

Abstract
Plastid mRNA stability is tightly regulated by external signals such as light. We have investigated the biochemical mechanism responsible for the dark-induced decrease of relative half-lives for mRNAs encoding photosynthetic proteins. Protein fractions isolated from plastids of light-grown and dark-adapted plants correctly reproduced an RNA degradation pathway in the dark that is downregulated in the light. This dark-dependent pathway is initiated by endonucleolytic cleavages in the petD mRNA precursor substrate proximal to a region that can fold into a stem–loop structure. Polynucleotide phosphorylase (PNPase) polyadenylation activity was strongly increased in the protein fraction isolated from plastids in dark-adapted plants, but interestingly PNPase activity was not required for the initiation of dark-induced mRNA degradation. A protein factor present in the protein fraction from plastids of light-grown plants could inactivate the endonuclease activity and thereby stabilize the RNA substrate in the protein fraction from plastids of dark-adapted plants. The results show that plastid mRNA stability is effectively controlled by the regulation of a specific dark-induced RNA degradation pathway.
INTRODUCTION
Essential proteins and enzymes in the chloroplast are encoded in both nuclear and the plastid genomes. The expression of plastid-encoded genes during chloroplast development is tightly controlled at different levels, including processing and accumulation of their mRNAs. Plastid RNA metabolism is regulated by mechanisms that depend on RNA secondary structures, nucleases and regulatory RNA-binding proteins (RNPs) (reviewed in 1–3). Similar to bacterial mRNAs, most plastid mRNAs contain an inverted repeat sequence in their 3′-untranslated region (UTR) that can fold into a stable stem–loop structure (1). The precursor RNA is processed to a mature mRNA, which terminates in the 3′ stem–loop structure (1).
The molecular mechanisms of RNA degradation in the chloroplast have been studied in detail during the last few years and in certain aspects resemble RNA degradation in Escherichia coli (2,3). RNA degradation in E.coli is initiated by endoribonucleolytic cleavage of the RNA molecule followed by addition of a poly(A) tail to the 3′ end of the resulting fragments (2–5). A similar mode of mRNA degradation has been proposed for higher plant chloroplasts based on the detection of internally polyadenylated fragments of psbA mRNA (3,6). The polyadenylated cleavage products are subsequently removed by exonucleolytic degradation involving polynucleotide phosphorylase (PNPase) and RNase II in E.coli, and PNPase in the chloroplast (6–8).
In E.coli, the enzymatic activities involved in the degradation process are associated with a high molecular-weight complex termed the ‘degradosome’ (4). They include PNPase, enolase, an RNA helicase and the endonuclease RNaseE, which appear to work together during RNA processing and degradation. RNaseE most likely initiates degradation, followed by PNPase degradation of the cleavage products. RNA helicase is most likely required for unwinding secondary structures (reviewed in 4). Recent studies in chloroplasts revealed that PNPase is present as a homo-multimer complex capable of degrading plastid RNAs (9). Several endonucleases have also been reported in the chloroplast, but none of them resembles bacterial RNaseE (10–12). Among the chloroplast endonucleases characterized in more detail are the p54 protein from mustard and the CSP41 protein from spinach plastids (10,11). The activities of both enzymes are controlled by their redox and phosphorylation states (11,13).
Chloroplast mRNA processing and stabilization is also dependent on the presence of nuclear-encoded RNPs (3,12). These RNPs have been identified from a variety of plant species, including tobacco (14,15), Arabidopsis thaliana (16), maize (17) and barley (18). Their structures are similar and consist of a transit peptide, an acidic N-terminal domain and two RNA-binding domains that are separated by a spacer region (reviewed in 2,3). Although their molecular function is not fully characterized, it appears that RNPs are required to stabilize ribosome-free mRNAs in the chloroplast stroma (19–21). The RNA-binding activity of the 28 RNP has been investigated in more detail and can be modulated by serine/threonine phosphorylation of the N-terminal acidic region (22). Thus, the 28 RNP may function in a light-dependent signal cascade that regulates plastid RNA stability by reversible protein phosphorylation.
Here we report an in vitro system that can reproduce light-dependent changes of relative half-lives of chloroplast mRNAs, which were previously measured in vivo (6,23). Using this system we analyzed activities of RNPs, PNPase and endonucleases that are involved in plastid mRNA degradation and processing. This approach revealed an endonuclease-initiated RNA degradation pathway in the dark that is inactivated by light.
MATERIALS AND METHODS
Plant growth conditions and plastid processing extract isolation
Spinach plants were grown for 7 weeks under greenhouse conditions in a 8/16 h light–dark cycle. After 7 weeks, 20 plants were transferred into darkness for 48 h. Control plants were continued in the 8/16 h light–dark cycle during this time. After 48 h, leaves from plants in the light cycle and from dark-grown plants were harvested and chloroplasts were isolated at 4°C as described (24). Chloroplasts from dark-adapted plants were isolated under green safe light conditions. A soluble chloroplast protein extract was isolated and fractionated as described previously (24,25). We refer to the protein fraction obtained from chloroplasts of illuminated plants as ‘light-protein fraction’ (LPF) and the corresponding protein fraction from plants transferred to darkness as ‘dark-protein fraction’ (DPF).
