


Abstract
Escherichia coli RNA polymerase associated with the σ54 factor (RNAP·σ54) is a holoenzyme form that transcribes a special class of promoters not recognized by the standard RNA polymerase·σ70 com plex. Promoters for RNAP·σ54 vary in their overall ‘strength’ and show differences in their response to the presence of DNA curvature between enhancer and promoter. In order to examine whether these effects are related to the promoter affinity, we have determined the equilibrium dissociation constant Kd for the binding of RNAP·σ54 to the three promoters glnAp2, nifH and nifL. Binding studies were conducted by monitoring the changes in fluorescence anisotropy upon titrating RNAP·σ54 to carboxyrhodamine-labeled DNA duplexes. For the glnAp2 and nifH promoters similar values of Kd = 0.94 ± 0.55 nM and Kd = 0.85 ± 0.30 nM were determined at physiological ionic strength, while the nifL promoter displayed a significantly weaker affinity with Kd = 8.5 ± 1.9 nM. The logarithmic dependence of Kd on the ionic strength I was –Δlog(Kd)/Δlog(I) = 6.1 ± 0.5 for the glnAp2, 5.2 ± 1.2 for the nifH and 2.1 ± 0.1 for the nifL promoter. This suggests that the polymerase can form fewer ion pairs with the nifL promoter, which would account for its weaker binding affinity.
INTRODUCTION
RNA polymerase from Escherichia coli complexed with the alternative σ factor σ54 (RNAP·σ54) recognizes a specific class of promoters with conserved sequence elements around position –24 and –12 upstream of the transcription start site at +1 (1). RNAP·σ54 can bind to the promoter and form a stable closed complex. However, the enzyme is unable to melt the DNA at the transcription start site, i.e. to undergo the transition into the open complex. This process involves specific transcription factors with ATPase activity as for example nitrogen regulatory protein C (NtrC or NRII) or nitrogen fixation protein A (NifA). The binding sites of these proteins are located at upstream enhancer sequences and require looping of the DNA to enable interaction with RNAP·σ54 (2–6). The mechanism of transcription activation in this system has been discussed in recent reviews (7–9). Several lines of evidence indicate that the activator protein targets the σ54 subunit and works by triggering a conformation change that involves the N-terminus of σ54 (10).
In a number of studies differences in the relative strength of RNAP·σ54 promoters have been reported with respect to the in vivo RNA levels (11), the activity of a β-galactosidase reporter gene (12,13) or by measuring the equilibrium amount of open complexes formed in vitro in single round transcription experiments (14–16). Here we define the overall promoter strength as the rate with which the open complex RPo of RNAP·σ54 (R) at a given promoter P is formed in a multi-step reaction according to equation 1.
k1
R + P ⇋ RPc ⇋… ⇋ RPo1
k–1
In this equation 1 the forward and backward rate constants for the formation/dissociation of the closed complex RPc are given by k1 and k–1 so that the equilibrium dissociation constant Kd = k–1/k1 in equation 2 reflects the affinity of the promoter for RNAP·σ54.
Kd = k–1/k1 = ([R]·[P])/[RPc]2
For the standard E.coli RNAP·σ70 holoenzyme a number of promoters have been analyzed in detail. It has been demonstrated that both the binding of the RNA polymerase to the promoter as well as the subsequent conversion of the closed complex into the open complex RPo can be rate limiting (17). For RNAP·σ54 no quantitative analysis of the promoter strength in terms of the relative contributions of the separate steps has been reported so far. From footprinting experiments a Kd of 3 nM at approximately physiological salt concentrations was estimated for the glnAp2 promoter (18). In contrast, the nifH (19,20) and nifL (21–23) promoters from Klebsiella pneumoniae showed no or only a very weak footprint under similar conditions. Accordingly, glnAp2 has been designated as a ‘strong’ promoter as opposed to the ‘weak’ nifH and nifL promoters. However, it should be noted that both glnAp2 and nifH are ‘strong’ promoters in the sense that they can be expressed at high levels under suitable physiological conditions (13,24). In contrast, the nifL promoter displayed an approximately 5-fold lower expression level than the nifH promoter (13). Here we have measured the equilibrium dissociation constant Kd dependence on the ionic strength for oligonucleotide duplexes with sequences from the glnAp2, nifH and nifL promoters. The binding of RNAP·σ54 to DNA was monitored by fluorescence anisotropy measurements using DNA oligonucleotide duplexes with a carboxyrhodamine end label.
MATERIALS AND METHODS
DNA and RNAP·σ54 preparation
HPLC-purified DNA oligonucleotides were purchased from PE-Applied Biosystems (Weiterstadt, Germany). The fluorescent dye 6-carboxy-X-rhodamine (ROX) was covalently attached to the 5′-end via a C6 linker. The extinction coefficients of the single DNA strands were determined as described previously (25). Equimolar amounts of complementary single strands were mixed in a buffer containing 10 mM Tris–HCl, pH 7.5, 10 mM NaCl, 0.1 mM EDTA and annealed by 2 min heating at 70°C followed by slow cooling to room temperature over several hours. Purification of the resulting DNA duplexes was done by extraction from native polyacrylamide gels according to Rippe et al. (26). From an analysis of various ROX-labeled DNAs of different lengths an average extinction coefficient of ROX attached to DNA of ε583 = 96 000 M–1 cm–1 at 25°C was calculated (J.F.Kepert and K.Rippe, unpublished results). This value was used to determine the concentration of the purified ROX-labeled DNA duplex stock solutions. The concentration of the unlabeled promoter duplexes was determined based on an extinction coefficient of ε260 = 559 000 M–1 cm–1 at 25°C. The sequences of the three ROX-labeled duplexes correspond to the glnAp2 promoter from E.coli and to the nifH and nifL promoters from K.pneumoniae (Fig. 1) (1). In the nifH and nifL promoters the 5 bp found adjacent to the ROX label were kept identical to the glnAp2 sequence in order to avoid differences in the spectroscopic properties of the ROX label.
