that this domain may have a role in the interaction of epithin and cytoskeletal proteins, and PMA may modulate this interaction to regulate the shedding of the protease.
In this study, we show that actin reorganization induced by PMA causes the local accumulation of epithin in a filamin-dependent manner, and that this is responsible for the release of epithin, possibly by facilitating its cleavage by a shedding enzyme.
Results And Discussion
Translocation and release of epithin is induced by PMA
Previously, we showed that epithin exists in a soluble or membrane-bound form in the mouse thymic epithelial cell line 427.1.86 cells (Fig 1Ba; Cho et al, 2001). Compared with the membrane-bound form, the soluble form has a smaller size, which indicates that a further cleavage other than that at Gly 149 is required for the release (Fig 1Bb). To investigate whether PMA can induce the release of epithin, as described for other transmembrane proteins (Fan & Derynck, 1999), we treated 427.1.86 cells with PMA. As shown in Fig 1C, this led to an increase in the amount of epithin recovered in trichloroacetic acid (TCA) precipitates of the culture medium (Fig 1C, lane 2). Moreover, epithin, which was dispersed throughout the plasma membrane in serum-free medium, was translocated to areas of cell–cell contact by PMA treatment (Fig 1D, left panels). Interestingly, PMA treatment also induced a rearrangement of the actin cytoskeleton from stress fibres to cortical actin filaments (Fig 1D, middle panels), and this new pattern overlapped with that of epithin at sites of cell–cell contact (Fig 1D, right panels). This coincidence led us to suggest that actin reorganization has an essential role in the accumulation of epithin at sites of cell–cell contact and also in its release. To test this, we pretreated cells with 0.5 μM cytochalasin D, a well-known F-actin-disrupting agent, before PMA treatment. This led to a marked inhibition (about 60% inhibition by densitometric quantification) of the PMA-induced release of epithin (Fig 1C, lane 3; also see supplementary Fig S1 online) and of its accumulation at cell–cell contacts (Fig 1D). These data indicate that actin reorganization is required for the local accumulation and release of epithin.
Filamin links epithin to actin filaments
We next inquired how rearrangement of the actin cytoskeleton is related to the accumulation of epithin at cell–cell contacts, and we considered that there might be a linker molecule that connects epithin to the PMA-induced cortical actin filaments. To identify the linker molecule, we carried out a yeast two-hybrid screen using the epithin intracellular domain as bait, and isolated a mouse complementary DNA encoding an 85-amino-acid sequence (Fig 2A), referred to as Y2. Y2 proved to be identical to the C terminus of an expressed sequence, AL024016 (accession number XP_127565), which is thought to be mouse filamin-B. It was 98% identical to the corresponding C terminus of human filamin-B (Takafuta et al, 1998) and 70% identical to that of human filamin-A (Fig 2A). Interaction of the two proteins was confirmed by showing that 6 × histidine (His)/thioredoxin-tagged Y2 was efficiently pulled down by the glutathione S-transferase (GST)-fused epithin intracellular domain (Fig 2B).
Figure 2.