Synthesis of the petD 3′-UTR mRNA precursor substrate
The petD 3′-UTR mRNA precursor substrate was synthesized by transcription from a plasmid carrying the petD 3′-UTR as described previously (26). The synthetic RNA contains 70 nt of the coding region and extends 58 nt 3′ proximal to the stem–loop, which itself is 46 nt long. The transcription reaction was performed for 1 h at 37°C with 0.5 µg of linearized plasmid as the DNA template in a total volume of 20 µl. The assay contained 40 mM Tris–HCl pH 8.0, 6 mM MgCl2, 2 mM spermidin, 10 mM DTT, 0.5 mM of GTP, CTP and ATP, 0.1 mM UTP, 10 U RNasin (Boehringer-Mannheim/Roche), 10 U T7-RNA polymerase (Boehringer-Mannheim/Roche) and 100 µCi [α-32P]UTP (20 µCi/µl, 800 Ci/mmol). Following transcription, the RNA products were purified by denaturing TBE electrophoresis and isolated by ethanol precipitation.
UV-crosslinking analysis
The indicated amount of LPF or DPF, 2 µl of RNA probe (50 000 c.p.m./µl), 3.75 mM MgCl2, 2 mM DTT and 10 mM KCl were combined in a total volume of 10 µl in an Eppendorf tube. The mixture was incubated on ice and then placed in an UV-Stratalinker (Stratagene) with the lids of the Eppendorf tubes opened. The RNA–protein mixture was then irradiated twice with 0.12 J. Following addition of 2 µg of RNaseA, the mixture was incubated at 37°C for 20 min. The reaction was terminated by the addition of 4 µl of SDS-sample buffer, the samples were heated for 5 min at 75°C and proteins were separated by 6 or 10% SDS–PAGE (27). Following electrophoresis, the gel was dried and subjected to autoradiography.
In vitro RNA-processing assays
The indicated amount of LPF or DPF, 2 µl of the labeled RNA substrate, 3.75 mM MgCl2, 2 mM DTT, 10 mM KCl and 2 mM sodium phosphate (pH 7) were combined in a total volume of 10 µl in an Eppendorf tube. If not stated otherwise, 2 mM sodium phosphate was included routinely in the assays to stimulate the phosphorolytic activity of the chloroplast PNPase. Samples were incubated for the indicated times at room temperature. The reaction was terminated by the addition of 90 µl of stop solution [6 M urea, 1% SDS, 4.5 mM aurintricarboxylic acid (Sigma)] and the RNA was purified by phenol/chloroform extraction. The RNA products were precipitated by the addition of 2 µl of 3 M sodium acetate (pH 5.5), 2 µg of E.coli tRNA and 200 µl of ethanol, incubated at –20°C for 30 min and analyzed by TBE gel electrophoresis and autoradiography. Polyadenylation assays were performed as described above except that 2 mM ATP and 20 U RNasin were added to, and phosphate omitted from, the in vitro processing reactions. Under these conditions the nucleases are inactivated and the polyadenylated precursor RNA can accumulate.
Assembly of a PNPase-specific antibody column and immunodepletion of LPF or DPF
The PNPase-specific antibody column was assembled as previously described (21). In brief, the PNPase-specific antibody (anti-rabbit) was linked to protein A beads as recommended by the manufacturer (Pierce). The immunodepletion was performed at 4°C in batch overnight. Approximately 500 µg of LPF or DPF were incubated with a 1 ml suspension of the protein A beads coupled to the PNPase-specific antibody. The column was washed extensively until the OD280 of the flow-through (FT) dropped to zero. The column FT was recovered, dialyzed against Buffer E containing 60 mM KCl and subsequently concentrated using Centricon tubes with a molecular weight cut-off (MWCO) of 3 kDa. The concentrated fractions were loaded onto a 10% SDS gel and afterwards transferred to nitrocellulose membranes. The membranes were decorated with a PNPase-specific antibody to test for the depletion efficiency and reacting proteins were visualized by ECL chemoluminescence.
Purification of the 28, 41 and 55 RNPs
The RNPs were purified from LPF by a combination of ion-exchange chromatography on MonoQ and affinity chromatography on single-stranded DNA (ssDNA). LPF was loaded onto the column at a flow rate of 1 ml/min, followed by extensive washing with six to eight column volumes of Buffer E with 60 mM KCl (see above). Washing of the bound protein fraction was considered complete when the OD280 of the FT was close to zero. The bound proteins were eluted with 20 ml of a linear salt gradient (60 mM to 1 M KCl) and collected in 20 1 ml fractions, which were dialyzed against Buffer E for 10 h. Each fraction was then analyzed for protein content and RNA-binding activity by crosslinking to the petD RNA 3′-end probe. The RNP-containing fractions were pooled (250– 750 mM KCl) and dialyzed against Buffer E containing 5 mM KCl. The protein sample was subsequently applied to a 2 ml column of ssDNA [1 cm column diameter, 3.5 mg ssDNA/g cellulose (Sigma)] equilibrated with Buffer E containing 5 mM KCl at a flow rate of 0.5 ml/min (peristaltic pump, 10 column volumes). The column was washed with approximately 10 column volumes of the equilibration buffer (Buffer E). Bound proteins were eluted with a linear salt gradient from 5 mM to 1 M KCl, and collected in 20 1 ml fractions that were subsequently dialyzed against Buffer E (containing 60 mM KCl).
