


Abstract
We find that nuclear protein extracts from mammalian cells contain an activity that allows DNA ends to associate with circular pUC18 plasmid DNA. This activity requires the catalytic subunit of DNA-PK (DNA-PKcs) and Ku since it was not observed in mutants lacking Ku or DNA-PKcs but was observed when purified Ku/DNA-PKcs was added to these mutant extracts. Purified Ku/DNA-PKcs alone did not produce association of DNA ends with plasmid DNA suggesting that additional factors in the nuclear extract are necessary for this activity. Competition experiments between pUC18 and pUC18 plasmids containing various nuclear matrix attachment region (MAR) sequences suggest that DNA ends preferentially associate with plasmids containing MAR DNA sequences. At a 1:5 mass ratio of MAR to pUC18, approximately equal amounts of DNA end binding to the two plasmids were observed, while at a 1:1 ratio no pUC18 end binding was observed. Calculation of relative binding activities indicates that DNA end-binding activities to MAR sequences was 7–21-fold higher than pUC18. Western analysis of proteins bound to pUC18 and MAR plasmids indicates that XRCC4, DNA ligase IV and scaffold attachment factor A preferentially associate with the MAR plasmid in the absence or presence of DNA ends. In contrast, Ku and DNA-PKcs were found on the MAR plasmid only in the presence of DNA ends suggesting that binding of these proteins to DNA ends is necessary for their association with MAR DNA. The ability of DNA-PKcs/Ku to direct DNA ends to MAR and pUC18 plasmid DNA is a new activity for DNA-PK and may be important for its function in double-strand break repair. A model for DNA repair based on these observations is presented.
INTRODUCTION
The ability to repair DNA double-strand breaks (DSBs) generated by ionizing radiation, chemical agents or endogenous cellular processes is important for survival and for maintaining genomic integrity of the cell. Both yeast and mammalian cells have homologous recombinational and non-homologous end joining (NHEJ) pathways for repairing DNA DSBs (1–5). In yeast most DSBs are repaired by homologous recombination, whereas in mammalian cells the NHEJ pathway appears to be the primary mechanism.
Studies using ionizing radiation sensitive and DSB repair defective rodent cell lines have identified at least five gene products involved in the repair of DNA DSBs by the NHEJ pathway. Three of these gene products encode components of a large protein complex known as the DNA-dependent protein kinase (DNA-PK). Two of the proteins in this complex consist of a tightly associated 70 kDa/86 kDa heterodimer known as Ku (6,7). The 86 kDa subunit of Ku is absent in the xrs series of CHO mutants (8–10). The third member of the DNA-PK consists of a 465 kDa catalytic subunit (DNA-PKcs) that is deficient in the CHO mutant cell line V3, the mouse SCID cell line and the human glioma cell line M059J (11,12). The fourth gene product is XRCC4, a 38 kDa nuclear phosphoprotein, which complements the DSB repair defect in the CHO mutant XR-1 (13) and tightly associates with the fifth gene product, DNA ligase IV, resulting in a stabilization of the protein and a stimulation of DNA ligase IV activity (14,15). Further studies using these mutants revealed that these proteins were also required for V(D)J recombination, the process in B and T cells which allows immunological diversity during antibody and T-cell receptor production (10,16), indicating that NHEJ and V(D)J recombination share common components.
A key element in NHEJ is the binding of the Ku heterodimer to DNA ends and the recruitment of the DNA-PKcs to the Ku-bound ends. Indeed, Ku has high affinity for binding to free DNA ends (6,7,17) as well as other DNA structural alterations including nicks, single strand gaps and hairpin loops (18–20). Recruitment of the DNA-PKcs to Ku-bound DNA results in the stimulation of its cognate protein kinase activity (19,21). The DNA-PKcs phosphorylates a number of proteins in vitro including p53, replication protein A, several transcription factors, both subunits of Ku, and XRCC4, as well as itself (22,23). Autophosphorylation of the DNA-PKcs is associated with inactivation of the protein kinase activity and dissociation of the DNA-PKcs from Ku (23). The kinase activity of DNA-PKcs appears to be important to its functioning in NHEJ since mutants showing no kinase activity are both defective in DSB repair and V(D)J recombination.
Many lines of evidence point to the nuclear matrix as the site for DNA replication and RNA transcription (24). The nuclear matrix is generally thought of as a three-dimensional filamentous protein network within the nucleus that is associated with the organization of the DNA into higher order chromosomal domains or territories (25). Attached to the nuclear matrix are repeating DNA loop domains of 50–200 kb which are anchored at their base through interaction with matrix attachment region (MAR) DNA sequences (26–28). MAR sequences are typically regions of AT-rich DNA, which are usually located both 5′ and 3′ of genes as well as within introns, and are considered to be the DNA sequences which delineate active transcriptional units within the nuclear matrix (29–31). RNA polymerases and many transcription factors are tightly associated with the nuclear matrix and form a complex that is associated with MAR sequences (32–35). Many MAR sequences are located close to or within transcription enhancer sequences (26,27,29) and MARs have been shown to enhance transcriptional activity in vivo (36,37). DNA synthesis also occurs in association with the nuclear matrix and many of the proteins involved in DNA synthesis exist as a complex tightly associated with the nuclear matrix (24,38,39). Thus, the current idea is that many of the macromolecular complexes involved in nuclear processes exist as ‘factories’ anchored to the nuclear matrix.
In this report, we present in vitro evidence indicating that DNA-PK bound to DNA ends preferentially associates with circular plasmid DNA containing nuclear MAR DNA sequences. In addition to DNA-PKcs and Ku heterodimer, this interaction requires other unknown factors in the nuclear extract. We also find that XRCC4, DNA ligase IV, poly(ADP-ribose) polymerase, scaffold attachment factor-A and topoisomerase II preferentially bind to MAR sequences suggesting that a repair complex may assemble on the nuclear matrix.
MATERIALS AND METHODS
Cell culture
The human malignant glioma cell lines, M059K and M059J, were maintained in DMEM (Life Technologies, Inc.) supplemented with 10% fetal bovine serum (FBS) and were kindly provided by Dr Allalunis-Turner. The Chinese hamster ovary cell lines CHO-K1, AA8, V-3, V3-transfectant and xrs5 were grown in Ham’s F12 medium (Life Technologies, Inc.) supplemented with 10% FBS. All cell lines were maintained in the absence of antibiotics.
Antibodies and enzymes
Mouse monoclonal antibody against DNA-PKcs was purchased from Kamiya Biomedical Co. and is a mixture of monoclonal antibodies from clones 25-4, 18-2 and 42-24. Mouse anti-Ku86 monoclonal antibody, clone Ku15 (19), was purchased from Sigma-Aldrich. Rabbit anti-XRCC4 (LN13) and anti-DNA ligase IV (LN16) antibodies were made as described in a previous study (40). The rabbit anti-SAF-A antibody was kindly provided by Dr Frank Fackelmeyer (University of Konstanz, Germany). The mouse anti-poly (ADPribose) polymerase, clone C2-10 and mouse anti-topoisomerase II were purchased from Oncogene Research Products. Purified DNA-PKcs/Ku and exonuclease III were obtained from Promega Corp.
Preparation of nuclear extracts
Nuclear extracts from all cell lines were prepared by a modification of the method of Dignam et al. (41) using 0.2% (v/v) NP-40 to lyse the cells. The concentration of MgCl2 in the extraction buffers was increased to 5 mM to prevent clumping of the nuclei. Nuclear extracts were aliquoted, flash frozen in dry ice and stored at –70°C until used in the mobility shift assays. The protein concentration of the nuclear extracts was determined using the Bradford method (Bio-Rad) using bovine serum albumin as a standard. Labeled nuclear extracts were prepared by culturing M059K cells for 4 h in DMEM containing 20 µM 35S-methionine at 1.75 Ci/mmol.
Probe preparation and radiolabeling
The 144 bp probe used in this study was produced by digesting pUC18 to completion with PvuII and EcoRI and polyacrylamide gel electrophoresis (PAGE) purified as described previously (9). This fragment was 32P-labeled using the Klenow fragment of DNA polymerase I (Roche Molecular Systems) in the presence of 100 µCi of [α-32P]dATP (3000 Ci/mmol; NEN Life Sciences) and 200 µM each of unlabeled dGTP, dCTP and dTTP (Roche Molecular Systems). Unincorporated deoxynucleotides were removed by chromatography through Sephadex G-50.