Figure 1.

Three different ROX-labeled DNA duplexes were used in the binding studies, the glnAp2 promoter sequence from E.coli and the nifH and nifL promoters from K.pneumoniae. The nucleotides that fit the –24/–12 consensus sequence for RNAP·σ54-specific promoters are in bold (1). The RNAP binding region from about –34 close to the transcription start site at position –2 is shaded in gray and has been derived from footprinting studies (44,45). Positions +1, –12 and –24 relative to the RNA transcript start site are indicated.
RNA polymerase core enzyme from E.coli was purchased from Epicentre Technologies (Madison, WI). It was mixed with σ54 in a ratio of 1:1.5 to form the RNAP·σ54 holoenzyme at a stock concentration of ∼1 µM. This preparation was stored at –20°C in a buffer with 50 mM Tris–HCl, pH 7.5, 250 mM NaCl, 0.1 mM EDTA, 1 mM DTT and 50% glycerol. The σ54 protein does not bind to the wild-type nifH and glnAp2 promoters in the absence of the core RNA polymerase (27,28). Only for a special mutant form of the nifH promoter (–17 to –15 sequence TTT) has binding of isolated σ54 (Kd ≈ 10–7 M) at moderate ionic strength been demonstrated (27). Thus, the presence of excess σ54 protein should not affect the association of RNAP·σ54 with the sequences studied here.
The activity of the purified DNA duplexes and the RNAP·σ54 holoenzyme was confirmed in native gel electrophoresis. The purified DNA duplexes and ROX-labeled single strands were analyzed on a 20% native polyacrylamide gel. For the gel mobility shift assay 245 nM of the respective promoter DNA duplexes in 0.5× RNAP·σ54 storage buffer were mixed with varying amounts of RNAP·σ54 holoenzyme. Complexes and free DNA were separated on a 5% polyacrylamide gel (29:1) and visualized by ethidium bromide staining.
Fluorescence anisotropy measurements
Fluorescence anisotropy measurements were performed with a SLM 8100 fluorescence spectrometer (SLM Aminco Inc.) using an L-format setup. The ROX excitation wavelength of 580 nm was selected with a double grated monochromator using an 8 or 16 nm slit width for high intensity. In the emission channel scattered light was suppressed with a 610 nm cut-off filter. Intensity variations of the lamp were corrected by normalization to a reference channel with a rhodamine quantum counter. All measurements were conducted at 25°C in a buffer containing 20 mM HEPES–KOH, pH 8.0, 5 mM magnesium acetate, 1 mM DTT, 0.1 mg/ml BSA, 0.01% NP-40 detergent (Roche Diagnostics, Germany), supplemented with potassium acetate at a concentration from 50 to 350 mM.
According to the Perrin equation (29), the anisotropy of a fluorescent complex increases with its volume and reflects its rotational mobility. The assay used here is based on the rationale that the free DNA has a relatively low fluorescence anisotropy. This signal increases upon protein binding, due to the reduced rotational diffusion time after formation of the protein–DNA complex. The same approach has been used successfully in a number of studies (see for example 25,30–35).
The RNAP·σ54 DNA binding activity was determined by stoichiometric titrations at a DNA concentration of 10 nM duplex in low salt binding buffer (50 mM potassium acetate) for high affinity binding. From the linear increase at low protein concentration and the plateau region obtained at saturation of the binding sites the equivalence point for the formation of a 1:1 complex was determined. In these experiments a DNA binding activity between 80 and 90% with respect to the core polymerase concentration given by the manufacturer was determined for different RNAP·σ54 holoenzyme preparations (data not shown).
For the determination of dissociation constants a DNA solution with 25–200 pM duplex in binding buffer was titrated with a RNAP·σ54 protein solution diluted into the same buffer. After addition of protein the sample was equilibrated for ∼3 min before measuring the equilibrium anisotropy value. For each anisotropy value the average of 20 measurements with an integration time of 5 s was determined. To check whether the quantum yield of the ROX dye changed upon binding of the polymerase the fluorescence intensity of the free promoter DNA and the intensity after saturation of the DNA binding sites was recorded under polarization-independent ‘magic angle’ conditions (vertically polarized excitation and emission polarizer oriented at 54.7°) for every titration.
Data analysis
Equilibrium binding data were analyzed according to the reaction given in equation 1 for the formation of a 1:1 complex between RNAP·σ54 and the ROX-labeled duplex with the promoter sequence. The DNA duplex concentration [P] in the experiments was chosen so that 10·[P] ≤ Kd. Under these conditions the concentration of free RNAP·σ54 [R] can be approximated by the total polymerase concentration [Rtot], i.e. [R] ≈ [Rtot], and the fractional saturation θ of the promoter duplex with RNAP·σ54 is given by
θ = [Rtot]/([Rtot] + Kd) = (r – rP)/(rRP – rP)3
In equation 3 r is the measured anisotropy at a given polymerase concentration. The anisotropy values of the free promoter DNA and that of the RPc complex are given by rP and rRP, respectively. Rearrangement of equation 3 leads to equation 4, which was used to determine Kd from a least squares fit of the binding curve obtained by plotting r versus the added RNA polymerase concentration [Rtot] with rP and rRP as additional fit parameters.
r = (rP·Kd + [Rtot]·rRP)/([Rtot] + Kd)4
The least squares fit was computed with the program Kaleidagraph v.3.5 (Synergy Software, PA). The use of equation 4 is only correct if the quantum yield of the ROX dye does not change upon binding of RNAP·σ54, which was the case in the experiments reported here (see below).