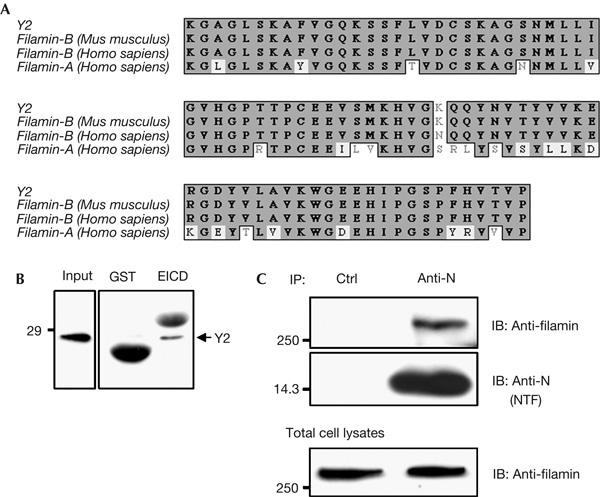
Filamin interacts with the intracellular domain of epithin. (A) Comparison of Y2 with the carboxyl termini of mouse filamin-B, human filamin-B and human filamin-A. Grey-boxed residues represent sequence identities, and residues inside the black line represent sequence homology. (B) 6 × histidine (His)/thioredoxin-tagged Y2 protein tested for interaction with glutathione S-transferase (GST) or GST–epithin intracellular domain (EICD) fusion protein. The arrows indicate the 6 × His/thioredoxin-tagged Y2 probed with anti-His tag monoclonal antibody. GST and GST–EICD proteins used for pull-down of Y2 were visualized nonspecifically with anti-His tag antibody owing to the large amounts of protein. (C) After transfection of epithin complementary DNA into COS7 cells, cell lysates were immunoprecipitated (IP) with normal rabbit serum (Ctrl) or anti-N antiserum, and blotted (IB) with anti-filamin antibody or anti-N antiserum, as indicated.
Filamin is known to interact with actin through the actin-binding domain at its N terminus, and to promote orthogonal branching of the actin filaments. It has also been reported recently that filamin interacts with several other transmembrane proteins through its C-terminal region, stabilizing them at the cell membrane (Stossel et al, 2001). We therefore suspected that filamin could be the molecular bridge that links transmembrane epithin to the actin cytoskeleton.
As Y2 and human filamin-A show significant sequence identity in the C-terminal region, and no commercial antibody against mouse filamin-B was available, we first tested the interaction between human filamin-A and the epithin intracellular domain by a co-immunoprecipitation assay. When cells expressing the C terminus of human filamin-A were transfected with epithin, we were able to detect filamin-A in the immunoprecipitated epithin complex (data not shown). Moreover, endogenous filamin-A in COS7 cells was efficiently co-immunoprecipitated with epithin (Fig 2C), confirming that the epithin intracellular domain interacts with filamin-A as well as filamin-B. We therefore used filamin-A in further experiments.
PMA enhances interaction between epithin and filamin
To examine the nature of the interaction between epithin and filamin, we investigated the interactions of the two proteins under various conditions by co-immunoprecipitation assays. The epithin and filamin interaction was greatly influenced by the presence of serum in culture medium (supplementary Fig S2 online), probably due to stimulation by serum factors. We observed a weak interaction between epithin and filamin in control epithin-transfected COS7 cells in the absence of serum (Fig 3A, lane 1). However, when the cells were treated with PMA, filamin was efficiently brought down with epithin (Fig 3A, lane 2). We also observed that epithin and filamin accumulated and colocalized at the cell margins after PMA treatment (Fig 3B, arrows), confirming the co-immunoprecipitation result. The PMA-induced interaction between epithin and filamin was abolished by cytochalasin D (Fig 3C; also see supplementary Fig S3 online), showing that the interaction is dependent on an intact actin cytoskeleton. These data indicate that filamin links epithin to the PMA-induced cortical actin filaments, resulting in the local accumulation of epithin in actin-rich cortical regions, such as cell–cell contacts and cell margins. Although the molecular mechanism by which PMA enhances interaction of the two proteins is not known, it is reasonable to assume that when filamin is bound to cortical actin it has improved access to epithin on the membrane.
Figure 3.