Competitor plasmids and MAR sequences
pUC18, 2.7 kb, was used for the majority of the studies. The 2.1 and 4.0 kb plasmids in Figure 2 are pBR322 constructs obtained from a supercoiled DNA ladder (Invitrogen) while the 6.2 kb plasmid is pcDNA3.1 containing a 0.8 kb fragment of hamster glucose-6-phosphate dehydrogenase gene. The plasmid pMII-MAR contains a 2974 bp intronic MAR sequence from the human topoisomerase I gene cloned into pUC18 and was was kindly provided by Dr Frank Fackelmeyer. The 671 bp human HGPRT MAR and the 623 bp mouse Igk MAR sequences were obtained by PCR amplification using the following primers from the published sequence (42,43): 5′-TGTAGTAGTTATGAGCCCATGTCCC-3′ (HGPRT MAR forward), 5′-CCAACACGGTGAAATCCTGTCTC-3′ (HGPRT MAR reverse), 5′-AAG GAAAGGGTGACTTATTGGAG-3′ (Igk MAR forward) and 5′-AACAGAAACTGAGAGTTCTTTACCAAG-3′ (Igk MAR reverse). Each MAR amplification product was isolated from low melting point agarose (FMC Bioproducts) and initially cloned into the TA cloning vector pCRIITopo (Invitrogen) using the manufacturer’s protocol. The presence of the correct sequence for both MAR-containing plasmids was confirmed by standard dideoxy sequencing. For the present study, each MAR sequence was subcloned into the EcoRI site of pUC18 by standard techniques. The high-AT control plasmid pB3/LIM-1 was obtained as a gift from Dr John Dunn (Brookhaven National Laboratory) and contained a 3569 bp insert from Borrelia burgdorferi cloned into the vector pSCANS (44). For our experiments, a 2566 bp BamHI–SacI fragment of pB3/LIM-1 was subcloned into the BamHI–SacI site of pUC18 and this plasmid was designated pB3/BS. All plasmids used in this study were purified using a combination of standard silica-based columns (Qiagen, Inc.) and CsCl density gradient centrifugation.
Figure 2.

Binding of radiolabeled probe to the complex comigrates with plasmid DNA and formation of the complex requires Ku-bound DNA ends. (A) Agarose mobility shift assays with or without M059K nuclear extract (0.5 µg) were performed as described in Figure 1. Mobility shift reactions contained closed circular plasmids of increasing molecular size as indicated above the lanes in the autoradiogram and ethidium bromide stained gel. White arrows indicate the position of the large DNA end-binding (EB) complex in the ethidium bromide stained gel. (B) The dependence of probe binding to closed circular pUC18 on DNA ends was determined by competition with unlabeled ends. Lane 1, closed circular pUC18 and a 32P-labeled 144 bp probe without nuclear extract; lane 2, labeled probe, pUC18, and M059K nuclear extract (0.5 µg). Lanes 3–9, competition agarose mobility shift assays containing fixed concentrations of 32P-labeled probe and total DNA, but with increasing concentrations of unlabeled DNA ends previously obtained by digesting pUC18 with restriction enzymes: EcoRI (lane 3), HindIII (lane 4), PvuII (lane 5), RsaI (lane 6), HaeII (lane 7), TaqI (lane 8) and AluI (lane 9). The number of restriction cuts in each reaction is indicated. Upper arrow, the position of the pUC18 end-binding complex; lower arrows, the position of the Ku-bound probe.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts (0.5–2 µg) were incubated with 1 ng of 32P-labeled probe for 15 min at room temperature in the presence of 1 µg of supercoiled pUC18 or 500 ng each of pUC18 and pMII-MAR in a final volume of 20 µl in binding buffer [10 mM Tris–HCl, pH 8.0, 0.1 mM EDTA, 150 mM NaCl, 5 mM DTT, 200 µM PMSF, 2.5 µg/ml leupeptin, 1 µg/ml pepstatin A and 10% (v/v) glycerol]. The standard polyacrylamide-based EMSA reaction mixtures were separated in a 5% polyacrylamide gel, dried, and subjected to autoradiography as described (29). For agarose-based EMSA reaction mixtures were separated in a 1.5 or 2.0% Seakem Gold agarose gel (FMC Bioproducts) at 8 V/cm for 2.5 h in 1× TBE buffer (90 mM Tris–HCl, pH 8.3, 90 mM boric acid, 1 mM EDTA). Agarose gels were dried on a ZetaProbe nylon membrane (Bio-Rad) and subjected to autoradiography using Kodak X-OMAT AR film. In most cases, a parallel agarose gel was run under the same conditions, stained with 1 µg/ml ethidium bromide, and photographed. Probe binding to plasmids was quantified using a Molecular Dynamics PhosphorImager to scan dried mobility shift gels and NIH Image software version 1.59 for data analysis. For experiments requiring plasmids pretreated with nuclear extract, supercoiled pUC18 and pMII-MAR plasmids were incubated with M059K nuclear extract under standard reaction conditions as described above, purified using phenol/chloroform and ethanol precipitated.
DNA end-binding activity relative to pUC18 was calculated using the equation:
Keq mar/Keq pUC = [(Bcomp/BpUC) × (pUC/Mcomp) – a]/b
where Keq mar/Keq pUC is the ratio of equilibrum constants for probe binding to the MAR versus pUC18; Mcomp is the nmoles (in base pairs) of the MAR competitor plasmid; pUC is the nmoles (in base pairs) of the pUC18 plasmid; Bcomp/BpUC is the observed ratio of counts (or pixels) bound to the MAR competitor plasmid (Figs 4 and 5, white arrows) versus pUC (Figs 4 and 5, black arrows); a is the proportion of the competitor plasmid that is pUC18 DNA; and b is the proportion of the competitor plasmid that is MAR insert DNA.
Figure 4.
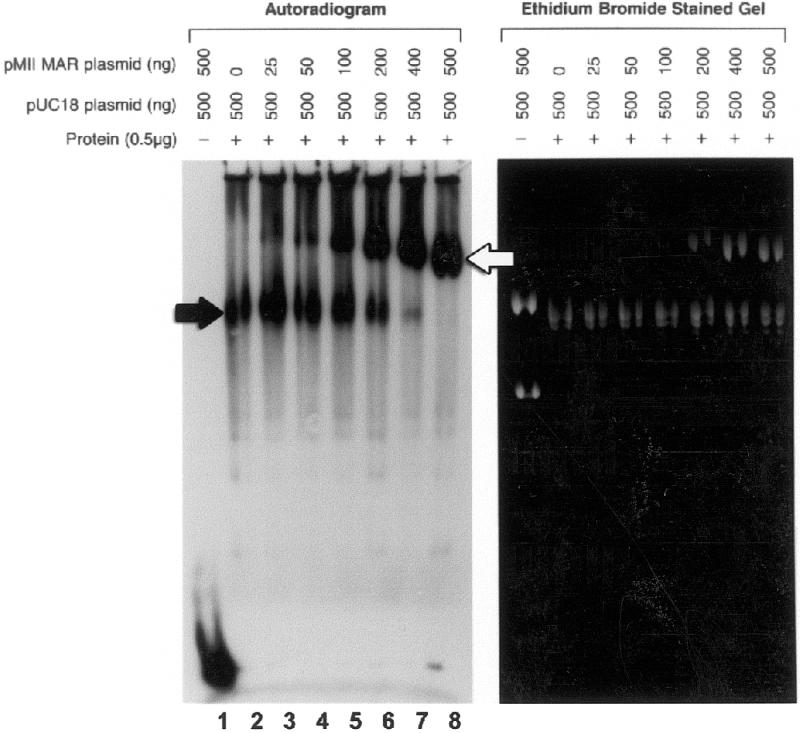
Binding of radiolabeled probe to pUC18 is specifically competed by pMII-MAR, a 2974 bp MAR sequence containing plasmid. To determine the relative specificity of probe binding to a MAR sequence versus pUC18, mobility shift reactions containing M059K nuclear extract (0.5 µg) and 32P-labeled probe (1 ng) were preincubated with closed circular pUC18 (0.5 µg) for 10 min before addition of closed circular pMII-MAR in increasing amounts as indicated. Duplicate sets of reactions were performed. Both sets were separated in parallel 1.5% agarose gels. One set (autoradiogram) was dried for autoradiography, while the other set (ethidium bromide stained) was stained with 1 mg/ml ethidium bromide and photographed. The position of pUC18 is indicated by a black arrow and the pMII-MAR by a white arrow.
Figure 5.

Binding of radiolabeled probe is also competed by other MAR-containing plasmids. Mobility shift reactions were performed exactly as described in Figure 4, except increasing amounts of a 671 bp human HGPRT MAR plasmid (A) or a 623 bp mouse Igk MAR plasmid (B) were used. The position of pUC18 and MAR plasmids are indicated by black arrow and white arrows, respectively.
The above relationship was derived assuming first order kinetics, a steady state equilibrium of probe binding to competitor plasmids, and three independent binding reactions: probe binding to pUC18 sequence in the MAR competitor plasmid, probe binding to MAR sequence in the MAR competitor plasmid and probe binding to pUC18 plasmid.