RESULTS
Gel analysis of ROX-labeled promoter DNA
The oligonucleotide duplexes of 43 bp length shown in Figure 1 were studied with respect to their binding affinity for RNAP·σ54. The sequences correspond to the glnAp2 promoter from E.coli and the nifH and nifL promoters from K.pneumoniae. All duplexes carried the fluorescent dye ROX at the 5′-end. Gel analysis of the purified duplexes is shown in Figure 2A (three left lanes) in comparison to the ROX-labeled single strands (Fig. 2A, three right lanes), which displayed a higher electrophoretic mobility. Both the ROX fluorescence signal as well as ethidium bromide staining of the duplexes revealed only a single band demonstrating that the synthesis and reconstitution of the promoter DNA sequences was successful.
Figure 2.
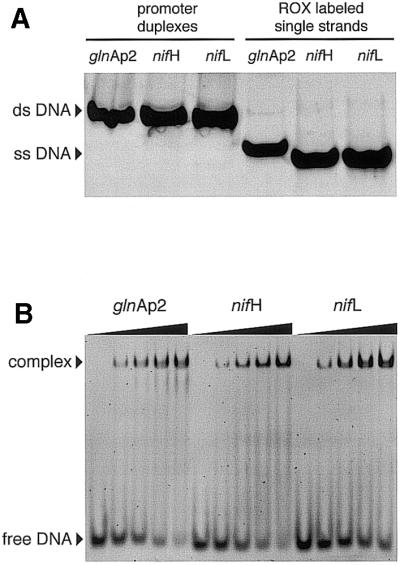
Gel electrophoretic analysis of ROX-labeled promoter duplexes and RNAP·σ54–DNA complexes. (A) Native polyacrylamide gel of promoter duplexes (three left lanes) and ROX-labeled single strands (three right lanes). (B) Gel shift analysis of RNAP·σ54 complexes with the three promoter DNA duplexes. The five lanes for each promoter sequence display an increasing ratio of RNAP·σ54 to the DNA duplex (245 nM concentration) from 0:1, 0.25:1, 0.5:1, 0.75:1 to 1:1.
The binding of RNAP·σ54 to these duplexes was qualitatively characterized in an electrophoretic gel mobility shift assay under conditions of stoichiometric binding (Fig. 2B). Both the RNAP·σ54 enzyme and the DNA promoter sequences were active with respect to binding as indicated by the almost fully shifted DNA fraction at an approximate 1:1 ratio of protein and DNA (Fig. 2B, highest protein concentrations).
Binding affinity of RNAP·σ54 for glnAp2, nifH and nifL promoters
In order to measure the dissociation constant Kd of RNAP·σ54 with the three different promoters under true equilibrium conditions and at defined ionic strength and pH the binding of RNAP·σ54 was followed by fluorescence anisotropy measurements. The polymerase was titrated into a solution of the ROX-labeled DNA duplex at a given salt concentration. Protein was added until all binding sites were saturated and the anisotropy reached a plateau value, which reflects the anisotropy of the 1:1 complex of RNAP·σ54 with the DNA. The resulting binding curve was fitted to equation 4 and Kd as well as the anisotropies of the free promoter DNA (rP) and the protein–DNA complex (rRP) were obtained. As expected, similar average values of both rP (0.165–0.169) and rRP (0.244–0.266) were obtained for the three promoters indicating similar rotational diffusion times for the free DNA and its complex with the polymerase (Table 1).
Table 1. Fluorescence anisotropy parameters for the binding of RNAP·σ54 to the glnAp2, nifH and nifL promoters.
| glnAp2 | nifH | nifL | |
|---|---|---|---|
| Anisotropy of free DNA (rP)a | 0.165 ± 0.02 | 0.169 ± 0.01 | 0.166 ± 0.01 |
| Anisotropy of complex (rRP)a | 0.244 ± 0.03 | 0.260 ± 0.01 | 0.266 ± 0.02 |
| Quenching factor qb | 1.13 ± 0.1 | 1.08 ± 0.1 | 1.0 ± 0.1 |
aThe anisotropies of free (rP) and complexed DNA (rRP) were derived from fitting the binding curves to equation 4.
bThe quenching factor q was determined for every titration under polarization-independent conditions and averaged.
The analysis of the binding curve according to equation 4 is only valid if the quantum yield of the ROX dye does not change upon binding of the polymerase (36). To test whether this was the case the fluorescence intensities of each sample before and after the titration were measured. After correction for dilution these intensity ratios corresponded to quenching factors q of 1.13 ± 0.1 (glnAp2), 1.08 ± 0.1 (nifH) and 1.02 ± 0.1 (nifL) (Table 1). Thus, within the accuracy of the measurements, the binding of RNAP·σ54 to the promoter DNA did not change the ROX quantum yield so that the analysis of the data according to equation 4 is valid.
Figure 3 displays representative binding curves recorded by titrating the glnAp2, nifH and nifL promoters at an approximately physiological ionic strength (200 mM potassium acetate and 5 mM magnesium acetate, I = 0.229 M). The data were fitted to equation 4 to obtain Kd, rP and rRP (Fig. 3A). The measured r values were then converted according to equation 3 into the fractional saturation of the DNA θ (Fig. 3B). A good agreement of the measured data according to the 1:1 binding model described by equations 3 and 4 was obtained yielding Kd values of 0.7 ± 0.1 (glnAp2), 1.2 ± 0.1 (nifH) and 10.1 ± 1.1 nM (nifL) for the titrations shown in Figure 3. While the glnAp2 and nifH promoters displayed a similar affinity at an ionic strength of I = 0.229 M, binding to the nifL sequence was about an order of magnitude weaker. Average values from multiple measurements are summarized in Table 2 with Kd values of 0.94 ± 0.55 (glnAp2), 0.85 ± 0.30 (nifH) and 8.5 ± 1.9 nM (nifL) at I = 0.229 M.
