

Abstract
The Wnt signalling pathway can activate transcription of genes such as c-myc through β-catenin. Here, we describe the protein breakpoint cluster region, Bcr, as a negative regulator of this pathway. Bcr can form a complex with β-catenin and negatively regulate expression of c-Myc. Knockdown of Bcr by short interfering RNA relieves the block and activates expression of c-Myc. Expression of Bcr in the human colon carcinoma cell line HCT116, which has a high level of endogenous β-catenin, leads to reduced c-Myc expression. The negative effect is exerted by the amino terminus of Bcr, which does not harbour the kinase domain. Bcr-Abl, the oncogene protein expressed in chronic myelogenous leukaemia (CML), does not bind to β-catenin. It phosphorylates Bcr in the first exon sequence on tyrosines, which abrogates the binding of Bcr to β-catenin. The inhibitor of the Bcr–Abl tyrosine kinase, STI-571 or Gleevec, a drug against CML, reverses this effect. Our data contribute to the understanding of Bcr as a tumour suppressor in the Wnt signalling pathway, as well as in CML.
Keywords: tumour suppressor, Bcr, Bcr–Abl, β-catenin, Myc, STI-571, Gleevec
Introduction
The human breakpoint cluster region protein, Bcr, is a multidomain protein with several enzymatic functions. Bcr contains a serine/threonine protein kinase domain (Maru & Witte, 1991), a guanine nucleotide exchange factor (GEF) function and a GTPase-activating protein (GAP) domain (Fig 1A). Furthermore, Bcr contains an oligomerization domain at its amino terminus, and a carboxy-terminal sequence (s-T-E-V), which is a ligand for a PDZ domain (Radziwill et al, 2003).
Figure 1.
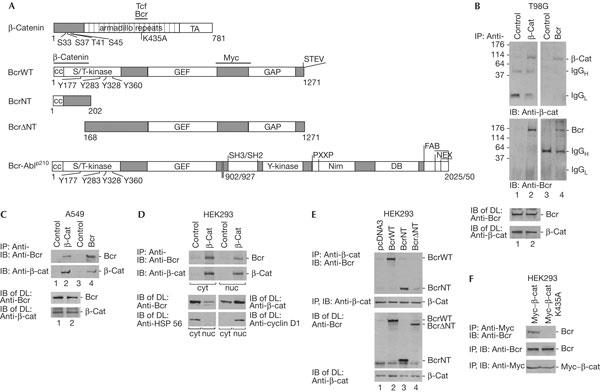
Interaction of Bcr with β-catenin. (A) Schematic diagrams of β-catenin, BcrWT, BcrNT, BcrΔNT and Bcr-Ablp210 constructs used. Bcr is a serine/threonine (S/T) kinase with four tyrosine phosphorylation sites. GEF is a guanine nucleotide exchange factor and GAP is a GTPase-activating protein. Bcr–Abl is a tyrosine (Y) kinase and a fusion protein with various breakpoints of Bcr at 902 or 927. p210 indicates the molecular mass (kDa); SH2 and SH3 are Src-homology domains; PXXP is the proline-rich region; NIM is the nuclear import region; DB is a DNA-binding domain; FAB is an actin binding site; NEX is the nuclear export region. Horizontal bars mark binding regions of the proteins indicated. TA denotes transactivator. (B) T98G is a glioblastoma cell line. Immunoprecipitation (IP) and subsequent immunoblot (IB) were carried out with antibodies as indicated, and with an irrelevant antibody as a control. IgG heavy and light chains (H, L) and molecular mass in kDa are indicated as markers. (C) Same as (B) except using an A549 human lung carcinoma cell line. Protein expression levels were analysed in direct lysates (DL) by IB. (D) (Top) Co-IP of endogenous Bcr with β-catenin from cytoplasmic (cyt) and nuclear (nuc) extracts from human embryonic kidney (HEK)293 cells. IP, IB and DL were carried out as indicated. Cyclin D1 served as a nuclear marker and heatshock protein HSP 56 as a cytoplasmic marker. (E) The plasmids coding for BcrWT, BcrNT and BcrΔNT were transfected into HEK293 cells; pcDNA3 is an empty plasmid control. IPs were carried out with anti-β-catenin, followed by IB with anti-Bcr (N-20 and C-20 together). Protein expression controls are shown below. (F) A Myc-tagged β-catenin wild type and a point mutant of β-catenin (K435A) were expressed ectopically in HEK293 cells. Co-IP and IB were carried out as indicated (top). Protein expression controls are shown below.