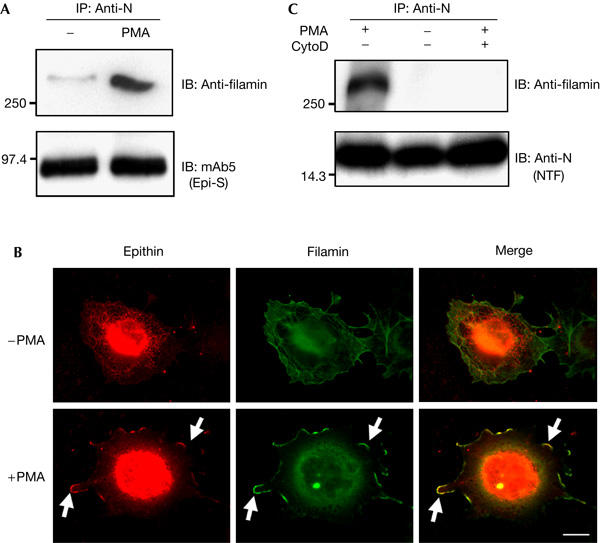
Phorbol myristate acetate induces the interaction of epithin and filamin. (A) COS7 cells were treated as indicated after transfection with epithin complementary DNA. Cell lysates were immunoprecipitated (IP) with anti-N antiserum and blotted (IB) with anti-filamin antibody or with mAb5. (B) The localization of epithin and filamin in the cells was analysed by fluorescence microscopy after staining with anti-epithin antiserum and anti-filamin antibody, respectively. Arrows indicate colocalization of epithin and filamin in cell marginal regions. Scale bar, 20 μm. (C) Epithin cDNA-transfected COS7 cells were treated with 1 μM phorbol myristate acetate (PMA) with or without 10 μM cytochalasin D (CytoD) pretreatment, as indicated. The cell lysates were immunoprecipitated with anti-N antiserum and blotted with anti-filamin antibody or anti-N antiserum.
Epithin is not released in filamin-deficient M2 cells
As discussed above, cortical actin formation is required for the PMA-induced release of epithin, and filamin can link epithin to the cortical actin. Therefore, we asked whether epithin can be released if its interaction with filamin is impaired. To answer this question, we examined the release of epithin from a filamin-deficient cell line, M2, originating from a human malignant melanoma that does not express detectable filamin-A messenger RNA or protein (Cunningham et al, 1992). As M2 cells do not express epithin, they were transfected with epithin cDNA. The expressed epithin was not released (Fig 4, lane 2) even after PMA treatment (Fig 4, lane 5). However, when filamin-A cDNA was introduced into the M2 cells by transfection, some release was observed in the absence of stimulation (Fig 4, lane 3), and this was greatly enhanced by PMA treatment (Fig 4, lane 6). The interaction of epithin with filamin is evidently essential for the release of epithin. The release was also dependent on the dose of filamin-A (supplementary Fig S4 online).
Figure 4.
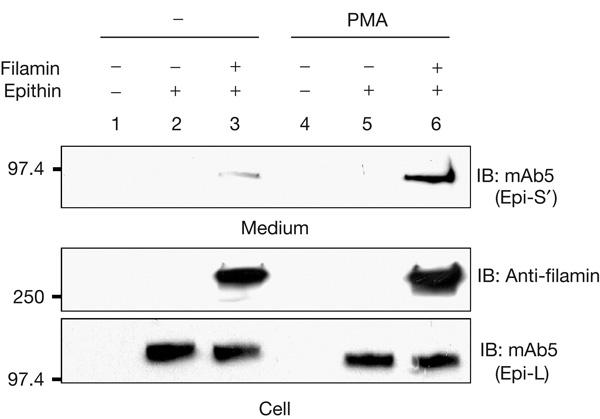
Filamin is essential for the release of epithin. Epithin complementary DNA was transfected into M2 cells with or without human filamin-A cDNA. At 24 h after transfection, the cells were treated as indicated. Trichloroacetic acid-precipitated proteins from the medium were analysed by western blotting (IB) with mAb5 (upper panel). The cell lysates were also examined for expression of filamin and epithin (lower panel). PMA, phorbol myristate acetate.