Preparation of Form II (nicked circular) plasmid standards and exonuclease III digestion
Form II (nicked circular) standards of pUC18 and pMII-MAR plasmid DNA were prepared by digestion with EcoRI (5 U/µg DNA) at 30°C in the presence of 100 µg/ml ethidium bromide (18). Digestion products were separated in a 1% low melting point agarose gel. The band containing the Form II plasmid DNA was excised from the gel and purified using β-agarose according to the manufacturer’s suggested protocol (Roche Molecular Systems).
Nuclear extract-pretreated pMII-MAR and pUC18 were incubated at 37°C with 300 U exonuclease III in a buffer containing 66 mM Tris–Cl, pH 8.0, 0.66 mM MgCl2. An aliquot (100 ng) of each reaction was removed at 0 and 1 h, terminated by the addition of EDTA to 100 mM and separated by agarose gel electrophoresis. Form II standards of both plasmids were similarly treated with exonuclease III and run in parallel.
Elution of binding proteins from plasmids and identification of proteins by western blot analysis
Mobility shift assays with or without DNA ends were scaled up to 100 µg of M059K nuclear extract and 50 µg each of pMII and pUC18 per 400 µl reaction and loaded in a preparative well of a 1.5% agarose gel prepared as above. After separation, plasmid bands were identified by brief ethidium bromide staining and strips containing pMII or pUC18 were excised from the agarose gels. Agarose strips were placed in dialysis bags containing 5 ml of elution buffer (1× TBE, 0.2% SDS and 25 mM 2-mercaptoethanol). Plasmid-bound proteins were eluted from the agarose at 175 V for 3–4 h in the same buffer. Eluted proteins were concentrated by ethanol precipitation (5 vol) at –20°C, dissolved in 100 µl of 1× Laemmli buffer and denatured at 95°C. Proteins were separated in a 10% SDS–PAGE gel (Novex) and electroblotted to a PVDF membrane (Novex) according to the manufacturer’s protocol. Immunoblots were probed with the following primary antibodies: (i) anti-DNA-PKcs (1:500); (ii) anti-Ku86, clone Ku15 (1:500) (19); (iii) anti-XRCC4 (1:500); (iv) anti-DNA ligase IV (1:500); (v) anti-SAF-A (1:1000); (vi) anti-poly(ADP-ribose) polymerase (PARP), clone C2-10 (1:100); and (vii) anti-topisomerase II (1:500). After washing, the immunoblots were incubated with horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit IgG secondary antibodies at 50 mU/ml (Roche). Detection was performed by enhanced chemiluminescence using a LumiLight substrate (Roche) according to the manufacturer’s protocol.
RESULTS
Detection of a large DNA-PKcs-dependent end-binding complex
In a previous study, we used a modified EMSA to detect DNA end-binding activity in hamster, mouse and human nuclear extracts and showed that this DNA end-binding activity is due to a heterodimeric protein known as Ku (9). In this assay, nuclear extracts are incubated with a radiolabeled 144 bp linear DNA fragment in the presence of unlabeled circular plasmid DNA to remove non-DNA end specific DNA binding proteins. The reaction mix is resolved by non-denaturing PAGE and DNA end-binding activity is detected as an upward shift in the mobility of the DNA probe as determined by autoradiography. Using a polyacrylamide-based EMSA, we observed binding activity which would not enter the polyacrylamide gel matrix suggesting a large molecular weight complex. This complex was present in nuclear extracts from a human glioma cell line, M059K (Fig. 1, lane 2), but not in nuclear extracts from another human glioma cell line, M059J (Fig. 1, lane 3). Lees-Miller et al. (12) have shown that the radiosensitive M059J cell line is deficient in DNA-PK activity in vitro and is devoid of any detectable DNA-PKcs by western blot analysis. In contrast, the radioresistant M059K cell line has both in vitro DNA-PK activity and detectable DNA-PKcs. The molecular basis for this difference is the rapid turnover of DNA-PKcs mRNA in the M059J cell line (45,46). Despite the absence of the large complex in M059J, normal Ku-DNA end-binding patterns were observed with both cell lines (Fig. 1, lanes 2 and 3, black arrows). These data suggested the presence of a DNA-PKcs-dependent high molecular weight complex. We were able to resolve this complex using an agarose gel-based EMSA and observed probe binding to a large slow mobility complex (Fig. 1, white arrow) using nuclear extract from the DNA-PKcs-positive M059K cell line, but not with the DNA-PKcs-negative M059J cell (Fig. 1, lanes 5 and 6). However, when purified DNA-PKcs/Ku was added to reactions containing M059J nuclear extract, formation of the slow mobility complex in this DNA-PK-negative cell line was reconstituted (Fig. 1, lane 7). In contrast, the addition of purified DNA-PKcs/Ku to reaction mixtures containing probe and competitor plasmid but without nuclear extract did not result in the formation of the slow mobility complex (Fig. 1, lane 8). In addition, preincubation of M059K nuclear extract with anti-DNA-PKcs antibody resulted in the complete inhibition of probe binding to the pUC18 (data not shown). These data strongly suggest that formation of the large slow mobility complex requires DNA-PKcs and other factors in the nuclear extract.
Figure 1.

A large DNA–protein complex is resolved by an agarose gel EMSA and its formation of the complex is DNA-PK dependent. EMSAs were performed using nuclear extracts (0.5 µg) from human glioblastoma cell lines M059K or M059J in the presence of closed circular pUC18 (1 mg) and 32P-labeled 144 bp probe (1 ng) derived from pUC18. Reactions were separated in a 5% PAGE gel (lanes 1–3) or 1.5% agarose gel (lanes 4–8), dried, and subjected to autoradiography. A parallel agarose gel was stained with 1 mg/ml ethidium bromide and photographed (lanes 9–13). Purified DNA-PKcs/Ku (30 ng) was added to reactions either alone (lanes 8 and 13) or containing M059J nuclear extract (lanes 7 and 12). Black arrows indicate the positions of Ku-bound probe; the white arrow the position of the large DNA-PK-dependent end-binding complex.
In these EMSAs, circular supercoiled plasmid DNA, pUC18, was used as a competitor DNA. In order to determine the migration of the competitor plasmid DNA in relation to this large complex, a parallel agarose gel was run and stained with ethidium bromide. As seen in Figure 1 (lanes 9 and 10), the mobility of the pUC18 plasmid is reduced in the presence of nuclear extract and appears to comigrate with the large slow mobility complex (Fig. 1, lane 10). The absence of probe binding in M059J nuclear extract was not due to some different effect of the extract on the pUC18 plasmid since M059J nuclear extract produced the same reduction in plasmid mobility as did the M059K nuclear extract (Fig. 1, lanes 10 and 11).
The DNA end-binding complex associates with plasmid DNA
Comigration of the large complex with the plasmid observed in Figure 1 raises the possibility that the linear probe is interacting with DNA-PKcs/Ku and other proteins bound to the circular competitor plasmid. To answer this question, plasmids of increasing molecular size (2.1–6.2 kb) were added to reaction mixtures containing M059K nuclear extract and radiolabeled probe before separation on a 1.5% agarose gel. Results in Figure 2A show that the end-binding activity is associated with plasmid DNA since probe binding moved concomitantly with the increasing molecular size of the plasmids (see arrows). In each case, an upward shift in the plasmid DNA mobility was observed after the addition of nuclear extract.
To determine if complex formation on the pUC18 plasmid is dependent on Ku bound to DNA ends, a competition experiment was performed with unlabeled DNA ends. Agarose mobility shift assays were done using M059K nuclear extract and a fixed amount of 32P-labeled probe and pUC18 competitor DNA (either circular or cut plasmid). A fixed amount of pUC18 (linear plasmid), previously cut with various restriction enzymes producing an increasing number of cut sites, was added to the reactions. The total mass of DNA was kept constant in all reactions while the number of unlabeled DNA ends varied. As shown in Figure 2B, as the ratio of 32P-labeled DNA ends to total ends decreases, the amount of labeled probe in the lower Ku-bound DNA bands (arrows) decreased, thus confirming results from a previous study (9). Similarly, the amount of labeled ends bound to the upper pUC18 end-binding complex (arrow) also decreased. These data suggest that the binding activity to the plasmid is dependent on the number of Ku-bound DNA ends.