RESULTS
The chloroplast DPF facilitates degradation of the petD 3′-UTR mRNA precursor substrate
Soluble chloroplast protein fractions were obtained from plants grown in 8/16 h light–dark conditions (LPF) or grown in the dark for 48 h (DPF). LPF and DPF were analyzed for RNA-processing activity using the petD 3′-UTR mRNA precursor as a gene-specific RNA substrate (26). As expected, LPF processed the petD precursor mRNA to a mature form, with the 3′ end terminating downstream of the stem–loop structure (Fig. 1) (26). This RNA product is stable in LPF for at least 90 min (data not shown). In DPF, however, the petD mRNA precursor substrate was rapidly degraded (Fig. 1), and only traces of the RNA could be detected after 10 min. This suggests that LPF and DPF contain proteins that can reproduce correct mRNA processing and dark-induced degradation of mRNAs. These properties of LPF and DPF allowed us to identify and characterize the proteins and enzymatic activities that are involved in processing and differential stabilization of plastid mRNAs.
Figure 1.

Dark-induced degradation of a petD 3′-UTR precursor probe. (A) Schematic depiction of the experimental set-up. Seven-week-old spinach plants were transferred to darkness for 48 h. For the same time, control plants were left in a growth chamber with a dark–light cycle. Plants were harvested under green light and the processing extracts were isolated in parallel as described (24,25). (B) In vitro processing assay of light and dark extracts. Five micrograms of the dark and the light extract were incubated for 60 min at room temperature with the petD 3′-UTR probe. After 60 min the reaction was stopped and the remaining RNA precipitated with ethanol and loaded onto a denaturing TBE gel. The gel was subsequently dried and RNA molecules analyzed by autoradiography. Lower panel, Kinetic of dark-induced RNA degradation. The petD probe was incubated with the light and the dark extract as described above. After the given time points the reaction was stopped and RNA products analyzed by subsequent denaturing TBE gel electrophoresis and autoradiography. NP, no protein control; LPF, light protein fraction; DPF, dark protein fraction.
The RNA-binding activity of the 100 RNP is different in LPF and DPF
RNPs of 24, 28 and 100 kDa were previously reported to participate in processing and degradation of chloroplast mRNAs (2,3,20,21). Using specific antibodies against the 24, 28 and 100 RNPs (reviewed in 3) we showed that these RNPs are present in LPF and DPF in equal amounts (Fig. 2A). We noticed, however, that the 100 RNP in DPF consistently showed a clear shift in electrophoretic mobility, suggesting that the protein might potentially be light-regulated by post-translational modification. All attempts to induce this electrophoretic mobility shift by phosphorylation/dephosphorylation with a heterologous kinase (catalytic subunit of PKA from bovine heart) and phosphatases (calf intestinal alkaline phosphatase and shrimp alkaline phosphatase), however, were not successful (data not shown).
Figure 2.

Analysis of the RNA-binding activities from the light and the dark extract. (A) Antibody detection of the 100, 28 and 24 RNPs. Five micrograms of protein from the dark and light extract were loaded onto an SDS gel (6% acrylamide for the 100 RNP and 10% acrylamide for the 28 and 24 RNPs). The resolved proteins were subsequently transferred to a nitrocellulose membrane and the membranes decorated with the designated antibody. Detection of reacting proteins was carried out using ECL chemoluminescence. (B) UV crosslinking of the RNPs to the petD probe. Three micrograms of protein from the dark and the light extract were incubated with 50 000 c.p.m. of the petD 3′-UTR probe for 7 min on ice. Where indicated the ribohomopolymeres polyA or polyC were added to the incubation mixture (–, 2, 5, 50 ng). The mixture was then transferred to an UV crosslinker (Stratagene) and irradiated twice with 0.12 J. RNA was subsequently degraded by RNaseA and proteins resolved by SDS–PAGE. Proteins bound to RNA (labeled UTP) were made visible by autoradiography of the dried SDS gel.