Next, we asked whether the interaction of epithin with filamin is dependent on actin filament and whether filamin-B can bind to epithin as efficiently as filamin-A does. For this, co-immunoprecipitation of the actin-binding-deficient filamin-B (filamin-B1704−2602) and the transfected epithin was examined in M2 cells. The filamin-B1704−2602 was efficiently co-immunoprecipitated with epithin (Fig 5A), which indicates that the interaction of the two proteins is independent of actin filaments. However, this interaction was not modulated by PMA or by cytochalasin D. These data indicate that the PMA-induced rearrangement of actin filaments and its binding to filamin are required for the PMA-induced interaction between epithin and intact filamin. In addition, the expression of filamin-B1704−2602 competitively inhibited the interaction between epithin and filamin-A in M2 cells (Fig 5B). The release of endogenous epithin in the 427.1.86 cells was also inhibited by the transfected filamin-B1704−2602 in a dose-dependent manner (Fig 5C). These results indicate that filamin-B can efficiently compete with filamin-A for binding to epithin. Similar experiments with the actin-binding-deficient filamin-A1534−2647 also showed essentially the same results (supplementary Fig S5 online).
Figure 5.
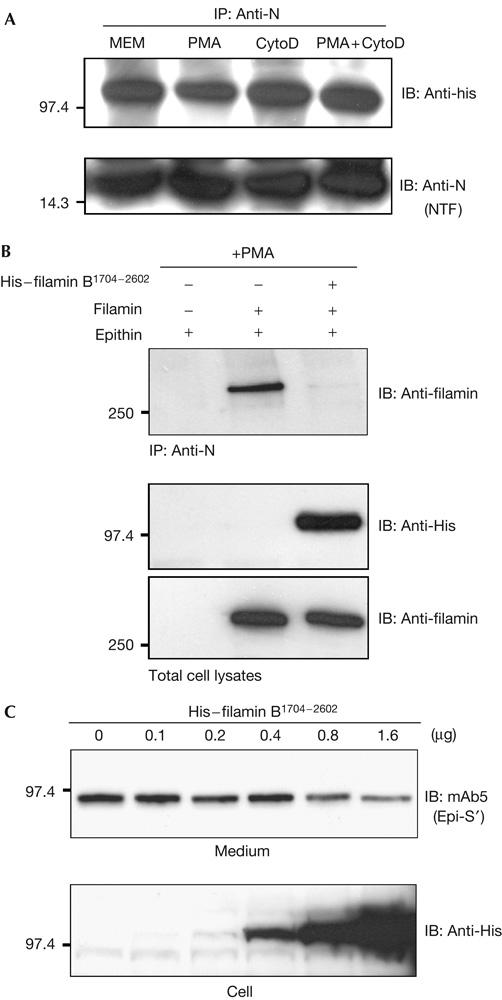
Interaction of filamin with epithin and release of epithin are inhibited by the actin-binding-deficient filamin-B. (A) Epithin and histidine (His)-tagged human filamin-B1704−2602 constructs were co-transfected into M2 cells, and the cells were treated with phorbol myristate acetate (PMA) and/or cytochalasin D (CytoD), as indicated. Cell lysates were immunoprecipitated (IP) with anti-N antiserum, and blotted (IB) with anti-His antibody or with anti-N antiserum. (B) M2 cells were transfected with indicated complementary DNAs and treated with PMA. Cell lysates were immunoprecipitated with anti-N antiserum, and blotted with anti-filamin antiserum for filamin-A (upper panel). The amounts of filamin-A and His–filamin-B1704−2602 in the cell lysates are shown (lower panel). (C) Release of endogenous epithin in 427.1.86 cells was examined after transfection of the increasing amounts of His–filamin-B1704−2602 cDNAs, as indicated.