Bcr is a negative regulator of the small GTPase Rac—owing to its GAP function—and blocks the p21-activating kinase, Pak (Voncken et al, 1995). In nonstimulated cells, Bcr is active as a serine/threonine kinase and it has recently been shown in these cells that Bcr negatively regulates the Ras–Raf–MEK–ERK (MEK, mitogen-activated protein kinase kinase; ERK, extracellular signal-regulated kinase) signalling pathway through phosphorylation of AF-6, a Ras-binding protein (Radziwill et al, 2003).
Bcr is a fusion partner of Abl in Bcr–Abl (Fig 1A), which is a hallmark of chronic myelogenous leukaemia (CML; for a review, see Arlinghaus, 2002). The Bcr–Abl fusion protein exhibits tyrosine kinase activity through Abl, which phosphorylates Bcr in cis or trans on tyrosines (Liu et al, 1996). Bcr has been described as a negative regulator of the pathogenic effects of Bcr–Abl (Wu et al, 1999; Arlinghaus, 2002). Expression of Bcr–Abl in CML is accompanied by high expression levels of c-Myc, which contributes to disease progression (Sawyers, 1993). c-Myc is overexpressed in several tumours and tumour cell lines (Marcu et al, 1992).
c-Myc is also induced by the Wnt/β-catenin signalling pathway, which regulates cell proliferation, differentiation and stem cell renewal and is linked to cancer. β-Catenin has a key role at the membrane, in the cytoplasm, as well as in the nucleus. In the absence of Wnt signalling, it is phosphorylated by glycogen synthase kinase-3β (GSK3-β) and targeted for degradation (Giles et al, 2003; Nelson & Nusse, 2004). In stimulated cells, it acts as a coactivator of transcription by binding to the transcription factors, T-cell factors (Tcfs; Moon et al, 2002). Interaction partners of β-catenin can modify the β-catenin/Tcf-dependent transcription, resulting in downregulation of expression of the target genes of the Wnt signalling pathway such as c-Myc (He et al, 1998). In numerous cancers, constitutively active β-catenin leads to expression of c-Myc (Giles et al, 2003).
We describe Bcr as a negative regulator of β-catenin. Bcr leads to downregulation of β-catenin/Tcf-dependent transcription. Furthermore, we show that Bcr–Abl counteracts the inhibitory effect accompanied by tyrosine phosphorylation of Bcr, which can be reverted by the drug Gleevec. Thus, our results contribute to the understanding of Bcr as a putative tumour suppressor, by negatively regulating the expression of proliferation-promoting genes.
Results
Bcr interacts with β-catenin
In view of the inhibitory function of Bcr on Rac–Pak and Ras–Raf signalling (Voncken et al, 1995; Lu & Mayer, 1999; Radziwill et al, 2003), we investigated whether Bcr might exert a similar function in the Wnt/β-catenin pathway. Here, we showed the interaction of Bcr and β-catenin in the absence of stimulation of the Wnt pathway. We selected two tumour cell lines, the glioblastoma tumour cell line T98G and the lung carcinoma cell line A549, to investigate Bcr and β-catenin complex formation. In both cell lines, we found Bcr co-precipitating with β-catenin and vice versa (Fig 1B,C). Additionally, we carried out cell fractionation of human embryonic kidney (HEK)293 cells and found complexes of Bcr and β-catenin in the cytoplasm as well as in the nucleus (Fig 1D). Protein expression levels of Bcr and β-catenin are shown as well as marker proteins for cytoplasm and nucleus, HSP 56 or cyclin D1 as controls (Fig 1D, lower panels). Bcr is mainly cytoplasmic and β-catenin is present in both the cytoplasm and nucleus.