Figure 3.

Fluorescence anisotropy measurement of RNAP·σ54 binding to different promoter sequences. A comparison of the binding affinity to three promoter duplexes at approximately physiological ionic strength is shown. The ROX-labeled DNA at a concentration of 50 pM for glnAp2 (filled triangles) and nifH (open squares) and of 100 pM for nifL (filled squares) was preincubated in a buffer supplemented with 200 mM potassium acetate and 5 mM magnesium acetate (I = 0.229 M). This solution was titrated with the indicated concentrations of RNAP·σ54. (A) The resulting binding curves of anisotropy r versus RNAP·σ54 concentration were analyzed according to equation 4 to determine Kd. Values of 0.7 ± 0.1 (glnAp2), 1.2 ± 0.1 (nifH) and 10.1 ± 1.1 nM (nifL) were obtained for the experiments shown in this figure. Average Kd values at various salt concentrations are given in Table 2. (B) To account for differences in the values of rP and rRP between the three promoters the measured anisotropy values can be converted into the fractional saturation of the DNA θ according to equation 3 for a direct comparison of the titrations.
Table 2. Dissociation constants Kd for the binding of RNAP·σ54 to the glnAp2, nifH and nifL promoters as determined by fluorescence anisotropy measurements.
| Salt concentration | Kd (nM) glnAp2 | Kd (nM) nifH | Kd (nM) nifL |
|---|---|---|---|
| 50 mM potassium acetate I = 0.079 M | 0.76 ± 0.56 (5) | ||
| 100 mM potassium acetate I = 0.129 M | 3.2 ± 1.7 (4) | ||
| 150 mM potassium acetate I = 0.179 M | 0.40 ± 0.04 (4) | 0.50 ± 0.36 (3) | 4.6 ± 0.9 (2) |
| 200 mM potassium acetate I = 0.229 M | 0.94 ± 0.55 (6) | 0.85 ± 0.30 (3) | 8.5 ± 1.9 (3) |
| 250 mM potassium acetate I = 0.279 M | 5.1 ± 2.3 (13) | 2.4 ± 0.7 (4) | 10 ± 8 (7) |
| 300 mM potassium acetate I = 0.329 M | 11 ± 9 (6) | 16 ± 10 (3) | 18 ± 13 (3) |
| 350 mM potassium acetate I = 0.379 M | 15 ± 11 (8) | 40 ± 11 (3) |
Average values for Kd and corresponding standard deviations are given in nanomolar concentrations and were determined in binding buffer (20 mM HEPES–KOH, pH 8.0, 5 mM magnesium acetate, 1 mM DTT, 0.1 mg/ml BSA, 0.01% NP-40), supplemented with the indicated potassium acetate concentrations, yielding the indicated ionic strength I. The number in parentheses after the value of the dissociation constant refers to the number of experiments averaged.
In addition, whether the ROX-fluorophore affected the differences in binding affinities observed in the experiments described above was tested at the same ionic strength with unlabeled DNA duplexes. A 10 nM solution of preformed RNAP·σ54 complex with a given ROX-labeled promoter DNA was titrated with unlabeled glnAp2, nifH or nifL duplex. The concentration of the unlabeled DNA which was required to displace 50% of the ROX–DNA from the complex with RNAP·σ54was determined. For titrating glnAp2–ROX with nifH and nifH–Rox with glnAp2 this concentration was the same within ∼10%. Thus, the unlabeled glnAp2 and nifH promoter fragments displayed essentially the same binding affinities at physiological ionic strength. In contrast, the nifL promoter (nifL–Rox versus glnAp2, nifL–Rox versus nifH and glnAp2–ROX versus nifL) displayed a significantly weaker association with RNAP·σ54.
Salt dependence of RNAP·σ54 promoter binding affinity
When the experiments were performed at different salt concentrations in the range I = 0.079–0.379 M K+ equivalents the value of Kd increased at higher ionic strength. This is due to a weakening of electrostatic interactions between protein and DNA as reviewed in Record et al. (37). An example of this type of experiment is given in Figure 4, which shows binding curves of the nifL promoter in buffer supplemented with potassium acetate at a concentration of 50 mM (Kd = 0.43 nM), 150 mM (Kd = 5.2 nM) and 250 mM (Kd = 22 nM). A plot of the logarithm of the average Kd values determined at a given salt concentration versus the logarithm of the ionic strength displayed an apparently linear relation (Fig. 5). The slopes of this plot –Δlog(Kd)/Δlog(I) were 6.1 ± 0.5 (glnAp2), 5.2 ± 1.2 (nifH) and 2.1 ± 0.1 (nifL). This means that the dissociation constant became weaker by a factor of 106.1 (glnAp2), 105.2 (nifH) and 102.1 (nifL) per decade of higher ionic strength. Again, the glnAp2 and nifH promoters were indistinguishable within the accuracy of the measurements, whereas the nifL promoter showed a much weaker salt dependence.
Figure 4.

Representative binding curves of nifL at different ionic strength. Data are shown for 50 mM (filled squares), 150 mM (open squares) and 250 mM (filled triangles) potassium acetate buffer. The concentration of DNA in the given buffer was 25 pM for the 50 mM potassium acetate buffer and 200 pM for the higher salt concentrations.
Figure 5.