The RNPs in both LPF and DPF were crosslinked to the petD 3′-UTR precursor substrate and analyzed by SDS–PAGE and autoradiography to determine their RNA-binding properties (25). Using this approach we detected a significant difference in the RNA-binding properties of the 100 RNP, which showed an enhanced binding in DPF (Fig. 2B). To confirm that the 100 RNP represents polynucleotide phosphorylase (PNPase), we performed competition experiments with the ribohomopolymeres polyA and polyC. Binding of PNPase to the petD precursor probe can be efficiently competed by polyA and to a weaker extent by polyC, whereas binding of the 28 and 24 RNPs is not affected by these competitors (8,28). In both extracts, the 100 RNP signal is completely abolished in the presence of 50 ng of polyA or polyC. As shown in Figure 2B, no significant competition could be detected with 5 ng of either competitor. Similarly, no competition was observed for the RNPs in the 24–33 kDa range. These results, together with our previous observation that no other 100 kDa RNP except PNPase is present in the chloroplast extract (9), confirm that the 100 RNP has the properties previously described for PNPase (8). Since PNPase is the only exonuclease that has been identified in chloroplasts to date, it seems reasonable that the light-dependent effect on the RNA-binding properties of this enzyme directly correlated with the increased DPF RNA degradation activity.
PNPase is responsible for dark-enhanced polyadenylation activity but is not the key nuclease in dark-induced mRNA degradation
Previous reports indicated that polyadenylation facilitates degradation of plastid mRNAs and that this activity is enhanced in dark-adapted plastids (6,7). PNPase has also been implicated in chloroplast RNA polyadenylation (29,30). These observations prompted us to investigate the polyadenylation activity in LPF and DPF. Following addition of 2 mM ATP and 20 U RNasin to the RNA-processing assay, we terminated the reaction after 3, 7, 18 and 40 min. Under these conditions, a significantly higher polyadenylation activity was detectable in DPF (Fig. 3). This result is consistent with the enhanced RNA-binding activity of PNPase in DPF shown in Figure 2. As a control, we added cordycepin, an efficient polyadenylation inhibitor, to LPF and DPF. Figure 3 shows that polyadenylation activity in DPF was significantly reduced and only a small amount of polyadenylated product was detectable after 40 min.
Figure 3.
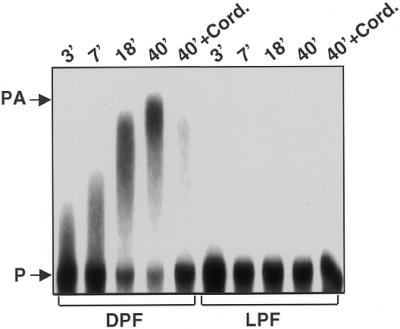
Elevated polyadenylation activity of the dark extract. Polyadenylation was assayed under the conditions described for the in vitro processing assays except that 2 mM ATP and 20 U RNasin were added to inactivate the endogenous nucleases. After the indicated times the reaction was stopped and the RNA products analyzed by electrophoresis and autoradiography. Control assays were incubated for 40 min in the presence of 4 mM cordycepin (Sigma; lanes 40′ + Cord.).
We subsequently immunodepleted PNPase from LPF and DPF using a PNPase-specific antibody column (Pierce) to confirm that the observed increased polyadenylation in DPF resulted from PNPase activity. The column FT was recovered and tested for PNPase protein, polyadenylation and RNA degradation activity. Figure 4A shows that essentially no PNPase was detectable in the FT fraction, indicating that the immunodepletion had been successful. Both PNPase-immunodepleted LPF and DPF had completely lost the polyadenylation activity (Fig. 4B). This observation is consistent with a previous report that PNPase can also function as a polyadenylating enzyme in chloroplasts (30). Furthermore, the results suggest that increased RNA binding of PNPase shown in Figure 2 is responsible for the elevated polyadenylation activity in DPF.
Figure 4.

Polyadenylation and RNA degradation activities of PNPase- immunodepleted processing extracts. The light (LPF) and the dark (DPF) processing extract were incubated in batch at 4°C overnight with a PNPase-specific antibody coupled to protein A beads (Pierce). (A) Immunodetection of remaining PNPase in the flow-through (FT) from the antibody column of the dark (FTDPF) and the light extract (FTLPF). (B) Polyadenylation activity of the immunodepleted light (FT-LPF) and dark extracts (FT-DPF) was performed as described. The polyadenylation reaction was stopped after 45 min incubation. Complete processing extract from dark-adapted plastids (DPF) was analyzed in parallel as a control. (C) RNA degradation activity of the dark (FT-DPF) and light (FT-LPF) immunodepleted extracts was assayed as described and the reactions were stopped after 45 min. RNA products were subsequently analyzed by electrophoresis and autoradiography. DPF, dark protein fraction; LPF, light protein fraction; FT-DPF, flow-through obtained with the dark protein fraction; FT-LPF, flow-through obtained with the light protein fraction; PA, polyadenylated product; P, precursor.