The site on Bcr required for interaction with β-catenin was analysed by studies on two Bcr mutants, the N-terminal fragment of Bcr (BcrNT, 1–202 amino acids (aa)) and an N-terminal deletion fragment (BcrΔNT, 168–1,271 aa; Fig 1A). Bcr wild type (BcrWT) and the mutants were transfected into HEK293 cells. BcrWT and BcrNT, but not BcrΔNT, co-precipitated with β-catenin, which indicated that the N terminus of Bcr is the binding site (Fig 1E).
To locate the interaction region of β-catenin with Bcr, we asked whether this might overlap with the Tcf4-binding site in the armadillo repeat region comprising the lysine residue K435 (Fig 1A). The mutant K435A seemed a possible candidate because of its defect in Tcf4 binding and its reduced transactivation capacity (von Kries et al, 2000). To this aim, we used a Myc-tagged β-catenin wild type and a K435A mutant, both of which were expressed in HEK293 cells. Indeed, the Myc-tagged β-catenin K435A mutant was not able to co-precipitate Bcr (Fig 1F). Thus, the binding site of β-catenin for Bcr overlaps with that of the Tcf4 transcription activators.
Bcr as a negative regulator of gene expression
We then analysed the effect of Bcr on β-catenin/Tcf-dependent gene expression in two cell lines, HEK293 and the human colon carcinoma cell line HCT116. HCT116 expresses a β-catenin mutant with a deletion of the amino acid serine at position 45. This mutation stabilizes the protein and was found to correlate with a high level of β-catenin/Tcf transcription protein complex and strong expression of c-Myc (Morin et al, 1997; He et al, 1998). β-Catenin/Tcf-dependent gene expression was tested with the Tcf reporter TOPflash together with the mutant reporter FOPflash as a control. In both cell lines, BcrWT or BcrNT led to reduced activation of β-catenin/Tcf-mediated transactivation of TOPflash in a concentration-dependent manner (Fig 2A). BcrΔNT did not significantly inhibit transactivation, indicating that the N terminus of Bcr, which binds to β-catenin (Fig 1E), regulates the inhibitory effect of β-catenin-dependent transcription. In addition, in HCT116 cells containing stabilized β-catenin, the strong endogenous expression level of c-Myc may be downregulated by the expression of BcrNT and BcrWT but not BcrΔNT (Fig 2B). In the case of BcrWT, the contribution of its kinase to transactivation cannot be excluded. To analyse the effect of endogenous Bcr as a suppressor of the function of β-catenin, we used short interfering RNA (siRNA) oligonucleotides to downregulate expression of Bcr in HEK293 cells. Two concentrations of siRNAs were used, and at both concentrations, Bcr protein expression was significantly reduced. Under conditions of reduced Bcr levels, expression of the c-Myc protein was increased, which indicated relief from the Bcr-dependent suppressive effect (Fig 2C).
Figure 2.
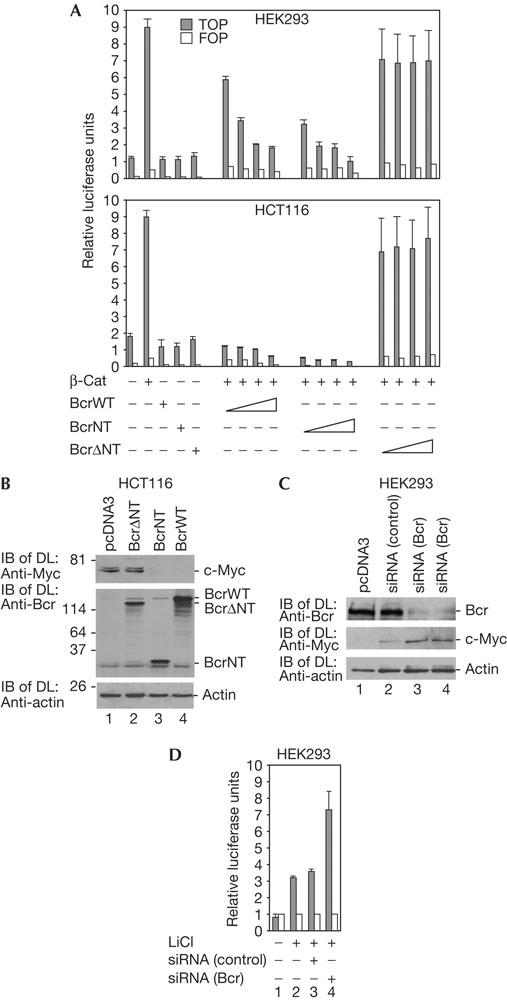
Effect of Bcr on gene expression. (A) Human embryonic kidney (HEK)293 cells or the human colon carcinoma cell line HCT116 was transfected with TOPflash and its control FOPflash (0.5 μg each) together with plasmid coding for β-catenin (1 μg) and BcrWT, BcrNT and BcrΔNT (1, 3, 4 and 7 μg each). Luciferase activity was determined by spectrophotometer. The error bars were obtained from four independent experiments. (B) BcrNT effect on c-Myc expression. HCT116 cells were transfected with plasmid DNA for protein expression. For immunoblot of direct lysates (IB of DL), a mixture of two antibodies against Bcr (N-20 and C-20) was used. (C) The effect of Bcr on expression of the c-Myc was analysed by means of short interfering RNA (siRNA) oligonucleotides. HEK293 cells were transfected with 50 or 100 nM of Bcrspecific siRNA duplexes and an irrelevant interleukin-12 (IL-12) siRNA as a control, which downregulated IL-12 but not Bcr messenger RNA. IB with DL are shown as indicated. (D) Reporter assay, as in (A), with HEK293 cells stimulated by LiCl with or without siRNA against Bcr or the control, as in (C).