Complex formation in DSB repair-deficient hamster mutants
To further characterize the involvement of DNA-PKcs and Ku in the association of DNA ends with plasmid DNA, we examined complex formation in hamster cells which have significantly reduced levels of Ku and DNA-PKcs protein (∼10-fold) and DNA-PK kinase activity (∼50-fold) compared with human cells (47–49). Nuclear extracts from hamster wild-type and DSB repair mutant cell lines were examined for plasmid DNA end-binding activity using the same agarose mobility shift assay described above. The binding of the probe to closed circular pUC18 was observed in both wild-type CHO and AA8 nuclear extracts (Fig. 3, lanes 2 and 3, white arrow), but not in the mutant cell line V3 which is deficient in DNA-PKcs (Fig. 3, lane 4) or in the DSB repair mutant cell line xrs5 which lacks Ku86 (Fig. 3, lane 6). In contrast, the V3-YAC147 cell line, which is a V3-YAC fusion hybrid expressing the human DNA-PKcs gene, produced an intense band at the position of the large end-binding complex (Fig. 3, lane 5, white arrow) and virtually no DNA end-binding activity at the position of the Ku bands (Fig. 3, lane 5, black arrows). Densitometer scans of bands at the position of the large complex indicated that the V3-YAC147 cell line had 11- and 25-fold greater binding activity than CHO and the parental AA8 cell, respectively. Previous studies have shown that the level of human DNA-PKcs protein and DNA-PK kinase activity in the V3-YAC147 cell line is similar to that observed in human cell extracts which is 10- and 40-fold greater than AA8, respectively (11,50). Thus, the large amount of plasmid end binding observed in the V3-YAC147 cell is most likely due to this increase in human PKcs protein. This suggests that the targeting of DNA ends to circular plasmid DNA is strongly dependent on the level of DNA-PKcs and/or its kinase activity and that hamster Ku and other proteins can interact with human DNA-PKcs to perform this reaction. The above observations combined with those in Figures 1 and 2 strongly suggest that the association of DNA ends with circular plasmid DNA requires both DNA-PKcs and Ku86.
Figure 3.
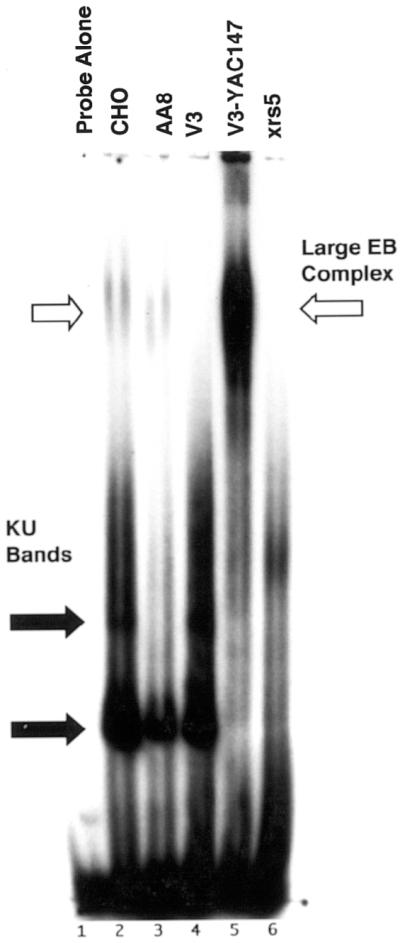
Complex formation in Chinese hamster DSB repair mutants. Agarose gel EMSAs were performed using nuclear extracts (5 µg) from Chinese hamster cell lines CHO, AA8, V3 (DNA-PKcs–), V3-YAC147 (DNA-PKcs+) and xrs5 (Ku86–) as described in Figure 1. A large complex was observed (large EB complex, upper black arrow) in extracts from parental lines CHO and AA8, but not in mutant lines lacking Ku86 (xrs5), or the catalytic subunit of DNA-PK (V3). In contrast, the V3-YAC147 cell line that contains a YAC expressing human DNA-PKcs produced an intense band at the position of the large complex. Lower black arrows identify the Ku bands.
Preferential binding of DNA ends to plasmids containing MAR sequences
The above results indicate that DNA ends interact in vitro with circular plasmid DNA and that this interaction is DNA-PKcs/Ku dependent. This raises the question: what is the in vivo significance of this interaction? As discussed in the Intro duction, the nuclear matrix plays an important role in many nuclear processes and MAR sequences play a critical role in many structural and functional aspects of the nuclear matrix. This raised the possibility that in this in vitro assay the binding of Ku-bound DNA ends to plasmid DNA might occur preferentially at MAR DNA sequences.
To test this possibility, we performed competition experiments using a plasmid, pMII-MAR, which contains a 2974 bp MAR sequence from the human topoisomerase I gene inserted into pUC18. DNA end-binding reaction mixtures were first preincubated with a fixed amount of pUC18 (500 ng) and then increasing amounts of pMII-MAR (0–500 ng) were added. As shown in Figure 4, as the mass ratio of pMII-MAR to pUC18 increases from 1:20 to 1:1, DNA end-binding activity clearly shifts from the position of pUC18 (2.7 kb) (black arrow) to the larger pMII-MAR plasmid (5.7 kb) (white arrow). At a 1:5 mass ratio of pMII-MAR to pUC18 approximately equal amounts of DNA end binding were observed, while at a 1:1 mass ratio no pUC18 end binding was observed. Also, there was a progressive increase in electrophoretic mobility of the pMII-MAR plasmid as the amount of this plasmid in the binding reaction is increased from 25 ng (Fig. 4, lane 3) to 500 ng (Fig. 4, lane 8); however, no change in mobility of the pUC18 plasmid was observed. This suggests that proteins responsible for DNA end binding on the pUC18 plasmid are a small fraction of the total proteins bound since their removal during competition does not affect the mobility of this plasmid. Increased electrophoretic mobility of the pMII-MAR plasmid could be due to an alteration in plasmid conformation and/or a significant decrease in the total mass of plasmid-bound protein; however, the molecular weight of this plasmid is 3.7 × 106 Da. Considering this and the unchanged mobility of the pUC18 plasmid, the most reasonable explanation for the change in pMII-MAR mobility is that, as proteins responsible for DNA end binding are removed from the pUC18 plasmid during competition and bind to the pMII-MAR plasmid, they induce a change in the conformation of pMII-MAR plasmid which increases its mobility.
It is possible that the above-mentioned preferential end binding may be unique to pMII since it is unusually large for a MAR sequence. To determine whether preferential binding is a general phenomenon, similar competition experiments were performed using a 623 bp mouse Igk MAR (Fig. 5A) and a 671 bp human HGPRT MAR (Fig. 5B) cloned into pUC18. Although these competitor plasmids contain only ∼20% MAR sequence compared to ∼50% for pMII, a similar shift of DNA end-binding activity from the position of pUC18 to the MAR competitor plasmid was observed as the mass ratio of MAR competitor to pUC18 increases from 1:20 to 1:1. The increase in DNA end-binding activity relative to pUC18 was calculated for each of these MAR sequences and the results are displayed in Table 1. DNA end-binding activity for the MII MAR was 7-fold higher than pUC18, while IgK and the HGPRT MARs were ∼21-fold higher than pUC18 and 3-fold greater than MII.
Table 1. DNA end-binding activity relative to pUC18 for MAR and high-AT sequences.
| Ratio of DNA end-binding constants for various DNA sequences inserted into pUC18 versus pUC18 plasmid without insert | |||
|---|---|---|---|
| Human MII (2974 bp) | Mouse IgK (623 bp) | Human HGPRT (671 bp) | B3/BS (2566 bp) |
| 7.1 ± 0.71a | 22.0 ± 4.6a | 20.7 ± 3.7a | 1.8 ± 0.76a |
aMean and standard deviation for at least three independent observations; the ratio of DNA end-binding constants was calculated using the equation in Materials and Methods.
As mentioned previously, many MAR sequences are AT-rich. Indeed many of the functional properties of MAR sequences can be attributable to AT-rich motifs within them, e.g. topoisomerase II binding and cleavage sites, origins of replication, and binding sites for several classical nuclear matrix binding proteins such as SATB1, SAF-A and Lamin B1 (51,52). Since the MAR sequences inserted into pUC18 are 64–72% AT, we explored whether the preferential binding of DNA ends to these sequences was a consequence of their high AT content. A 2566 bp AT-rich (69%) non-MAR sequence, B3/BS, from B.burgdorferi was inserted into pUC18 and used in competition experiments to compare its binding activity relative to pUC18 as described above (44). As shown in Table 1, DNA end-binding activity to the AT-rich B3/BS sequence was 1.8-fold higher than pUC18; however, binding to the MII MAR was still 4-fold higher than the AT-rich B3/BS sequence and 12-fold higher than the IgK and HGPRT MARs. As we have seen, in the absence of MAR DNA (Figs 1 and 2), a significant amount of DNA end-binding activity is observed using the pU18 plasmid which does have two 300 bp high (>60%) AT regions that theoretically can form secondary hairpin structures (53).