To clarify whether elevated PNPase-mediated polyadenylation of the RNA substrate is a prerequisite for the observed DPF-dependent degradation of the petD 3′-UTR mRNA substrate, we analyzed RNA degradation activity in immunodepleted DPF and LPF using the RNA-processing assay. Figure 4C shows that after 45 min incubation, DPF had completely degraded the petD 3′-UTR mRNA substrate, while the LPF did not process the mRNA precursor to any significant extent. The mock immunodepletions with pre-immune serum substantiate these observations. The mock-depleted LPF produced a correctly processed RNA, but the mock-depleted DPF completely degraded the RNA precursor (data not shown). These results confirm that PNPase is important for chloroplast RNA processing in the light, but also suggest that PNPase is not a major prerequisite for dark-induced mRNA degradation.
An endonuclease is activated in chloroplasts of dark-adapted plants
The above results suggest that ribonucleases other than PNPase direct the rapid degradation of chloroplast mRNA in the dark. To test this possibility, we inhibited exonuclease(s) in LPF and DPF by the addition of tRNA. Excess tRNA binds exonucleases, but prevents efficient degradation of the highly structured regions in the tRNA molecule, resulting in unproductive interactions (31). LPF and DPF were incubated with a uniformly labeled petD 3′-UTR mRNA substrate in the presence of 5 µg of tRNA, and the reaction products were analyzed after 0, 10, 20 and 50 min. No significant RNA cleavage products were found with LPF, except for a high-molecular-weight product that was 10–15 nt shorter than the 3′-UTR mRNA substrate (Fig. 5). In contrast, DPF produced a distinctive pattern of short RNA cleavage products over a broad molecular weight range (Fig. 5), indicating the activity of a specific endonuclease or the combined activity of several endonucleases. To determine where the endonuclease(s) in the dark extract cleaves the petD precursor, the petD 3′-UTR mRNA substrate was labeled with [γ-32P]ATP and polynucleotide kinase. As a control, the 5′-labeled 3′-UTR mRNA substrate was also incubated with LPF. No difference was observed in RNA-processing activity with LPF using either the 5′-labeled substrate or the uniformly labeled substrate (compare Figs 5 and 1B). When the 5′-labeled probe was incubated with DPF in the presence of tRNA, two major cleavage products were detected, both terminating upstream of the predicted stem–loop structure (Fig. 5). These results confirm that DPF contains an active endonuclease, which initiates mRNA degradation by cleavage of the precursor RNA substrate proximal to the stem–loop structure.
Figure 5.

Dark-induced activation of endonucleolytic cleavages upstream of the stem–loop structure. In vitro processing analysis with the dark and the light extracts in the presence of 5 µg of tRNA. Five micrograms of the light- and dark-processing extract proteins were incubated with a uniformly labeled or 5′-labeled petD 3′-UTR probe in the presence of 5 µg of tRNA. After the given times (bottom) the reaction was stopped and RNA products analyzed by denaturing TBE gel electrophoresis and subsequent autoradiography. The 5′-labeled probe was incubated with 5 µg of the light-extract proteins in the absence of tRNA to control the processing activity of this extract on 5′-monophosphate-carrying RNAs. To check for the endonucleolytic cleavage sites used by the dark extract the 5′-labeled probe was incubated with 5 µg of the dark extract in the presence of tRNA. RNA products were analyzed as described above.
A protein factor from LPF protects mRNA degradation in DPF
It is possible that the endonuclease present in DPF is expressed only after transfer of plants to the dark. Alternatively, the endonuclease may also be present in the light, but its activity is regulated to increase the half-life of mRNAs for photosynthetic proteins. To distinguish between these possibilities, we tested in reconstitution experiments if DPF was able to degrade the petD 3′-UTR mRNA substrate in the presence of LPF proteins. As control assays, we incubated 2 µg of LPF and DPF separately with the petD 3′-UTR mRNA substrate for 1 h using conditions described previously for the RNA-processing assays (25). As expected, the light extract processes the RNA precursor substrate to the stable mature RNA product and DPF completely degrades the 3′-UTR mRNA substrate. After mixing 2 µg of LPF and 2 µg of DPF protein, while maintaining the same ratio of nuclease to RNA as in the control assays, the 3′-UTR mRNA substrate was again processed into a mature, stable mRNA product (Fig. 6A). This result suggests that LPF contains one or more protein factors, which can inactivate the endonucleolytic activity and prevent cleavages that initiate RNA degradation in DPF.
Figure 6.

(A) A protein factor from the light extract inhibits RNA degradation by the dark extract. Two micrograms of the dark and the light extract were tested for their RNA degradation activity either separately or as a mixture. The extracts were pre-incubated for 5 min at room temperature prior to the addition of the RNA probe. The extracts were then incubated with the petD precursor probe for 45 min under in vitro processing assay conditions. The RNA products were subsequently analyzed by TBE gel electrophoresis and autoradiography. (B) The 28, 55 and 41 RNPs were partially purified from the light extract by ion-exchange chromatography on MonoQ (41 RNP), or a combination of ion-exchange and ssDNA chromatography (55 and 28 RNPs). From this column the RNPs were eluted with a linear KCl gradient from 5 mM to 1 M KCl, resulting in the separation of the RNPs. The RNP-containing fractions were concentrated using Centricon tubes (MWCO 3 kDa) and dialyzed against Buffer E (60 mM KCl). The fractions were tested for the presence of the RNPs by UV-crosslinking analysis as described. (C) Two micrograms of the dark extract were pre-incubated with the RNPs or with Buffer E for 10 min at room temperature before the mixture was incubated with the petD 3′-UTR probe. After 45 min, the reaction was stopped and the RNA products analyzed by TBE gel electrophoresis and subsequent autoradiography.