c-Myc expression is low in HEK293 cells, as long as the Wnt pathway is not stimulated. In another approach, we therefore used LiCl for stimulation of the β-catenin/Tcf-dependent transcription which activates the luciferase reporter assay described in Fig 2A. Similar results were obtained in the presence of LiCl with and without siRNA against Bcr. siRNA reduces the suppressive effect of Bcr on β-catenin-mediated transcription (Fig 2D).
Bcr–Abl tyrosine kinase regulates Bcr and β-catenin
We then investigated whether the Bcr–Abl oncoprotein expressed in leukaemic cells can influence Bcr and β-catenin complex formation. The Bcr–Abl fusion protein exhibits tyrosine kinase activity through the Abl moiety, whereas the serine kinase activity of Bcr is repressed in the fusion and wild-type protein (Arlinghaus, 2002). The oncogenicity of Bcr–Abl is reduced by Bcr overexpression, in a dose-dependent manner (Lin et al, 2001; Arlinghaus, 2002; Mahon et al, 2003). The N terminus of Bcr (1–160 aa) is sufficient to suppress colony formation induced by Bcr-Ablp210 (Guo et al, 1998). We expressed the Bcr–Abl fusion protein p210 in HEK293 cells (Fig 1A) and tested for co-precipitation of β-catenin with Bcr–Abl and Bcr (Daley et al, 1990). As can be seen, Bcr–Abl did not co-precipitate with β-catenin, whereas Bcr did. Furthermore, expression of Bcr–Abl slightly reduced the precipitation of Bcr with β-catenin (Fig 3A). We then tested whether Bcr–Abl with its tyrosine kinase activity was able to phosphorylate endogenous Bcr and Bcr–Abl in HEK293 cells and whether the Bcr–Abl tyrosine kinase activity was blocked by the inhibitor STI-571 (Gleevec), which is a drug developed against CML (Roskoski, 2003). As can be seen in Fig 3B, Bcr–Abl and Bcr are phosphorylated on tyrosine and this effect is strongly reduced by STI-571. The effect of Bcr–Abl-dependent tyrosine phosphorylation of Bcr on complex formation with β-catenin was then analysed in HEK293 cells. Bcr–Abl was overexpressed in HEK293 cells, which were later fractionated into nuclear and cytoplasmic fractions. Bcr is more strongly expressed in the cytoplasm in this system and β-catenin is enriched in the nucleus (Fig 3C). In the presence of STI-571, co-precipitation of Bcr with β-catenin occurred mainly in the cytoplasm and less in the nucleus (lanes 1 and 2). In the absence of STI-571, the amount of Bcr co-precipitating with β-catenin was reduced in both the fractions (compare lanes 3 and 4 with lanes 1 and 2). Protein expression of Bcr and Bcr–Abl has been controlled and is shown in Fig 3B (lower panel). Furthermore, we used a cell line, MEG-01, derived from a CML patient, which expresses endogenous Bcr-Ablp210, to test the effect of STI-571 on Bcr and β-catenin complex formation. We first confirmed the inhibitory effect of STI-571 on Bcr–Abl-dependent tyrosine phosphorylation of Bcr–Abl and Bcr (Fig 3D). Reduced phosphorylation of Bcr in the presence of STI-571 led to an increase in the association of β-catenin with Bcr (Fig 3E). This result indicates that the presence of Bcr–Abl leads to disruption of the Bcr/β-catenin complex, and that STI-571 reverts this effect.