Altered plasmid conformation does not produce plasmid association of DNA ends by purified DNA-PKcs/Ku
To investigate the reason for the reduced mobility of pUC18 and MAR-containing plasmids in the presence of nuclear extract, pUC18 and MII-MAR plasmid bands were excised from agarose gels after a mobility shift assay with 1:1 mixtures of pMII-MAR and pUC18 plasmids as described in Figure 4. Both sets of plasmids were subjected to purification by proteinase-K digestion, organic solvent extraction and ethanol precipitation. When these purified pUC18 and MII-MAR plasmids were again subjected to agarose gel electrophoresis, they had reduced mobility compared with the supercoiled pUC18 and MII-MAR standards suggesting that the nuclear extract had produced a change in supercoiling (data not shown). This observation suggests the possibility that DNA-PKcs and Ku are the only proteins necessary for the observed binding of DNA ends to plasmid DNA. The only role other proteins in nuclear extract play in this process would be to alter the topological conformation of the plasmid DNA (change in supercoiling) and allow DNA-PKcs/Ku bound to DNA ends to associate with the MAR plasmid without additional nuclear proteins. To test this possibility, 1:1 mixtures of pMII-MAR and pUC18 plasmids were preincubated with or without 0.5 mg of M059K nuclear extract and both sets of plasmids purified as described above. Equal amounts of untreated or nuclear extract-pretreated plasmid DNAs were added to end-binding reaction mixtures containing increasing amounts of purified DNA-PKcs/Ku and the products were separated in agarose gels. As we observed previously, preincubation of pMII-MAR and pUC18 plasmids with nuclear extract produced a conformational change (Fig. 6B, ethidium bromide stained gel). However, neither the preincubated nor the non-preincubated plasmids showed any 32P-probe binding at the position of the pUC18 or the pMII-MAR plasmids even at 120 ng of purified DNA-PKcs/Ku (Fig. 6, lanes 2–9). The preincubated plasmids were fully capable of binding DNA ends in the presence of 30 ng of DNA-PKcs/Ku and 0.5 µg of M059J nuclear extract (Fig. 6A, lane 10). These data support the conclusion that protein factors in addition to DNA-PKcs/Ku are required for binding of DNA ends to plasmid DNA. Although alterations in plasmid conformation may be necessary for this interaction, it is not sufficient to explain probe binding to the plasmids.
Figure 6.

Effect of plasmid conformation on probe binding to plasmid DNA in the presence of purified DNA-PKcs/Ku. Mixtures of pUC18 and pMII-MAR at 0.5 µg each were preincubated without or with 0.5 µg of M059K nuclear extract for 10 min at room temperature, purified using phenol/chloroform and ethanol precipitated. Agarose mobility shift assays containing labeled probe (1 ng) and untreated or nuclear extract pretreated plasmid DNA were performed with increasing amounts of purified DNA-PKcs/Ku as indicated. A separate reaction containing labeled probe, M059J nuclear extract (0.5 µg) and 30 ng of DNA-PKcs/Ku [(A), lane 10] was included as a positive control. Reaction mixtures were separated in a 1.5% agarose gel, dried, and subjected to autoradiography (A). A duplicate set of reactions was separated in parallel but stained with 1 µg/ml ethidium bromide and photographed (B).
Binding of plasmid-associated DNA ends is not due to nicked plasmids
The Ku heterodimer binds to nicks and other single-strand gaps in DNA and the binding of Ku to these structures has been shown to recruit DNA-PKcs and to stimulate its kinase activity in vitro (18,20). Using an exonuclease III based assay, we investigated the possibility that our observed binding of DNA ends to pMII-MAR and/or pUC18 is due to the generation of single-strand nicks during incubation with nuclear extract. pMII-MAR and pUC18 plasmids were pretreated separately with M059K nuclear extract and purified as described in the previous section. The pretreated plasmids were reacted with exonuclease III for 1 h at 37°C. As a positive control, Form II (nicked circular) standards of pMII-MAR and pUC18 were reacted with exonuclease III in parallel. All reactions were separated in a 1% agarose gel and visualized by ethidium bromide staining. As shown in Figure 7, the majority of the pUC18 Form II standard (lane 6) was digested by exonuclease III after 1 h, while the Form II standard of pMII-MAR (lane 2) was completely digested. However, neither pMII-MAR (lane 4) nor pUC18 (lane 8) pretreated with nuclear extract showed any significant digestion by exonuclease III after 1 h. Both of these plasmids migrated at the same positions as nuclear extract pretreated pMII-MAR and pUC18 plasmids that were not treated with exonuclease III (data not shown). These results indicate that treatment with nuclear extract does not produce significant numbers of nicks in the plasmids and suggest that single-strand breaks are not involved in the binding of DNA ends to either plasmid.
Figure 7.

Exonuclease III detection of DNA nicks in plasmids treated with nuclear extract. Form II (nicked, relaxed) plasmid standards of pMII-MAR and pUC18 were prepared as described in Materials and Methods. Nuclear extract treated pMII-MAR (lanes 3 and 4) and pUC18 (lanes 7 and 8) plasmids were prepared separately as described in Figure 6 and were incubated for 0 (lanes 3 and 7) and 1 h (lanes 4 and 8) with 300 U exonuclease III at 37°C. Nicked standards of pMII-MAR (lanes 1 and 2) and pUC18 (lanes 5 and 6) were similarly treated for 0 (lanes 1 and 5) and 1 h (lanes 2 and 6) with 300 U exonuclease III. Aliquots (100 ng) were removed at the indicated times and reactions were terminated by the addition of EDTA to 100 mM, plasmids purified, and separated by agarose gel electrophoresis.
Identification of proteins preferentially associated with the pMII-MAR plasmid
The inability of purified DNA-PKcs/Ku to direct ends to the pMII-MAR when added to the probe in the absence of nuclear extract suggests that other proteins are required to affect this interaction. In an initial effort to identify other proteins that may be associated with the DNA-PKcs/Ku-dependent binding to the pMII-MAR plasmid, we examined known DSB repair and MAR interacting proteins. Agarose bands containing the pMII-MAR or pUC18 plasmids were excised after electrophoretic separation of DNA end-binding reactions containing M059K nuclear extract, unlabeled probe, and a 1:1 mass ratio of these plasmids. Agarose bands containing the pMII-MAR or pUC18 plasmids were also excised after similar reactions performed in the absence of probe. Proteins associated with these plasmids were purified by electroelution from equal volumes (by weight) of agarose and subjected to SDS–PAGE and western blot analysis. As seen in Figure 8, significantly greater amounts of XRCC4 (Fig. 8B), the MAR-binding protein SAF-A (Fig. 8C), DNA ligase IV (Fig. 8D), topoisomerase II (Topo II) (Fig. 8E), and PARP (Fig. 8F) were bound to the MII-MAR plasmid than pUC18 in binding reactions either with or without DNA ends. Control experiments using 35S-labeled nuclear proteins indicated that proteins were recovered from the MII-MAR and pUC18 plasmids with about equal efficiency; 96.2 ± 2.9% and 92.8 ± 7.2%, respectively. Furthermore, no immunoreactive bands were observed after western analysis of proteins isolated from agarose bands at the pMII-MAR and pUC18 positions in reactions that contained M059K nuclear extract but no probe or plasmid DNA. These observations suggest that the above proteins associate with the pMII-MAR plasmid in the presence or absence of DNA ends and that this preferential association is not due to differences in recovery of protein from the two plasmids. The preferential association of XRCC4, DNA ligase IV, SAF-A, Topo II and PARP on the MAR-containing plasmid in the presence or absence of DNA ends suggests that a pre-formed repair complex may be present on the nuclear matrix. In contrast to the above proteins, Ku86 and DNA-PKcs (Fig. 8A) were found only on the pMII-MAR in the presence of DNA ends.
Figure 8.

Identification of proteins preferentially associated with the pMII-MAR plasmid. Mobility shift reactions with (+Ends) or without (–Ends) unlabeled 144 bp probe were separated in a preparative 1.5% agarose gel, pUC18 and MII plasmid bands excised, and proteins electroeluted as described in Materials and Methods. Proteins eluted from equal amounts (weight) of agarose were subjected to SDS–PAGE and western blot analysis. Western blots were probed with antibodies directed against DNA-PKcs and Ku86 (A), XRCC4 (B), SAF-A (C), DNA ligase IV (D), Topoisomerase II (E) or PARP (F). Lane 5 of (A)–(F) contained 25 µg of M059K nuclear extract. For clarity, lanes in western blots (B) and (F) were rearranged; however, each blot contained proteins from the same experiment treated under identical conditions. Proteins extracted from three independent mobility shift reactions produced similar band patterns.