Effect of ionic strength on binding affinity. All Kd values for the glnAp2 (filled triangles), nifH (open squares) and nifL (filled squares) promoters are displayed in a double-logarithmic plot against the ionic strength I (see Table 1). The lines correspond to linear regressions according to log(Kd) = –5.6 + 5.2 log(I) (glnAp2), log(Kd) = –5.0 + 6.1 log(I) (nifH) and log(Kd) = –6.8 + 2.1 log(I) (nifL).
DISCUSSION
The promoter binding affinity of RNAP·σ54 as expressed by the dissociation constant Kd is an important parameter for analyzing the strength of different promoters. In addition, other steps involved in the activation pathway leading to the melting of the DNA at the transcription start site (open complex formation) could also be rate limiting (equations 1 and 2). These involve the interaction of the closed complex with the activator protein at the enhancer, the release of the block imposed by σ54 to open complex formation as well as the kinetics of the isomerization step itself. Here we report data for the binding affinity of RNAP·σ54 to the E.coli glnAp2 promoter and the two promoters nifH and nifL from K.pneumoniae (Fig. 1). Promoters for RNAP·σ54 are characterized by the –24 consensus sequence 5′-(T/C)TGGCACG-3′ (–27 to –20) and a conserved 5′-TTGC(A/T)-3′ motif (–15 to –11) at position –12 (1). All three promoters have conserved –25/–24 and –13/–12 residues, changes of which have been shown to strongly reduce binding of RNAP·σ54 (11,27). The glnAp2 promoter most closely resembles the above consensus sequence (two deviations at –20 and at –14) whereas nifH (–23, –21 and –15) and nifL (–26, –22 and –20) have differences in three positions. A change of the G·C base pair at position –22 in the nifL sequence into the consensus A·T increased the expression level more than 2-fold (38). However, the simple assumption that the consensus sequence with conserved residues from –27 to –20 and –15 to –11 provides the highest rate of transcription initiation appears not to be correct, as deduced from an in vivo comparison of 17 promoter sequences (11). In this context it is noteworthy that the glnAp2 promoter has a consecutive tract of seven A·T base pairs from –5 to +2 whereas for the nifH and nifL promoter only two or three A·T base pairs are found in this region (Fig. 1). This might lead to differences in the kinetics of strand separation during open complex formation.
It has been shown for RNAP·σ70 holoenzyme that the promoter affinity is strongly dependent on the ionic strength (39,40). Accordingly, the Kd for RNAP·σ54 was quantitated at different salt concentrations. The ionic strength in E.coli varies between 0.17 and 0.3 M K+ equivalents (41,42) and an in vivo activity of divalent cations such as Mg2+ between 1 and 10 mM has been estimated (43). Since Mg2+ is also essential for the synthesis activity of RNA polymerase it was included at a concentration of 5 mM in all fluorescence anisotropy binding measurements. Typical physiological conditions should correspond to an ionic strength around I = 0.23 M, which is indicated in Figure 5. At this ionic strength average Kd values of 0.94 ± 0.55 and 0.85 ± 0.3 nM for the nifH and glnAp2 promoters and 8.5 ± 1.9 nM for the nifL promoter were measured (Table 2). Thus, the nifL promoter displayed an about 10-fold lower binding affinity as compared to the two other promoters. The results for the glnAp2 and nifL promoters are in good agreement with previous footprinting experiments (18,21–23). However, the nifH promoter showed an unexpectedly high affinity under these conditions very similar to that of glnAp2 (Table 2). This is in contrast to the footprinting data, which revealed a much lower occupancy of the nifH promoter as compared to the glnAp2 promoter (19,20). Furthermore, a mutation of the CCC residues in the nifH promoter from –15 to –17 to TTT as in the glnAp2 sequence (Fig. 1) increased the footprinting protection (19). Mutations of C to T at –15 and –16 enhanced integration host factor (IHF)-independent gene expression in an in vitro transcription/translation assay (12) indicating that the binding affinity is also related to the promoter strength. In the fluorescence anisotropy binding experiments described here the glnAp2 and nifH promoters showed comparable Kd values over the whole range of salt concentrations whereas the nifL promoter displayed a much weaker salt dependence. This excludes the possibility that the apparently very similar binding affinities of the glnAp2 and nifH promoters is restricted to a certain ionic strength, and it is unclear how the differences between our results and those of the footprinting experiments described in Morret and Buck (19) and Buck and Cannon (20) can be explained. The determination of binding affinities by monitoring changes in the fluorescence anisotropy of dye-labeled DNA duplexes as it has been used here is a true equilibrium method. It is applicable for accurate determinations of Kd values over a large range of solution conditions (25,30–35). A potential source of errors could be an effect of the fluorescent dye on the binding affinity as compared to the unlabeled DNA. For the present set of duplexes a spacer sequence of 5 bp (∼17 Å length) separates the dye attached to a flexible alkyl linker of ∼9 Å length from the RNAP·σ54 binding region as determined by footprinting (44,45). Accordingly, a direct interaction between dye and polymerase appears unlikely but cannot be excluded, since the high resolution structure of the RNAP·σ54 closed complex is presently unknown. However, when preformed complexes of the ROX-labeled promoters with RNAP·σ54 were titrated with unlabeled glnAp2 and nifH duplexes no significant differences in their affinities were observed. Thus, we infer that the dye label had no effect on the relative binding affinities. It is also conceivable that residues outside the region protected in the footprinting experiments affect the binding affinity. In addition, the apparent contradiction between the fluorescence anisotropy and footprinting analysis of binding to the nifH promoter could be explained by a different mode of binding to nifH as opposed to glnAp2, which could lead to a change in the protection pattern.