Release of epithin is blocked by GM6001
Recently, it has been reported that the release of epithin is inhibited by serine protease inhibitors, ecotin and leupeptin, and that an epithin variant, epithin(Δ), which lacks 66 amino acids including the protease activation site, is incapable of being shed. On the basis of these findings, it was speculated that the serine protease activity of epithin itself and of other serine proteases are important factors for epithin release (Cho et al, 2005). Conversely, many PMA-induced shedding events have been known to be mediated by metalloproteases (Arribas et al, 1996). Therefore, to investigate the nature of the shedding enzyme for epithin, we tested whether the PMA-induced release of epithin is sensitive to a serine protease inhibitor, ecotin, or to a metalloprotease inhibitor, GM6001. To our surprise, the PMA-induced release of the endogenous epithin in 427.1.86 cells was inhibited by both ecotin and GM6001 (Fig 6A). To clarify whether the catalytic activity of epithin is essential for its own release, we examined the release of a catalytically inactive epithin mutant (S805A; Takeuchi et al, 1999) following PMA treatment in M2 cells. The release of the S805A mutant was triggered by filamin transfection and was greatly enhanced by PMA treatment in M2 cells, as observed with wild-type epithin (Fig 6B, lanes 1–3), suggesting that the release of epithin is independent of its own activity. Interestingly, whereas the release of wild-type epithin was inhibited by both ecotin and GM6001 in M2 cells (data not shown), the release of the S805A mutant was inhibited only by GM6001 and not by ecotin (Fig 6B, lanes 4,5). These results indicate that the shedding enzyme responsible for the PMA-induced release of epithin may be a metalloprotease. They also indicate that ecotin could inhibit the release of wild-type epithin not by inhibiting protease activity, but by binding to the active site of the protease and preventing its maturation to a form with the ability to be shed. In this regard, the shedding defect of epithin(Δ) observed by Cho et al (2005) could be due to a structural change that prevents the shedding enzyme from accessing it, rather than owing to the absence of protease activity.
Figure 6.
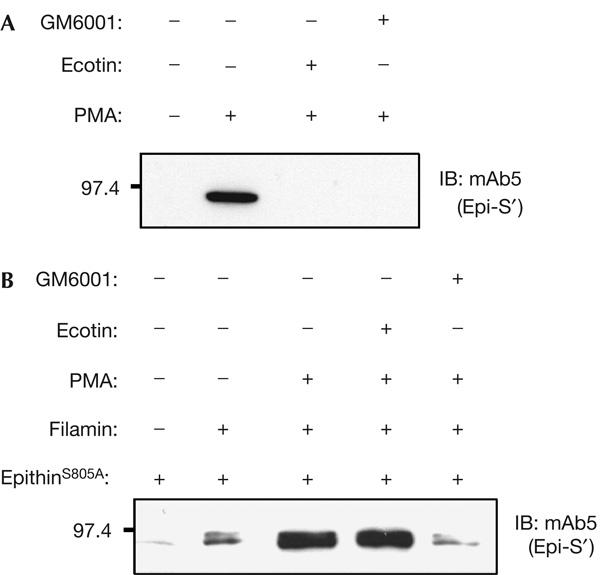
The metalloprotease inhibitor, GM6001, inhibits phorbol myristate acetate-induced release of epithin. (A) 427.1.86 cells in serum-free medium were treated with 500 nM ecotin or 50 μM GM6001, and incubated for 1 h with 1 μM phorbol myristate acetate (PMA), as indicated. Trichloroacetic acid-precipitated proteins in the medium were analysed by western blotting (IB) with mAb5. (B) A catalytically inactive epithin mutant (S805A) was coexpressed with human filamin-A in M2 cells, and release of the mutant epithin into the medium was analysed as in (A).
Methods
Cell lines, antibodies and cDNAs. COS7 and 427.1.86 cells were maintained in DMEM with 10% FBS. The filamin-A cDNA and M2 cells were kindly provided by Dr Thomas P. Stossel (Harvard Medical School). The human filamin-B cDNA was a gift from Dr Toshiro Takafuta (University of Yamanashi). Anti-N antiserum and monoclonal antibody, mAb5, have been described previously (Cho et al, 2001). Mouse monoclonal anti-filamin antibody, mAb1678, was purchased from Chemicon Inc. (Temecula, CA, USA) and the anti-His tag monoclonal antibody was purchased from Novagen (Madison, WI, USA).