DISCUSSION
Using a modified mobility shift assay, we have found that mammalian nuclear extracts contain an activity that allows DNA ends to associate with closed circular plasmid DNA. This activity was shown to require both DNA-PKcs and Ku since it was not observed in rodent mutants lacking Ku86 or in rodent or human mutants lacking the DNA-PKcs. However, plasmid DNA end-binding activity was restored when purified DNA-PKcs/Ku was added to extracts from cells lacking DNA-PKcs or when nuclear extract was prepared from DNA-PKcs-deficient mutants containing the complementing DNA-PKcs gene (Figs 1 and 3). In contrast, the addition of purified DNA-PKcs/Ku in the absence of nuclear extract did not produce an interaction of DNA ends with the plasmid DNA suggesting that additional factors in nuclear extract are required for this activity. This ability of DNA-PK/Ku to direct DNA ends to circular plasmid DNA is a new activity for DNA-PK that may be necessary for its function in DSB repair.
As discussed previously, MAR DNA sequences are known to anchor DNA to the nuclear matrix. In competition experiments between pUC18 and pUC18 containing various inserted MAR sequences, we observed a 7–21-fold higher DNA end-binding activity to MAR sequences than pUC18. Since many MAR sequences are AT rich, similar competition experiments were performed using plasmids containing an inserted AT-rich, non-MAR sequence. When plasmid DNA end-binding activity between the MAR and AT-rich non-MAR sequences was compared, binding activity to the MAR plasmids was 4–12-fold higher than the AT-rich, non-MAR plasmid. Thus, preferential binding of DNA ends to MAR DNA is not simply due to their high AT content but may be due to other characteristics unique to MAR sequences. Bode et al. (52) have shown that several MAR sequences become stably base-unpaired under negative superhelical stress. MAR sequences containing base-unpairing regions exhibit strong affinity for the nuclear matrix, stimulate promoter activity of a reporter gene in stable transfectants, and specifically bind certain nuclear matrix proteins (51,52,54,55). These observations suggest that some specific DNA structural conformations, possibly base-unpairing regions, may direct to MAR sequences those proteins responsible for binding DNA ends to circular plasmids. Support for this concept is the observation in Figure 4 that the mobility of the pMII-MAR plasmid progressively increases as the amount of labeled probe bound to this plasmid increased. This suggests that the proteins responsible for this end-binding activity produce a conformational change in the MAR plasmid that increases mobility. This conformational change could be the generation of protein-bound hairpin loops that generate a more compact plasmid structure (Fig. 9). A change in the topological conformation, supercoiling, of the pUC and MAR plasmid was also observed after treatment with nuclear extract (Fig. 6). This may be due to the presence of topoisomerase I or II in nuclear extract. Indeed, we did find that topoisomerase II preferentially associates with MAR plasmid DNA (Fig. 8E). However, a change in topology in either plasmid alone was shown to be insufficient to explain the binding of ends in the presence of purified DNA-PKcs/Ku (Fig. 6) suggesting that additional proteins are necessary for DNA end binding. Furthermore, covalent ligation of the probe to the pUC18 or MAR plasmid or a recombinational insertion of the probe is not the explanation for this binding activity since excision of pUC18 and MAR plasmid bands containing 32P-labeled probe, DNA purification, and re-electrophoresis of these plasmids produced a single band the size of the free probe and no observable radioactivity at the MAR and pUC18 positions in the gel (data not shown).
Figure 9.

A proposed model for the repair of DSBs on the nuclear matrix.
Using western blot analysis, we examined the proteins associated with the MAR plasmid versus those binding to pUC18 both in the presence or absence of DNA ends. We observed that XRCC4, DNA ligase IV, SAF-A, PARP and topoisomerase II preferentially associated with the MAR plasmid in the presence or absence of DNA ends. Although we have no direct evidence of physical association, these observations raise the possibility that these proteins may assemble into a MAR-associated DNA repair complex in the absence of DNA ends. In contrast to the above proteins, DNA-PKcs and Ku86 were not found on the pUC18 plasmid but were found on the MAR plasmid only in the presence of DNA ends. These results suggest that DNA-PKcs/Ku is bound to DNA ends prior to its association with the MAR plasmid and that this binding to DNA ends is necessary for MAR association. This observation is consistent with the findings of Ting et al. (56) who used protein cross-linking reagents and mobility shift assays to show that DNA-PKcs present in whole cell extracts is recruited to Ku-bound DNA ends.
Our observation that PARP, but not Ku86, is present on the MAR plasmid without DNA ends appears to be inconsistent with the results of Galande and Kohwi-Shigematsu (57). Using DNA affinity chromatography with a linear MAR sequence containing base-unpairing regions, they observed co-purification of DNA-PKcs, Ku heterodimer and PARP. They also showed that PARP and Ku form a complex in the absence of DNA and that both the complex and PARP and Ku individually were able to bind to MAR sequences containing base-unpairing regions in a DNA end-independent manner. The isolation of PARP on MAR sequences was observed in several breast cancer cell lines, but not in a non-tumorigenic breast epithelial cell line (58). The absence of end- independent Ku binding in our studies may be due to additional factors present in M059K nuclear extract, but absent in breast cancer nuclear extract, that allow Ku to associate with MAR sequences only if bound to DNA ends. Additionally, the presence of PARP on the MAR sequence without ends may have functional significance since the protein kinase activity of DNA-PKcs is stimulated by the ADP-ribosylation activity of PARP (59).
Our results suggest the following model for DNA DSB repair as seen in Figure 9. We postulate that topoisomerase II, MAR-binding proteins, and other proteins produce a secondary change in the DNA structure of the MAR sequence, illustrated in the model as hairpin loops and cruciform structures. This structure allows XRCC4, ligase IV, PARP and other proteins to assemble into a repair complex. A DSB that occurs in loop DNA is first bound by Ku70/Ku86 that then recruits DNA-PKcs and possibly other proteins to the ends. This DNA end-bound complex then associates with a MAR-bound repair complex where repair of the DSB occurs. Although the data suggest a repair function for targeting of DNA ends to the MAR-bound complex, this targeting activity might also function in regulating expression of genes involved in DNA repair and/or a DNA damage-induced signaling pathway for cell-cycle checkpoint control or apoptosis.
Support for the repair model comes from the observations of Johnston et al. (60,61). They found that loop DSBs were repaired with slow kinetics (t1/2 ∼5 h) while rapid repair (t1/2 ∼15 min) was observed using conventional techniques that measure total breaks, i.e. DSBs in both loop and nuclear matrix DNA. When loop repair and total repair were examined in the DSB repair-deficient xrs5 mutant, which lacks the Ku86 subunit and has no DNA end-binding activity in nuclear extracts, no significant repair of loop DSBs was observed, while total DSBs were repaired with normal rapid repair kinetics. When loop versus matrix repair was examined in the DNA-PKcs-deficient V3 cell line (11), again no repair of DSBs in loop DNA was observed, while the majority of DSBs in nuclear matrix attached DNA were repaired. Also, Yavuzer et al. (62) have found using a yeast two-hybrid screen that DNA-PKcs interacts with the nuclear matrix protein C1D (63).
Further support for this idea of recruitment of damaged DNA to a nuclear matrix repair complex has been found for ultraviolet light (UV) damaged DNA. Koehler and Hanawalt (64) found that the frequency of cyclobutane pyrimidine dimers (the major UV-induced lesion) in matrix-bound DNA increases 50% 1 h after UV irradiation in normal cells, but not in ERCC1 mutants deficient in repair of this lesion. Thus, cyclobutane pyrimidine dimers appear to be targeted to the nuclear matrix. Mullenders et al. (65) have shown that repair patches induced after UV irradiation are enriched in a nuclear matrix fraction. Further studies have shown that some of the enzymatic activities involved with the DNA resynthesis step after incision of the damaged DNA are tightly associated with the nuclear matrix fraction (66). Lastly, the association of DNA ends with MAR sites on the nuclear matrix might serve as focal points for the recruitment of repair proteins to assemble the repair complex. Indeed, clustering of repair proteins, hMre11, hRAD50 and hRAD51 in nuclei after γ-irradiation have been observed (67,68).
The preferential association of proteins involved in the NHEJ pathway on MAR sequences and the association of DNA-PKcs/Ku-bound DNA ends to this repair complex strengthens the argument that the nuclear matrix is important to the process of DNA repair. The precise role that the nuclear matrix and MAR sequences plays in DSB repair and what other proteins are involved in this process remain to be elucidated. Studies using 35S-labeled nuclear extract and two-dimensional gel electrophoresis are currently underway to identify other proteins that are preferentially associated with MAR sequences.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Janet Sawicki for critical reading of this manuscript and Loretta Rossino and Rosalyn Gilchrist for editorial assistance. This work was supported by the Lankenau Hospital Foundation and NIH grants CA877144, CA67095 and CA45277 from the National Cancer Institute.