Figure 3.
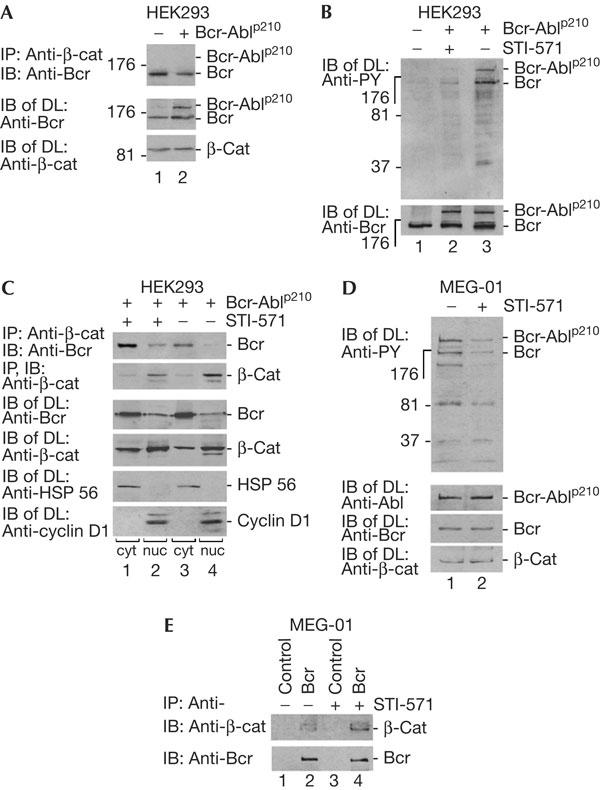
Tyrosine phosphorylation regulates Bcr and β-catenin complex formation. (A) Human embryonic kidney (HEK)293 cells were transfected with Bcr-Ablp210-expressing construct and treated for co-immunoprecipitation (Co-IP), immunoblotting (IB) and direct lysates (DL), as indicated, using Bcr antibodies (N-20), which would also recognize Bcr–Abl. (B) Cells as shown in (A) were treated with STI-571 (2 μM, 1 h). IB was carried out with phosphotyrosinespecific antibodies (PY). For control, Bcr–Abl and Bcr proteins are shown after IB with a Bcr-specific antibody (N-20) in DL. (C) Complex formation regulated by Bcr–Abl kinase. Cells as in (A) were treated with STI-571, fractionated as in Fig 1D and then IP and IB were carried out using Bcr antibodies (C-20). Cell fractionation was controlled by antibodies against HSP 56 and cyclin D1. (D) Human chronic myelogenous leukaemia (CML) cell analysis. The human patient cell line MEG-01 expressing endogenous Bcr and Bcr-Ablp210 was used to detect the phosphorylated forms of proteins by P-Y antibodies in the absence or presence of STI-571 (2 μM, 24 h). Bcr was detected by C-20 antibodies. (E) Co-IP of Bcr (C-20 antibodies) with β-catenin in MEG-01 cells in the presence or absence of STI-571.