A characteristic feature of protein–nucleic acid interactions is the strong dependence of Kd on the ion concentration. The slope of the regression line –Δlog(Kd)/Δlog(I) in the double logarithmic plot with log(Kd) versus the log of the salt concentration (Fig. 5) can be used to calculate the number of salt bridges between a protein and its DNA binding site in the absence of divalent ions (37,39,40,46,47). For the RNAP·σ70 closed complex at 0°C a value of 10.5 ± 1.5 has been determined with the T7 A1 promoter, which corresponds to about 12 salt bridges formed between polymerase and DNA (39). If Mg2+ is present in addition to monovalent ions, it acts as a competitor with the negatively charged phosphate groups of the DNA backbone. This leads to a reduction in the apparent slope and some curvature in the log-log plot, especially at higher Mg2+ concentrations (39,40). This effect is relevant for the binding studies described here, which were conducted in the presence of 5 mM MgCl2. Since our data set did not display a significant curvature in the range of salt concentration studied the data were fitted with a linear regression line. Values of the slope of 6.1 ± 0.5 (glnAp2) and 5.2 ± 1.2 (nifH) were obtained (Table 1). From the data reported in Strauss et al. (39) and Shaner et al. (40) a value of –Δlog(Kd)/Δlog(I) ≈ 6 in the range 0.1–0.4 M NaCl is estimated for RNAP·σ70 for a concentration of 5 mM Mg2+. This suggests that similar numbers of salt bridges are formed in the closed complex of RNAP·σ54 with the glnAp2 and nifH promoters as compared to the RNAP·σ70 T7 A1 promoter. In contrast, the slope of the salt dependence of the nifL promoter was clearly reduced and a value of 2.1 ± 0.1 was measured (Fig. 5 and Table 1). This demonstrates that the number of ion pairs between RNAP·σ54 and the nifL sequence is significantly smaller as compared to the two other promoters. We conclude that the reduced strength of the nifL promoter can be explained by its weak binding affinity due to a reduced number of electrostatic interactions with this promoter DNA.
Apart from overall strength, the three promoters for RNAP·σ54 are also different in their response to intrinsic DNA curvature and bending induced by binding of IHF. On the basis of the available data it appears that transcription activation of a ‘strong’ promoter like glnAp2 by NtrC or NifA on superhelical templates is not facilitated by DNA curvature (48). In contrast, the nifH and nifL promoters showed a 3- to 20-fold increase in the equilibrium amount of open complexes in single round transcription experiments if a curved sequence or IHF-induced bending was present between enhancer and promoter (12,49–51). This observation can be explained by a model in which the strong glnAp2 promoter has a higher affinity than the nifH and nifL promoters (48), which is supported by footprinting experiments (18–23). The Kd measurements reported here clearly demonstrate that the glnAp2 promoter has a significantly higher binding affinity than the nifL promoter. However, the differences in promoter occupation reported previously between the glnAp2 and nifH promoters were not detected in our experiments, as discussed above. The about 20-fold stimulation of open complex formation by IHF or by an intrinsically curved DNA sequence at the nifH promoter on supercoiled DNA is not observed with the glnAp2 promoter (12,48,49,51). If the two promoters indeed have very similar binding affinities for RNAP·σ54 other steps in the activation reaction that leads to open complex formation must be responsible for the observed differences with respect to promoter strength and the effect of DNA bending.
Acknowledgments
ACKNOWLEDGEMENTS
We acknowledge the support of Jörg Langowski. Part of the work by K.R. was done at the division Biophysik der Makromoleküle of the Deutsches Krebsforschungszentrum. The project was funded by the DFG (grant Ri 828-1) and by the Volkswagen Foundation in the program Junior Research Groups at German Universities.
REFERENCES
- 1.Barrios H., Valderrama,B. and Morett,E. (1999) Compilation and analysis of sigma(54)-dependent promoter sequences. Nucleic Acids Res., 27, 4305–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wedel A., Weiss,D.S., Popham,D., Dröge,P. and Kustu,S. (1990) A bacterial enhancer functions to tether a transcriptional activator near a promoter. Science, 248, 486–490. [DOI] [PubMed] [Google Scholar]
- 3.Reitzer L.J. and Magasanik,B. (1986) Transcription of glnA in E. coli is stimulated by activator bound to sites far from the promoter. Cell, 45, 785–792. [DOI] [PubMed] [Google Scholar]
- 4.North A.K., Klose,K.E., Stedman,K.M. and Kustu,S. (1993) Prokaryotic enhancer-binding proteins reflect eukaryote-like modularity: the puzzle of nitrogen regulatory protein C. J. Bacteriol., 175, 4267–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collado-Vides J., Magasanik,B. and Gralla,J.D. (1991) Control site location and transcriptional regulation in Escherichia coli. Microbiol. Rev., 55, 371–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rippe K., Guthold,M., von Hippel,P.H. and Bustamante,C. (1997) Transcriptional activation via DNA-looping: visualization of intermediates in the activation pathway of E. coli RNA polymerase-sigma 54 holoenzyme by scanning force microscopy. J. Mol. Biol., 270, 125–138. [DOI] [PubMed] [Google Scholar]