REFERENCES
- 1.Teo S.-H. and Jackson,S.P. (1997) Identification of Saccharomyces cerevisiae DNA ligase IV: involvement in DNA double strand break repair. EMBO J., 16, 4788–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boulton S.J. and Jackson,S.P. (1996) Identification of a Saccharomyces cerevisiae Ku80 homolog: roles in DNA double strand break repair rejoining and telomeric maintenance. Nucleic Acids Res., 24, 4639–4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boulton S.J. and Jackson,S.P. (1996) S. cerevisiae Ku70 potentiates illegitimate DNA double strand break repair and serves as a barrier to error prone DNA repair pathways. EMBO J., 15, 5093–5103. [PMC free article] [PubMed] [Google Scholar]
- 4.Herrmann G., Lindahl,T. and Schar,P. (1998) Saccharomyces cerevisiae LIF1: a function involved in DNA double-strand break repair related to mammalian XRCC4. EMBO J., 17, 4188–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takata M., Sasaki,M.S., Sonoda,E., Morrison,C., Hashimoto,M., Utsumi,H., Yamaguchi-Iwai,Y. and Shinohara,A. (1998) Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J., 17, 5497–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mimori T. and Hardin,J.A. (1986) Mechanism of interaction between Ku protein and DNA. J. Biol. Chem., 261, 10375–10379. [PubMed] [Google Scholar]
- 7.Zhang W.W. and Yaneva,M. (1992) On the mechanisms of Ku protein binding to DNA. Biochem. Biophys. Res. Commun., 186, 574–579. [DOI] [PubMed] [Google Scholar]
- 8.Smider V., Rathmell,W.K., Lieber,M.R. and Chu,G. (1994) Restoration of x-ray resistance and V(D)J recombination in mutant cells by Ku cDNA. Science, 266, 288–291. [DOI] [PubMed] [Google Scholar]
- 9.Getts R.C. and Stamato,T.D. (1994) Absence of a Ku-like DNA end binding activity in the xrs double-strand DNA repair-deficient mutant. J. Biol. Chem., 269, 15981–15984. [PubMed] [Google Scholar]
- 10.Taccioli G.E., Gottlieb,T.M., Blunt,T., Priestley,A., Demengeot,J., Mizuta,R., Lehmann,A.R., Alt,F.W. and Jackson,S.P. (1994) Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science, 265, 1442–1445. [DOI] [PubMed] [Google Scholar]
- 11.Blunt T., Finnie,N.J., Taccioli,G.E., Smith,G.C., Demengeot,J., Gottlieb,T.M., Mizuta,R., Varghese,A.J., Alt,F.W., Jeggo,P.A. et al. (1995) Defective DNA-dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell, 80, 813–823. [DOI] [PubMed] [Google Scholar]
- 12.Lees-Miller S.P., Godbout,R., Chan,D.W., Weinfeld,M., Day,R.S.,III, Barron,G.M. and Allalunis-Turner,J. (1995) Absence of p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science, 267, 1183–1185. [DOI] [PubMed] [Google Scholar]
- 13.Li Z., Otevrel,T., Gao,Y., Cheng,H.L., Seed,B., Stamato,T.D., Taccioli,G.E. and Alt,F.W. (1995) The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell, 83, 1079–1089. [DOI] [PubMed] [Google Scholar]
- 14.Critchlow S.E., Bowater,R.P. and Jackson,S.P. (1997) Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr. Biol., 7, 588–598. [DOI] [PubMed] [Google Scholar]
- 15.Grawunder U., Wilm,M., Wu,X., Kulesza,P., Wilson,T.E., Mann,M. and Lieber,M.R. (1997) Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature, 388, 492–495. [DOI] [PubMed] [Google Scholar]
- 16.Pergola F., Zdzienicka,M.Z. and Lieber,M.R. (1993) V(D)J recombination in mammalian cell mutants defective in DNA double-strand break repair. Mol. Cell. Biol., 13, 3464–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paillard S. and Strauss,F. (1991) Analysis of the mechanism of interaction of simian Ku protein with DNA. Nucleic Acids Res., 19, 5619–5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blier P.R., Griffith,A.J., Craft,J. and Hardin,J.A. (1993) Binding of Ku protein to DNA. Measurement of affinity for ends and demonstration of binding to nicks. J. Biol. Chem., 268, 7594–7601. [PubMed] [Google Scholar]
- 19.Gottlieb T.M. and Jackson,S.P. (1993) The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell, 72, 131–142. [DOI] [PubMed] [Google Scholar]
- 20.Morozov V.E., Falzon,M., Anderson,C.W. and Kuff,E.L. (1994) DNA-dependent protein kinase is activated by nicks and larger single-stranded gaps. J. Biol. Chem., 269, 16684–16688. [PubMed] [Google Scholar]
- 21.Dvir A., Peterson,S.R., Knuth,M.W., Lu,H. and Dynan,W.S. (1992) Ku autoantigen is the regulatory component of a template-associated protein kinase that phosphorylates RNA polymerase II. Proc. Natl Acad. Sci. USA, 89, 11920–11924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lees-Miller S.P., Chen,Y.R. and Anderson,C.W. (1990) Human cells contain a DNA-activated protein kinase that phosphorylates simian virus 40 T antigen, mouse p53, and the human Ku autoantigen. Mol. Cell. Biol., 10, 6472–6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan D.W. and Lees-Miller,S.P. (1996) The DNA-dependent protein kinase is inactivated by autophosphorylation of the catalytic subunit. J. Biol. Chem., 271, 8936–8941. [DOI] [PubMed] [Google Scholar]
- 24.Berezney R., Mortillaro,M.J., Ma,H., Wei,X. and Samarabandu,J. (1995) The nuclear matrix: a structural milieu for genomic function. Int. Rev. Cytol., 162A, 1–65. [DOI] [PubMed] [Google Scholar]
- 25.Ma H., Siegel,A.J. and Berezney,R. (1999) Association of chromosome territories with the nuclear matrix. Disruption of human chromosome territories correlates with the release of a subset of nuclear matrix proteins. J. Cell Biol., 146, 531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gasser S.M. and Laemmli,U.K. (1986) Cohabitation of scaffold binding regions with upstream/enhancer elements of three developmentally regulated genes of D. melanogaster. Cell, 46, 521–530. [DOI] [PubMed] [Google Scholar]
- 27.Cockerill P.N. and Garrard,W.T. (1986) Chromosomal loop anchorage of the kappa immunoglobulin gene occurs next to the enhancer in a region containing topoisomerase II sites. Cell, 44, 273–282. [DOI] [PubMed] [Google Scholar]
- 28.Roberge M. and Gasser,S. (1992) DNA loops: structural and functional properties of scaffold-attached regions. Mol. Microbiol., 6, 419–423. [DOI] [PubMed] [Google Scholar]
- 29.Forrester W., van Genderen,C., Jenuwein,T. and Grosschedl,R. (1994) Dependence of enhancer-mediated transcription of the immunoglobulin mu gene on nuclear matrix attachment regions. Science, 265, 1221–1225. [DOI] [PubMed] [Google Scholar]
- 30.Bode J. and Maass,K. (1988) Chromatin domain surrounding the human interferon-beta gene as defined by scaffold-attached regions. Biochemistry, 27, 4706–4711. [DOI] [PubMed] [Google Scholar]
- 31.Blasquez V., Xu,M., Moses,S. and Garrard,W. (1989) Immunoglobulin kappa gene expression after stable integration. I. Role of the intronic MAR and enhancer in plasmacytoma cells. J. Biol. Chem., 264, 21183–21189. [PubMed] [Google Scholar]
- 32.Dickinson P., Cook,P.R. and Jackson,D.A. (1990) Active RNA polymerase I is fixed within the nucleus of HeLa cells. EMBO J., 9, 2207–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cook P.R. (1994) RNA polymerase: structural determinant of the chromatin loop and the chromosome. Bioessays, 16, 425–430. [DOI] [PubMed] [Google Scholar]
- 34.Cook P.R. (1989) The nucleoskeleton and the topology of transcription. Eur. J. Biochem., 185, 487–501. [DOI] [PubMed] [Google Scholar]
- 35.Wei X., Somanathan,S., Samarabandu,J. and Berezney,R. (1999) Three-dimensional visualization of transcription sites and their association with splicing factor-rich nuclear speckles. J. Cell Biol., 146, 543–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klehr D., Maass,K. and Bode,J. (1991) Scaffold-attached regions from the human interferon beta domain can be used to enhance the stable expression of genes under the control of various promoters. Biochemistry, 30, 1264–1270. [DOI] [PubMed] [Google Scholar]