It is possible that one or more of the previously identified 28 RNP, 41 RNP and 55 RNP RNA-binding activities are responsible for the inactivation of the endonucleolytic activity in LPF. Therefore, we fractionated the LPF proteins to enrich the 28, 55 and 41 kDa RNPs (Fig. 6B) using a combination of ion-exchange (41 RNP) and affinity chromatography on ssDNA (28 and 55 RNPs). The protein fractions with the enriched RNPs were subsequently added to DPF and assayed under processing conditions for the 3′-UTR mRNA substrate. Figure 6C shows that protein fractions enriched for the 28 RNP or 55 RNP had no significant effect on the RNA degradation activity in DPF. In contrast, the protein fraction enriched for the 41 RNP effectively inhibited endonucleolytic RNA degradation in DPF. Therefore, it is possible that the 41 RNP or other proteins in this fraction are responsible for inactivating the endonucleolytic activity in plants exposed to light.
DISCUSSION
Post-transcriptional control of RNA processing and degradation are important for the regulation of gene expression. For higher plant chloroplasts, it was suggested that control of gene expression in response to changes of environmental conditions occurs primarily at the post-transcriptional level. Post-transcriptional control has been documented for several mRNAs encoding photosynthetic proteins (3,12,32–37). These mRNAs have a decreased half-life in plants that are grown in the dark and in which photosynthetic proteins are only synthesized at maintenance levels (3,23,38,39). In this report we demonstrate, using the petD mRNA as a model transcript for photosynthetic proteins, that stabilization or accelerated degradation of plastid mRNAs can be reproduced in vitro using partially fractionated extracts isolated from light- and dark-grown plants.
Analysis of LPF and DPF revealed two different pathways of chloroplast RNA metabolism: processing and stabilization of the mRNA in light-grown plants, and rapid and complete degradation of the mRNA after plants were transferred to the dark. The degradation pathway is initiated by endonucleolytic cleavages upstream of the petD 3′-UTR stem–loop structure. The observed degradation of the mRNA in DPF is reminiscent of the situation in E.coli, where RNA degradation is initiated by RNaseE cleavage, followed by exonucleolytic degradation (reviewed in 4). At present no RNaseE has been isolated from chloroplasts, suggesting that a different type of endonuclease is likely to be responsible for the initiation of mRNA degradation in chloroplasts. We previously reported a 67 kDa protein that specifically cross-reacted with antibodies against E.coli RNaseE (21) but this protein was subsequently identified as GroEL, which shows immunoreactivity with the RNaseE antibody (9). Interestingly, a partially purified fraction containing this protein cleaves the petD precursor at two positions upstream of the predicted stem–loop structure, similar to the cleavages observed with DPF (Fig. 5) (6). It is not known whether GroEL itself has endonucleolytic activity or whether another protein in this fraction catalyzes the observed cleavages (6,21). Enzymatic activities of two endonucleases, CSP41 from spinach and p54 from mustard, have been reported previously. These enzymes appear to be regulated by phosphorylation and redox control, thus making them good candidates for signal-dependent regulation of chloroplast mRNA stability and degradation (10,11,13). It seems reasonable that an endonuclease, which initiates mRNA degradation in chloroplasts, can be regulated by control mechanisms that are linked to the photosynthetic activity of the organelle. However, direct proof that these or another endonuclease facilitates the dark-induced degradation of mRNAs in chloroplasts has not been available. Further experiments are now required to purify the dark-activated endonuclease.
Several mechanisms can be considered to explain the difference between processing/stability and degradation of the petD mRNA precursor in LPF and DPF. First, the DPF endonuclease that facilitates mRNA degradation could be expressed or activated only after plants were transferred to darkness. Secondly, the DPF endonuclease is constitutive, but is inactivated in the light by phosphorylation or interaction with another protein, a modification that is preserved in LPF. Finally, the endonucleolytic cleavage sites in the petD mRNA substrate could be inaccessible in DPF, perhaps by binding of one or more specific regulatory proteins that are expressed or activated in the light. Our results suggest that a 41 kDa RNP in LPF could regulate access of the endonuclease to the mRNA substrate by masking the endonucleolytic cleavage sites. A similar mechanism has been proposed for the barley psbA mRNA, which appears to interact with a 37 and a 38 kDa protein specifically in the 3′-UTR upstream of the predicted stem–loop structure. In this case the authors concluded that these RNA-binding proteins increase the stability of psbA mRNA in the light (40).