Discussion
Bcr is here shown to negatively regulate β-catenin-mediated gene activation, including c-myc (Fig 4, model). Bcr negatively regulates transcription activation through its N terminus. Previously, this region has been shown to counteract Bcr–Abl-induced biological effects (Guo et al, 1998). The Bcr moiety of Bcr–Abl did not bind to β-catenin, as shown here. Also, in the presence of Gleevec, almost no binding was detectable (data not shown), presumably owing to conformational changes by fusion to Abl. The accessibility of Bcr to β-catenin may be regulated by the Bcr–Abl tyrosine kinase or other tyrosine kinases, such as c-Fes (Liu et al, 1993; Li & Smithgall, 1996).
Figure 4.
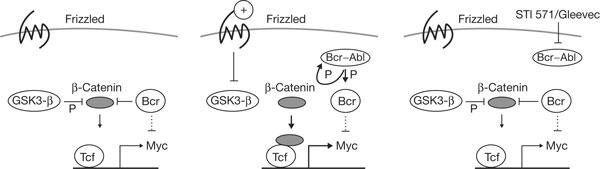
Model on Bcr effects on β-catenin. Negative regulation is indicated by bars and positive regulation by arrows. Dotted lines indicate downregulation of c-Myc protein levels by Bcr (Mahon et al, 2003). The figure depicts an unstimulated cell (left), Bcr–Abl-expressing tumour cell (middle) and STI-571 effect (right). The thick arrows indicate strong activation of c-Myc. For more details, see text.
Bcr can affect c-Myc level through its C terminus, which binds to c-Myc protein and targets it to degradation (Mahon et al, 2003). This may limit high c-Myc expression levels. It would be interesting to know whether this in turn negatively influences β-catenin-mediated transcription as a negative feedback loop. In a quiescent state of the cell, not only Bcr but also GSK3-β or membrane localization of β-catenin maintains a reduced level of β-catenin-mediated transcription. Thus, β-catenin and Bcr complex formation may be one of several negative regulatory effects. Such a complex negative control suggests biological importance that may be relevant for maintenance of the basal state of cells, possibly of stem cells (Reya et al, 2003). We have recently shown that Bcr inhibits Ras–Raf signalling by Bcr-mediated phosphorylation of AF-6—another Bcrspecific negative regulator effect besides binding to β-catenin or Myc (Radziwill et al, 2003).
Stimulation and activation of a tyrosine kinase may override this AF-6/Bcr effect similarly to the β-catenin/Bcr effect, which is reduced by the Bcr-Abl tyrosine kinase, as shown here (Liu et al, 1993; Li & Smithgall, 1996). Previously, it has been shown for c-Raf-1 that it can induce kinase activity-independent inhibition of apoptosis by protein–protein interaction by means of its N terminus (Chen et al, 2001).
Thus, in summary, we have shown a Bcr-mediated inhibitory effect on β-catenin-dependent gene expression, including c-Myc. This is important for understanding the Wnt pathway and the maintenance of the quiescent state of a cell, as well as the role of Bcr–Abl in cancer.
Methods
Plasmids. Plasmids coding for haemagglutinin (HA)–BcrWT and BcrΔNT have been described elsewhere (Radziwill et al, 2003). The plasmid HA–BcrNT carries the coding sequence for residues 1–202 of Bcr cloned into pcDNA3 (G. Radziwill, unpublished). Flag–β-catenin(K435A) was a gift from H.C. Clevers (Utrecht University, Utrecht, Netherlands) and used to clone Myc–β-catenin(K435A). PGD210 Bcr–Abl was a gift from G. Daley (Whitehead Institute for Biomedical Research, Cambridge, UK).
Cell culture. All cell lines were obtained from ATCC (USA) or DSMZ (Germany). HEK293 (ATCC; CRL-1573) is a human embryonic kidney cell line. HCT116 (ATCC; CCL-247) is a human colorectal carcinoma cell line. A-549 (ATCC; CCL-185) is a human lung carcinoma cell line. T98G (ATCC; CRL-1690) is a human glioblastoma cell line. MEG-01 (DSMZ; ACC-364) is a human chronic myeloid leukaemia in megakaryocytic blast crisis. MEG-01 cells were cultured in RPMI (Invitrogen, Basel, Switzerland). All other cells were cultured in DMEM (Invitrogen). The media were supplemented with 10% fetal bovine serum, penicillin and streptomycin. Transfections were carried out using Lipofectin (Invitrogen) according to the recommended procedure. STI-571 (imatinib mesylate; Novartis, Basel, Switzerland) was added to HEK293 cells at a concentration of 2 μM, for 1 h or 24 h, and to MEG01 cells 1 μM for 16 h. LiCl treatment (10 μM, 16 h) mimics wingless signalling and was performed as described previously (Stambolic et al, 1996).