Detection of soluble epithin. 427.1.86 cells were serum starved for 1 h with or without 0.5 μM cytochalasin D (Sigma, St Louis, MO, USA) and incubated for a further 1 h with or without 1 μM PMA (Sigma) in serum-free medium. Proteins in the medium were precipitated with TCA (10%, final concentration). The precipitates were dissolved in Laemmli SDS reducing sample buffer, and subjected to SDS–polyacrylamide gel electrophoresis and western blotting. pcDNA3/epithin and filamin cDNAs were introduced into M2 cells using LipofectAMINE Plus reagent (Life Technologies Inc., Gaithersburg, MD, USA). At 24 h after transfection, the cells were incubated with serum-free medium for 2 h, and samples were prepared as described above.
Yeast two-hybrid screening and plasmid constructs. The PCR-amplified epithin intracellular domain was cloned in-frame into pGBKT7 to produce a fusion between the Gal4-binding domain and the epithin intracellular domain. This plasmid was used to screen a pretransformed mouse 17-day embryo Matchmaker cDNA library (3.5 × 106 independent clones; average cDNA size, 2.0 kb; cDNA size range, 0.4–4.0 kb; CLONTECH Laboratories Inc., Palo Alto, CA, USA) according to the manufacturer's instructions. A total of 3.5 × 106 independent clones were screened, and the resulting nine interacting plasmids were subsequently transformed into Escherichia coli, purified and sequenced. They were digested with EcoR1 and Xho1, and ligated in-frame into 6 × His/thioredoxin-tagged pET32c(+) (Novagen) for use in pull-down analysis.
Pull-down analysis and immunoprecipitation. The expression of candidate epithin-interacting proteins in E. coli was induced with 1 mM isopropyl-β-D-thiogalactoside, and the cells were lysed by sonication in lysis buffer (20 mM Tris–HCl, pH 8.0, 200 mM NaCl, 1 mM EDTA, 0.5% NP-40 and protease inhibitors). Pull-down of the interacting proteins in the lysates with a GST–epithin intracellular domain fusion protein was carried out as described (Kim et al, 2001). Out of the nine candidate epithin-interacting proteins, only one, Y2, was positive in the pull-down assay.
For immunoprecipitation, pcDNA3/epithin was transfected into COS7 cells in 100 mm dishes using LipofectAMINE Plus reagent. At 1 day after transfection, epithin was immunoprecipitated by anti-N antiserum (Cho et al, 2001).
Immunofluorescence staining. Immunocytochemistry was carried out as described elsewhere (Kim et al, 2001). Epithin and filamin were detected by incubation with purified anti-epithin antiserum and mouse monoclonal anti-filamin antibody, respectively, followed by fluorescein isothiocyanate-labelled or tetramethyl rhodamine isothiocyanate-labelled secondary antibody (Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA). Cells were observed with a fluorescence microscope (Axioplan2, Zeiss) equipped with a × 63 (numerical aperture, 1.4) Planapochromat objective lens. Fluorescence images were collected using a digital camera with a cooled charge-coupled device (Axio-Cam, Zeiss). Individual images were processed and assembled with Photoshop 6.0 (Adobe system).
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
Supplementary Information
Acknowledgments
This work was supported by grant M1-0203-00-0072 of the National Research Laboratory Program and grant M103KV010013-04K2201-01310 from Brain Research Center of the 21st century Frontier Research Program, funded by the Ministry of Science and Technology, Republic of Korea (to D.P.). C.K. and Y.C. were supported by BK21 fellowships.