- 7.Gralla J.D. (2000) Signaling through sigma. Nature Struct. Biol., 7, 530–532. [DOI] [PubMed] [Google Scholar]
- 8.Buck M., Gallegos,M.T., Studholme,D.J., Guo,Y. and Gralla,J.D. (2000) The bacterial enhancer-dependent sigma(54) (sigma(N)) transcription factor. J. Bacteriol., 182, 4129–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu H. and Hoover,T.R. (2001) Transcriptional regulation at a distance in bacteria. Curr. Opin. Microbiol., 4, 138–144. [DOI] [PubMed] [Google Scholar]
- 10.Cannon W.V., Gallegos,M.T. and Buck,M. (2000) Isomerization of a binary sigma-promoter DNA complex by transcription activators. Nature Struct. Biol., 7, 594–601. [DOI] [PubMed] [Google Scholar]
- 11.Wang L. and Gralla,J.D. (1998) Multiple in vivo roles for the –12-region elements of sigma 54 promoters. J. Bacteriol., 180, 5626–5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoover T.R., Santero,E., Porter,S. and Kustu,S. (1990) The integration host factor stimulates interaction of RNA polymerase with NIFA, the transcriptional activator for nitrogen fixation operons. Cell, 63, 11–22. [DOI] [PubMed] [Google Scholar]
- 13.Dixon R., Eady,R.R., Espin,G., Hill,S., Iaccarino,M., Kahn,D. and Merrick,M. (1980) Analysis of regulation of Klebsiella pneumoniae nitrogen fixation (nif) gene cluster with gene fusions. Nature, 286, 128–132. [DOI] [PubMed] [Google Scholar]
- 14.Carmona M. and Magasanik,B. (1996) Activation of transcription of σ54-dependent promoters on linear templates requires intrinsic or induced bending of the DNA. J. Mol. Biol., 261, 348–356. [DOI] [PubMed] [Google Scholar]
- 15.Carmona M., Claverie-Martin,F. and Magasanik,B. (1997) DNA bending and the initiation of transcription at σ54-dependent bacterial promoters. Proc. Natl Acad. Sci. USA, 94, 9568–9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whitehall S., Austin,S. and Dixon,R. (1992) DNA supercoiling response of the σ54-dependent Klebsiella pneumoniae nifL promotor in vitro. J. Mol. Biol., 225, 591–607. [DOI] [PubMed] [Google Scholar]
- 17.McClure W.R. (1980) Rate-limiting steps in RNA chain initiation. Proc. Natl Acad. Sci. USA, 77, 5634–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Popham D.L., Keener,J. and Kustu,S. (1991) Purification of the alternative σ factor, σ54, from Salmonella thyphimurium and characterisation of σ54-holoenzyme. J. Biol. Chem., 266, 19510–19518. [PubMed] [Google Scholar]
- 19.Morret E. and Buck,M. (1989) In vivo studies on the interaction of RNA polymerase-σ54 with the Klebsiella pneumoniae and Rhizobium meliloti nifH promoters. J. Mol. Biol., 210, 65–77. [DOI] [PubMed] [Google Scholar]
- 20.Buck M. and Cannon,W. (1992) Activator-independent formation of a closed complex between σ54-holoenzyme and nifH and nifU promoters of Klebsiella pneumoniae. Mol. Microbiol., 6, 1625–1630. [DOI] [PubMed] [Google Scholar]
- 21.Wong P.K., Popham,D., Keener,J. and Kustu,S. (1987) In vitro transcription of the nitrogen fixation regulatory operon nifLA of Klebsiella pneumoniae. J. Bacteriol., 169, 2876–2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Minchin S.D., Austin,S. and Dixon,R.A. (1989) Transcriptional activation of the Klebsiella pneumoniae nifLA promoter by NTRC is face-of-the-helix dependent and the activator stabilizes the interaction of sigma 54-RNA polymerase with the promoter. EMBO J., 8, 3491–3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Austin S., Kundrot,C. and Dixon,R. (1991) Influence of a mutation in the putative nucleotide binding site of the nitrogen regulatory protein NTRC on its positive control function. Nucleic Acids Res., 19, 2281–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weiss D.S., Klose,K.E., Hoover,T.R., North,A.K., Porter,S.C., Wedel,A.B. and Kustu,S. (1992) Prokaryotic transcriptional enhancers. In McKnight,S.L. and Yamamoto,K.R. (eds), Transcriptional Regulation. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, Vol. 2, pp. 667–694.
- 25.Sevenich F.W., Langowski,J., Weiss,V. and Rippe,K. (1998) DNA binding and oligomerization of NtrC studied by fluorescence anisotropy and fluorescence correlation spectroscopy. Nucleic Acids Res., 26, 1373–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rippe K., Mücke,N. and Schulz,A. (1998) Association states of the transcription activator protein NtrC from E. coli determined by analytical ultracentrifugation. J. Mol. Biol., 278, 915–933. [DOI] [PubMed] [Google Scholar]
- 27.Buck M. and Cannon,W. (1992) Specific binding of the transcription factor sigma-54 to promoter DNA. Nature, 358, 422–424. [DOI] [PubMed] [Google Scholar]
- 28.Atkinson M.R. and Ninfa,A.J. (1994) Mechanism and regulation of transcription from bacterial σ54-dependent promoters. In Conaway,R.C. and Conaway,J.W. (eds), Transcription: Mechanism and Regulation. Raven Press, New York, NY, pp. 323–342.