Cell fractionation, immunoprecipitation and immunoblots. Immunoprecipitation and immunoblotting have been described previously (Radziwill et al, 2003). Nuclear extracts were prepared from HEK293 cells as described previously (Andrews & Faller, 1991). Primary antibodies used were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA): anti-Bcr (N-20, C-20), anti-β-catenin (E-5), anti-cyclin D1 (M-20), anti-HSP 56 (N-17), anti-Myc (A-14), anti-Myc-tagged protein (9E10) and anti-p27 (F-8). Secondary antibodies used were donkey anti-rabbit and sheep anti-mouse horseradish peroxidase-conjugated IgG (Amersham, Otelfingen, Switzerland).
Short interfering RNA preparation and transfection. Oligonucleotides for Bcr-specific siRNA duplex (Dharmacon, Lafayette, CO, USA) were as follows: sense strand 5′-UGUCAUCGUCCACUCAGCCTT-3′; antisense strand 5′-GGCUGAGUGGACGAUGACATT-3′. As a control, a sequence targeting interleukin-12 (IL-12) was used: sense strand 5′-AGGUCCAGGUGAUGUCAUCTT-3′; antisense strand 5′-GAUGACAUCACCUGGACCUTT-3′. HEK293 cells were transfected twice with 50 and 100 nM siRNA duplex, using oligofectamine (Invitrogen).
Reporter assay. HEK293 cells were transfected with a plasmid coding for β-catenin and Bcr together with the Tcf reporter plasmid TOPflash or the insensitive reporter FOPflash (Upstate, Luzern, Switzerland). To normalize for transfection efficiency, pRL-TK (Upstate) coding for Renilla luciferase was co-transfected. Luciferase activity was analysed using the Dual-Luciferase Reporter Assay system (Promega, Wallisellen, Switzerland) and a TD-20/20 luminometer (Turner Design, Sunnyvale, CA, USA).
Acknowledgments
We thank Dr G. Radziwill (University of Zurich, Institute of Medical Virology) for help and productive discussion. Some support was also given by Dr M. Baumgartner and Dr J. Pavlovic, Dr S. Rohrer and Dr J. Rossi (University of Zurich). F. Metz (Berlin) helped with the graphs. We thank Dr G. Daley (Whitehead Institute for Biomedical Research, Cambridge, UK) for providing the Bcr-Ablp210 construct and Professor Dr H.C. Clevers (Hubrecht Laboratory, Center for Biomedical Genetics, Utrecht, Netherlands) for the β-catenin constructs. Part of this work was supported by the Swiss National Science Foundation.