- 37.Phi-Van L., von Kries,J.P., Ostertag,W. and Stratling,W.H. (1990) The chicken lysozyme 5′ matrix attachment region increases transcription from a heterologous promoter in heterologous cells and dampens position effects on the expression of transfected genes. Mol. Cell. Biol., 10, 2302–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cook P.R. (1991) The nucleoskeleton and the topology of replication. Cell, 66, 627–635. [DOI] [PubMed] [Google Scholar]
- 39.Berezney R. (1991) Visualizing DNA replication sites in the cell nucleus. Semin. Cell Biol., 2, 103–115. [PubMed] [Google Scholar]
- 40.Bryans M., Valenzano,M.C. and Stamato,T.D. (1999) Absence of DNA liagse IV in XR-1 cells: evidence for stabilization by XRCC4. Mutat. Res., 433, 53–58. [DOI] [PubMed] [Google Scholar]
- 41.Dignam J., Lebovitz,R. and Roeder,R. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res., 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sykes R.C., Lin,D., Hwang,S.J., Framson,P.E. and Chinault,A.C. (1988) Yeast ARS function and nuclear matrix association coincide in a short sequence from the human HPRT locus. Mol. Gen. Genet., 212, 301–309. [DOI] [PubMed] [Google Scholar]
- 43.Max E.E., Maizel,J.V.J. and Leder,P. (1981) The nucleotide sequence of a 5.5-kilobase DNA segment containing the mouse kappa immunoglobulin J and C region genes. J. Biol. Chem., 256, 5116–5120. [PubMed] [Google Scholar]
- 44.Dunn J.J., Buchstein,S.R., Butler,L.L., Fisenne,S., Polin,D.S., Lade,B.N. and Luft,B.J. (1994) Complete nucleotide sequence of a circular plasmid from the Lyme disease spirochete, Borrelia burgdorferi. J. Bacteriol., 176, 2706–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Galloway A.M., Spencer,C.A., Anderson,C.W. and Allalunis-Turner,M.J. (1999) Differential stability of the DNA-activated protein kinase catalytic subunit mRNA in human glioma cells. Oncogene, 18, 1361–1368. [DOI] [PubMed] [Google Scholar]
- 46.Anderson C.W., Dunn,J.J., Freimuth,P.I., Galloway,A.M. and Allalunis-Turner,M.J. (2001) Frameshift mutation in PRKDC, the gene for DNA-PKcs, in the DNA repair-defective, human, glioma-derived cell line M059J. Radiat. Res., 156, 2–9. [DOI] [PubMed] [Google Scholar]
- 47.Finnie N.J., Gottlieb,T.M., Blunt,T., Jeggo,P.A. and Jackson,S.P. (1995) DNA-dependent protein kinase activity is absent in xrs-6 cells: implications for site-specific recombination and DNA double-strand break repair. Proc. Natl Acad. Sci. USA, 92, 320–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anderson C.W. and Lees-Miller,S.P. (1992) The nuclear serine/threonine protein kinase DNA-PK. Crit. Rev. Eukaryot. Gene Expr., 2, 283–314. [PubMed] [Google Scholar]
- 49.Wang J., Chou,C.H., Blankson,J., Satoh,M., Knuth,M.W., Eisenberg,R.A., Pisetsky,D.S. and Reeves,W.H. (1993) Murine monoclonal antibodies specific for conserved and non-conserved antigenic determinants of the human and murine Ku autoantigens. Mol. Biol. Rep., 18, 15–28. [DOI] [PubMed] [Google Scholar]
- 50.Priestley A., Beamish,H.J., Gell,D., Amatucci,A.G., Muhlmann-Diaz,M.C., Singleton,B.K., Smith,G.C., Blunt,T., Schalkwyk,L.S., Bedford,J.S. et al. (1998) Molecular and biochemical characterisation of DNA-dependent protein kinase-defective rodent mutant irs-20. Nucleic Acids Res., 26, 1965–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Belle I., Cai,S. and Kohwi-Shigematsu,T. (1998) The genomic sequences bound to special AT-rich sequence-binding protein 1 (SATB1) in vivo in Jurkat T cells are tightly associated with the nuclear matrix at the bases of the chromatin loops. J. Cell Biol., 141, 335–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bode J., Kohwi,Y., Dickinson,L., Joh,T., Klehr,D., Mielke,C. and Kohwi-Shigematsu,T. (1992) Biological significance of unwinding capability of nuclear matrix-associated DNAs. Science, 255, 195–197. [DOI] [PubMed] [Google Scholar]
- 53.SantaLucia J.J. (1998) A unified view of polymer, dumbbell and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl Acad. Sci. USA, 95, 1460–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kohwi-Shigematsu T., Maass,K. and Bode,J. (1997) A thymocyte factor SATB1 suppresses transcription of stabley integrated matrix-attachment region-linked reporter genes. Biochemistry, 36, 12005–12010. [DOI] [PubMed] [Google Scholar]
- 55.Dickinson L.A. and Kohwi-Shigematsu,T. (1995) Nucleolin is a matrix attachment region DNA-binding protein that specifically recognizes a region with with base-unpairing potential. Mol. Cell. Biol., 15, 456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ting N.S., Chan,D.W., Lintott,L.G., Allalunis-Turner,J. and Lees-Miller,S.P. (1999) Protein–DNA complexes containing DNA-dependent protein kinase in crude extracts from human and rodent cells. Radiat. Res., 151, 414–422. [PubMed] [Google Scholar]
- 57.Galande S. and Kohwi-Shigematsu,T. (1999) Poly(ADP-ribose) polymerase and Ku autoantigen form a complex and synergistically bind to matrix attachment sequences. J. Biol. Chem., 274, 20521–20528. [DOI] [PubMed] [Google Scholar]
- 58.Yanagisawa J., Ando,J., Nakayama,J., Kohwi,Y. and Kohwi-Shigematsu,T. (1996) A matrix attachment region (MAR)-binding activity due to a p114 kilodalton protein is found only in human breast carcinomas and not in normal and benign breast disease tissues. Cancer Res., 56, 457–462. [PubMed] [Google Scholar]
- 59.Ruscetti T., Lehnert,B.E., Halbrook,J., Le Trong,H., Hoekstra,M.F., Chen,D.J. and Peterson,S.R. (1998) Stimulation of the DNA-dependent protein kinase by poly(ADP-ribose) polymerase. J. Biol. Chem., 273, 14461–14467. [DOI] [PubMed] [Google Scholar]
- 60.Johnston P.J. and Bryant,P.E. (1994) A component of DNA double-strand break repair is dependent on the spatial orientation of the lesions within the higher-order structures of chromatin. Int. J. Radiat. Biol., 66, 531–536. [DOI] [PubMed] [Google Scholar]
- 61.Johnston P.J., MacPhail,S.H., Stamato,T.D., Kirchgessner,C.U. and Olive,P.L. (1998) Higher-order chromatin structure-dependent repair of DNA double-strand breaks: involvement of the V(D)J recombination double-strand break repair pathway. Radiat. Res., 149, 455–462. [PubMed] [Google Scholar]
- 62.Yavuzer U., Smith,G.C., Bliss,T., Werner,D. and Jackson,S.P. (1998) DNA end-independent activation of DNA-PK mediated via association with the DNA-binding protein C1D. Genes Dev., 12, 2188–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nehls P., Keck,T., Greferath,R., Spiess,E., Glaser,T., Rothbarth,K., Stammer,H. and Werner,D. (1998) cDNA cloning, recombinant expression and characterization of polypetides with exceptional DNA affinity. Nucleic Acids Res., 26, 1160–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Koehler D.R. and Hanawalt,P.C. (1996) Recruitment of damaged DNA to the nuclear matrix in hamster cells following ultraviolet irradiation. Nucleic Acids Res., 24, 2877–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mullenders L.H.F., van Kasteran-van Leeuwen,A.C., van Zeeland,A.A. and Natarajan,A.T. (1988) Nuclear matrix associated DNA is preferentially repaired in normal human fibroblasts, exposed to a low dose of ultraviolet light but not in Cockayne’s syndrome fibroblasts. Nucleic Acids Res., 16, 10607–10623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bouayadi K., van der Leer-van Hoffen,A., Balajee,A.S., Natarajan,A.T., van Zeeland,A.A. and Mullenders,L.H. (1997) Enzymatic activities involved in the DNA resynthesis step of nucleotide excision repair are firmly attached to chromatin. Nucleic Acids Res., 25, 1056–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haaf T., Golub,E.I., Reddy,G., Radding,C.M. and Ward,D.C. (1995) Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc. Natl Acad. Sci. USA, 92, 2298–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li M.J. and Maizels,N. (1997) Nuclear Rad51 foci induced by DNA damage are distinct from Rad51 foci associated with B cell activation and recombination. Exp. Cell Res., 237, 93–100. [DOI] [PubMed] [Google Scholar]