- 29.Perrin F. (1926) Polarisation de la lumière de fluorescence. Vie moyenne des molécules dans l’état excité. J. Phys. Radiat. V Ser. 6, 7, 390–401. [Google Scholar]
- 30.Heyduk T. and Lee,J.C. (1990) Application of fluorescence energy transfer and polarization to monitor Escherichia coli cAMP receptor protein and lac promoter interaction. Proc. Natl Acad. Sci. USA, 87, 1744–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heyduk T., Lee,J.C., Ebright,Y.W., Blatter,E.E., Zhou,Y. and Ebright,R.H. (1993) CAP interacts with RNA polymerase in solution in the absence of promoter DNA. Nature, 364, 548–549. [DOI] [PubMed] [Google Scholar]
- 32.LeTilly V. and Royer,C.A. (1993) Fluorescence anisotropy assays implicate protein-protein interactions in regulating trp repressor DNA binding. Biochemistry, 32, 7753–7758. [DOI] [PubMed] [Google Scholar]
- 33.Guest C.R., Hochstrasser,R.A., Dupuy,C.G., Allen,D.J., Benkovic,S.J. and Millar,D.P. (1991) Interaction of DNA with the Klenow fragment of DNA polymerase I studied by time-resolved fluorescence spectroscopy. Biochemistry, 30, 8759–8770. [DOI] [PubMed] [Google Scholar]
- 34.Reedstrom R.J., Brown,M.P., Grillo,A., Roen,D. and Royer,C.A. (1997) Affinity and specificity of trp repressor-DNA interactions studied with fluorescent oligonucleotides. J. Mol. Biol., 273, 572–585. [DOI] [PubMed] [Google Scholar]
- 35.Boyer M., Poujol,N., Margeat,E. and Royer,C.A. (2000) Quantitative characterization of the interaction between purified human estrogen receptor alpha and DNA using fluorescence anisotropy. Nucleic Acids Res., 28, 2494–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Senear D.F., Ross,J.B. and Laue,T.M. (1998) Analysis of protein and DNA-mediated contributions to cooperative assembly of protein-DNA complexes. Methods, 16, 3–20. [DOI] [PubMed] [Google Scholar]
- 37.Record M.T. Jr, Zhang,W. and Anderson,C.F. (1998) Analysis of effects of salts and uncharged solutes on protein and nucleic acid equilibria and processes: a practical guide to recognizing and interpreting polyelectrolyte effects, Hofmeister effects and osmotic effects of salts. Adv. Protein Chem., 51, 281–353. [DOI] [PubMed] [Google Scholar]
- 38.Khan H., Buck,M. and Dixon,R. (1986) Deletion loop mutagenesis of the nifL promoter from Klebsiella pneumoniae: role of the –26 to –12 region in promoter function. Gene, 45, 281–288. [DOI] [PubMed] [Google Scholar]
- 39.Strauss H.S., Burgess,R.R. and Record,M.T.,Jr (1980) Binding of Escherichia coli ribonucleic acid polymerase holoenzyme to a bacteriophage T7 promoter-containing fragment: evaluation of promoter binding constants as a function of solution conditions. Biochemistry, 19, 3504–3515. [DOI] [PubMed] [Google Scholar]
- 40.Shaner S.L., Melancon,P., Lee,K.S., Burgess,R.R. and Record,M.T.,Jr (1983) Ion effects on the aggregation and DNA-binding reactions of Escherichia coli RNA polymerase. Cold Spring Harb. Symp. Quant. Biol., 47, 463–472. [DOI] [PubMed] [Google Scholar]
- 41.Kao-Huang Y., Revzin,A., Butler,A.P., O’Conner,P., Noble,D.W. and von Hippel,P.H. (1977) Nonspecific DNA binding of genome-regulating proteins as a biological control mechanism: measurement of DNA-bound Escherichia coli lac repressor in vivo. Proc. Natl Acad. Sci. USA, 74, 4228–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cayley S., Lewis,B.A., Guttman,H.J. and Record,M.T.,Jr (1991) Characterization of the cytoplasm of Escherichia coli K-12 as a function of external osmolarity. Implications for protein-DNA interactions in vivo. J. Mol. Biol., 222, 281–300. [DOI] [PubMed] [Google Scholar]
- 43.Schlax P.J., Capp,M.W. and Record,M.T.,Jr (1995) Inhibition of transcription initiation by lac repressor. J. Mol. Biol., 245, 331–350. [DOI] [PubMed] [Google Scholar]
- 44.Popham D.L., Szeto,D., Keener,J. and Kustu,S. (1989) Function of a bacterial activator protein that binds to transcriptional enhancers. Science, 243, 629–635. [DOI] [PubMed] [Google Scholar]
- 45.Tintut Y., Wong,C., Jiang,Y., Hsieh,M. and Gralla,J.D. (1994) RNA polymerase binding using a strongly acidic hydrophobic-repeat region of σ54. Proc. Natl Acad. Sci. USA, 91, 2120–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Record M.T. Jr, Lohman,M.L. and De Haseth,P. (1976) Ion effects on ligand-nucleic acid interactions. J. Mol. Biol., 107, 145–158. [DOI] [PubMed] [Google Scholar]
- 47.Lohman T.M. and Mascotti,D.P. (1992) Thermodynamics of ligand-nucleic acid interactions. Methods Enzymol., 212, 400–425. [DOI] [PubMed] [Google Scholar]
- 48.Schulz A., Langowski,J. and Rippe,K. (2000) The effect of the DNA conformation on the rate of NtrC activated transcription of E. coli RNA polymerase σ54 holoenzyme. J. Mol. Biol., 300, 709–725. [DOI] [PubMed] [Google Scholar]
- 49.Santero E., Hoover,T.R., North,A.K., Berger,D.K., Porter,S.C. and Kustu,S. (1992) Role of integration host factor in stimulating transcription from the σ54-dependent nifH promoter. J. Mol. Biol., 227, 602–620. [DOI] [PubMed] [Google Scholar]
- 50.Cheema A.K., Choudhury,N.R. and Das,H.K. (1999) A- and T-tract-mediated intrinsic curvature in native DNA between the binding site of the upstream activator NtrC and the nifLA promoter of Klebsiella pneumoniae facilitates transcription. J. Bacteriol., 181, 5296–5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Molina-López J.A., Govantes,F. and Santero,E. (1994) Geometry of the process of transcription activation at the σ54-dependent nifH promoter of Klebsiella pneumoniae. J. Biol. Chem., 269, 25419–25425. [PubMed] [Google Scholar]