References
- Andrews NC, Faller DV (1991) A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res 19: 2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlinghaus RB (2002) Bcr: a negative regulator of the Bcr–Abl oncoprotein in leukemia. Oncogene 21: 8560–8567 [DOI] [PubMed] [Google Scholar]
- Chen J, Fujii K, Zhang L, Roberts T, Fu H (2001) Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK–ERK independent mechanism. Proc Natl Acad Sci USA 98: 7783–7788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley GQ, Van Etten RA, Baltimore D (1990) Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science 247: 824–830 [DOI] [PubMed] [Google Scholar]
- Giles RH, van Es JH, Clevers H (2003) Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta 1653: 1–24 [DOI] [PubMed] [Google Scholar]
- Guo XY, Cuillerot JM, Wang T, Wu Y, Arlinghaus R, Claxton D, Bachier C, Greenberger J, Colombowala I, Deisseroth AB (1998) Peptide containing the bcr oligomerization domain (AA 1–160) reverses the transformed phenotype of p210bcr-abl positive 32D myeloid leukemia cells. Oncogene 17: 825–833 [DOI] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW (1998) Identification of c-Myc as a target of the APC pathway. Science 281: 1509–1512 [DOI] [PubMed] [Google Scholar]
- Li J, Smithgall TE (1996) Co-expression with BCR induces activation of the FES tyrosine kinase and phosphorylation of specific N-terminal BCR tyrosine residues. J Biol Chem 271: 32930–32936 [DOI] [PubMed] [Google Scholar]
- Lin F, Monaco G, Sun T, Liu J, Lin H, Stephens C, Belmont J, Arlinghaus RB (2001) BCR gene expression blocks Bcr–Abl induced pathogenicity in a mouse model. Oncogene 20: 1873–1881 [DOI] [PubMed] [Google Scholar]
- Liu J, Campbell M, Guo JQ, Lu D, Xian YM, Andersson BS, Arlinghaus RB (1993) Bcr–Abl tyrosine kinase is autophosphorylated or transphorsphorylates P160 Bcr on tyrosine predominantly within the first Bcr exon. Oncogene 8: 101–109 [PubMed] [Google Scholar]
- Liu J, Wu Y, Ma GZ, Lu D, Haataja L, Heisterkamp N, Groffen J, Arlinghaus RB (1996) Inhibition of Bcr serine kinase by tyrosine phosphorylation. Mol Biol 16: 998–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Mayer BJ (1999) Mechanism of activation of Pak1 kinase by membrane localization. Oncogene 18: 797–806 [DOI] [PubMed] [Google Scholar]
- Mahon GM, Wang Y, Korus M, Kostenko E, Cheng L, Sun T, Arlinghaus RB, Whitehead IP (2003) The c-Myc oncoprotein interacts with Bcr. Curr Biol 13: 437–441 [DOI] [PubMed] [Google Scholar]
- Marcu KB, Bossone SA, Patel AJ (1992) Myc function and regulation. Annu Rev Biochem 61: 809–860 [DOI] [PubMed] [Google Scholar]
- Maru Y, Witte ON (1991) The BCR gene encodes a novel serine/threonine kinase activity within a single exon. Cell 67: 459–468 [DOI] [PubMed] [Google Scholar]
- Moon RT, Bowermann B, Boutros M, Perrimon N (2002) The promise and perils of Wnt signaling through β-catenin. Science 296: 1644–1646 [DOI] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW (1997) Activation of β-catenin–Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 275: 1787–1790 [DOI] [PubMed] [Google Scholar]
- Nelson WJ, Nusse R (2004) Convergence of Wnt, β-catenin, and cadherin pathway. Science 303: 1483–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radziwill G, Erdmann RA, Margelisch U, Moelling K (2003) The Bcr kinase downregulates Ras signaling by phosphorylating AF-6 and binding to its PDZ domain. Mol Cell Biol 23: 4663–4672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Duncan AW, Allies L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissmann IL (2003) A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 423: 409–414 [DOI] [PubMed] [Google Scholar]
- Roskoski R (2003) STI-571: an anticancer protein-tyrosine kinase inhibitor. Biochem Biophys Res Commun 309: 709–717 [DOI] [PubMed] [Google Scholar]
- Sawyers CL (1993) The role of myc in transformation by Bcr–Abl. Leuk Lymphoma 11(Suppl 1): 45–46 [DOI] [PubMed] [Google Scholar]
- Stambolic V, Ruel L, Woodgett JR (1996) Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol 6: 1664–1668 [DOI] [PubMed] [Google Scholar]
- Voncken JW et al. (1995) Increased neutrophil respiratory burst in Bcr-null mutants. Cell 80: 719–728 [DOI] [PubMed] [Google Scholar]
- von Kries JP, Winbeck G, Asbrand C, Schwarz-Romond T, Sochnikova N, Dell'Oro A, Behrens J, Birchmeier W (2000) Hot spots in β-catenin for interactions with LEF-1, conductin and APC. Nat Struct Biol 7: 800–807 [DOI] [PubMed] [Google Scholar]
- Wu Y, Ma G, Lu D, Lin F, Xu HJ, Liu J, Arlinghaus RB (1999) Bcr: a negative regulator of the Bcr–Abl oncoprotein. Oncogene 18: 4416–4424 [DOI] [PubMed] [Google Scholar]