The extent to which endonucleolytic cleavages in the coding region, followed by polyadenyation and exonucleolytic degradation, contribute to the chloroplast mRNA degradation pathway remains an unresolved problem. It is also unclear which of these enzymatic activities represent the limiting factor in the RNA degradation process (12). In earlier work and the results presented here we could demonstrate that chloroplast polyadenylation activity resides in PNPase, which is also the only exonuclease identified in chloroplasts to date (Fig. 4B) (30). Immunodepletion of PNPase from DPF or tRNA-inactivation of its nucleolytic activity does not abolish RNA degradation activity in DPF (Figs 4 and 5), suggesting that polyadenylation and exonucleolytic degradation by PNPase are not absolute prerequisites for dark-induced mRNA degradation (Fig. 4). Endonucleolytic cleavage of the petD mRNA substrate upstream of the 3′-UTR stem–loop structure, however, initiate and facilitate mRNA degradation in DPF, whereas this process can be blocked by a protein present in LPF (Fig. 6). Once the endonucleolytic cleavage has occurred, RNA degradation can proceed even in the absence of PNPase activity (Fig. 4C). These results strongly suggest that an alternative exonuclease, or a non-specific endonuclease, is a component of the chloroplast mRNA degradation pathway. Further studies will reveal the identity of the key players in this regulatory system, consisting of an endonuclease, an RNP and probably an exonuclease distinct from PNPase.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank Drs Johannes Fütterer and Gadi Schuster for critical reading of the manuscript and for helpful and encouraging comments. S.B. was supported by a DFG Postdoctoral Fellowship (Ba 1902/1-1). The research was supported by the Swiss Federal Institute of Technology.
REFERENCES
- 1.Barkan A. and Stern,D.B. (1998) Chloroploast mRNA processing: intron splicing and 3′-end metabolism. In Bailey-Serresa,J. and Gallie,D.R. (eds), A Look Beyond Transcription: Mechanisms Determining mRNA Stability and Translation in Plants. American Society of Plant Physiologists, Rockville, MD, pp. 162–173.
- 2.Schuster G., Lisitsky,I. and Klaff,P. (1999) Polyadenylation and degradation of mRNA in the chloroplast. Plant Physiol., 120, 937–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayes R., Kudla,J. and Gruissem,W. (1999) Degrading chloroplast mRNA: the role of polyadenylation. Trends Biochem. Sci., 24, 199–202. [DOI] [PubMed] [Google Scholar]
- 4.Carpousis A.J., Vanzo,N.F. and Raynal,L.C. (1999) mRNA degradation. A tale of poly(A) and multiprotein machines. Trends Genet., 15, 24–28. [DOI] [PubMed] [Google Scholar]
- 5.Steege D.A. (2000) Emerging features of mRNA decay in bacteria. RNA, 6, 1079–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kudla J., Hayes,R. and Gruissem,W. (1996) Polyadenylation accelerates degradation of chloroplast mRNA. EMBO J., 15, 7137–7146. [PMC free article] [PubMed] [Google Scholar]
- 7.Lisitsky I., Klaff,P. and Schuster,G. (1996) Addition of destabilizing poly (A)-rich sequences to endonuclease cleavage sites during the degradation of chloroplast mRNA. Proc. Natl Acad. Sci. USA, 93, 13398–13403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lisitsky I., Kotler,A. and Schuster,G. (1997) The mechanism of preferential degradation of polyadenylated RNA in the chloroplast. The exoribonuclease 100RNP/polynucleotide phosphorylase displays high binding affinity for poly(A) sequence. J. Biol. Chem., 272, 17648–17653. [DOI] [PubMed] [Google Scholar]
- 9.Baginsky S., Shteiman-Kotler,A., Liveanu,V., Yehudai-Resheff,S., Bellaoui,M., Settlage,R.E., Shabanowitz,J., Hunt,D.F., Schuster,G. and Gruissem,W. (2001) Chloroplast PNPase exists as a homo-multimer enzyme complex that is distinct from the E.coli degradosome. RNA, 7, 1464–1475. [PMC free article] [PubMed] [Google Scholar]
- 10.Nickelsen J. and Link,G. (1993) The 54 kDa RNA-binding protein from mustard chloroplasts mediates endonucleolytic transcript 3′ end formation in vitro. Plant J., 3, 537–544. [DOI] [PubMed] [Google Scholar]
- 11.Yang J., Schuster,G. and Stern,D.B. (1996) CSP41, a sequence-specific chloroplast mRNA binding protein, is an endoribonuclease. Plant Cell, 8, 1409–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monde R.A., Schuster,G. and Stern,D.B. (2000) Processing and degradation of chloroplast mRNA. Biochimie, 82, 573–582. [DOI] [PubMed] [Google Scholar]