References
- Arribas J, Borroto A (2002) Protein ectodomain shedding. Chem Rev 102: 4627–4638 [DOI] [PubMed] [Google Scholar]
- Arribas J, Coodly L, Vollmer P, Kishimoto TK, Rose-John S, Massague J (1996) Diverse cell surface protein ectodomains are shed by a system sensitive to metalloprotease inhibitors. J Biol Chem 271: 11376–11382 [DOI] [PubMed] [Google Scholar]
- Benaud C, Dickson RB, Lin CY (2001) Regulation of the activity of matriptase on epithelial cell surfaces by a blood-derived factor. Eur J Biochem 268: 1439–1447 [DOI] [PubMed] [Google Scholar]
- Cho EG, Kim MG, Kim C, Kim SR, Seong IS, Chung C, Schwartz RH, Park D (2001) N-terminal processing is essential for release of epithin, a mouse type II membrane serine protease. J Biol Chem 276: 44581–44589 [DOI] [PubMed] [Google Scholar]
- Cho EG, Schwartz RH, Kim MG (2005) Shedding of membrane epithin is blocked without LDLRA4 and its protease activation site. Biochem Biophys Res Commun 327: 328–334 [DOI] [PubMed] [Google Scholar]
- Cunningham CC, Gorlin JB, Kwiatkowski DJ, Hartwig JH, Janmey PA, Byers HR, Stossel TP (1992) Actin-binding protein requirement for cortical stability and efficient locomotion. Science 255: 325–327 [DOI] [PubMed] [Google Scholar]
- Fan H, Derynck R (1999) Ectodomain shedding of TGF-α and other transmembrane proteins is induced by receptor tyrosine kinase activation and MAP kinase signaling cascades. EMBO J 18: 6962–6972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper JD, Clements JA, Quigley JP, Antalis TM (2001) Type II transmembrane serine proteases. Insights into an emerging class of cell surface proteolytic enzymes. J Biol Chem 276: 857–860 [DOI] [PubMed] [Google Scholar]
- Kim MG, Chen C, Lyu MS, Cho EG, Park D, Kozak C, Schwartz RH (1999) Cloning and chromosomal mapping of a gene isolated from thymic stromal cells encoding a new mouse type II membrane serine protease, epithin, containing four LDL receptor modules and two CUB domains. Immunogenetics 49: 420–428 [DOI] [PubMed] [Google Scholar]
- Kim S, Lee SH, Park D (2001) Leucine zipper-mediated homodimerization of the Pak kinase interacting factor, βPix: implication for a role in cytoskeletal reorganization. J Biol Chem 276: 10581–10584 [DOI] [PubMed] [Google Scholar]
- Lee SL, Dickson RB, Lin CY (2000) Activation of hepatocyte growth factor and urokinase/plasminogen activator by matriptase, an epithelial membrane serine protease. J Biol Chem 275: 36720–36725 [DOI] [PubMed] [Google Scholar]
- Lin CY, Wang JK, Torri J, Dou L, Sang QA, Dickson RB (1997) Characterization of a novel, membrane-bound, 80-kDa matrix-degrading protease from human breast cancer cells. Monoclonal antibody production, isolation, and localization. J Biol Chem 272: 9147–9152 [PubMed] [Google Scholar]
- Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M, Shapiro SS (2001) Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol 2: 138–145 [DOI] [PubMed] [Google Scholar]
- Takafuta T, Wu G, Murphy GF, Shapiro SS (1998) Human β-filamin is a new protein that interacts with the cytoplasmic tail of glycoprotein Ibα. J Biol Chem 273: 17531–17538 [DOI] [PubMed] [Google Scholar]
- Takeuchi T, Shuman MA, Craik CS (1999) Reverse biochemistry: use of macromolecular protease inhibitors to dissect complex biological processes and identify a membrane-type serine protease in epithelial cancer and normal tissue. Proc Natl Acad Sci USA 96: 11054–11061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, Harris JL, Huang W, Yan KW, Coughlin SR, Craik CS (2000) Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J Biol Chem 275: 26333–26342 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information