- 13.Liere K. and Link,G. (1997) Chloroplast endoribonuclease p54 involved in RNA 3′-end processing is regulated by phosphorylation and redox state. Nucleic Acids Res., 25, 2403–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y. and Sugiura,M. (1990) Three distinct ribonucleoproteins from tobacco chloroplasts: each contains a unique amino terminal acidic domain and two ribonucleoprotein consensus motifs. EMBO J., 9, 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye L., Li,Y., Fukami-Kobayashi,K., Go,M., Konishi,T., Watanabe,A. and Sugiura,M. (1991) Diversity of a ribonucleoprotein family in tobacco chloroplasts: two new chloroplast ribonucleoproteins and a phylogenetic tree of ten chloroplast RNA-binding domains. Nucleic Acids Res., 19, 6485–6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohta M., Sugita,M. and Sugiura,M. (1995) Three types of nuclear genes encoding chloroplast RNA-binding proteins (cp29, cp31 and cp33) are present in Arabidopsis thaliana: presence of cp31 in chloroplasts and its homologue in nuclei/cytoplasms. Plant Mol. Biol., 27, 529–539. [DOI] [PubMed] [Google Scholar]
- 17.Cook W.B. and Walker,J.C. (1992) Identification of a maize nucleic acid-binding protein (NBP) belonging to a family of nuclear-encoded chloroplast proteins. Nucleic Acids Res., 20, 359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Churin Y., Hess,W.R. and Borner,T. (1999) Cloning and characterization of three cDNAs encoding chloroplast RNA-binding proteins from barley (Hordeum vulgare L.): differential regulation of expression by light and plastid development. Curr. Genet., 36, 173–181. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura T., Ohta,M., Sugiura,M. and Sugita,M. (2001) Chloroplast ribonucleoproteins function as a stabilizing factor of ribosome-free mRNAs in the stroma. J. Biol. Chem., 276, 147–152. [DOI] [PubMed] [Google Scholar]
- 20.Schuster G. and Gruissem,W. (1991) Chloroplast mRNA 3′ end processing requires a nuclear-encoded RNA-binding protein. EMBO J., 10, 1493–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayes R., Kudla,J., Schuster,G., Gabay,L., Maliga,P. and Gruissem,W. (1996) Chloroplast mRNA 3′-end processing by a high molecular weight protein complex is regulated by nuclear encoded RNA binding proteins. EMBO J., 15, 1132–1141. [PMC free article] [PubMed] [Google Scholar]
- 22.Lisitsky I. and Schuster,G. (1995) Phosphorylation of a chloroplast RNA-binding protein changes its affinity to RNA. Nucleic Acids Res., 23, 12506–12511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klaff P. and Gruissem,W. (1991) Changes in chloroplast mRNA stability during leaf development. Plant Cell, 3, 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gruissem W., Greenberg,B.M., Zurawski,G. and Hallick,R.B. (1986) Chloroplast gene expression and promoter identification in chloroplast extracts. Methods Enzymol., 118, 253–270. [DOI] [PubMed] [Google Scholar]
- 25.Baginsky S. and Gruissem,W. (2001) The chloroplast mRNA 3′-end nuclease complex. Methods Enzymol., 342, 408–419. [DOI] [PubMed] [Google Scholar]
- 26.Stern D.B. and Gruissem,W. (1987) Control of plastid gene expression: 3′ inverted repeats act as mRNA processing and stabilizing elements, but do not terminate transcription. Cell, 51, 1145–1157. [DOI] [PubMed] [Google Scholar]
- 27.Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 28.Lisitsky I. and Schuster,G. (1999) Preferential degradation of polyadenylated and polyuridinylated RNAs by the bacterial exoribonuclease polynucleotide phosphorylase. Eur. J. Biochem., 261, 468–474. [DOI] [PubMed] [Google Scholar]
- 29.Li Q.S., Gupta,J.D. and Hunt,A.G. (1998) Polynucleotide phosphorylase is a component of a novel plant poly(A) polymerase. J. Biol. Chem., 273, 17539–17543. [DOI] [PubMed] [Google Scholar]
- 30.Yehudai-Resheff S., Hirsh,M. and Schuster,G. (2001) Polynucleotide phosphorylase (PNPase) functions both as an exonuclease and a poly(A) polymerase in spinach chloroplast. Mol. Cell. Biol., 21, 5408–5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klaff P. (1995) mRNA decay in spinach chloroplasts: psbA mRNA degradation is initiated by endonucleolytic cleavages within the coding region. Nucleic Acids Res., 23, 4885–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gruissem W. and Tonkyn,J.T. (1993) Control mechanisms of plastid gene expression. Crit. Rev. Plant Sci., 12, 19–55. [Google Scholar]
- 33.Sugita M. and Sugiura,M. (1996) Regulation of gene expression in chloroplasts of higher plants. Plant Mol. Biol., 32, 315–326. [DOI] [PubMed] [Google Scholar]
- 34.Stern D.B., Higgs,D.C. and Yang,J. (1997) Transcription and translation in chloroplasts. Trends Plant Sci., 2, 308–315. [Google Scholar]
- 35.Maliga P. (1998) Two plastid RNA polymerases of higher plants: an evolving story. Trends Plant Sci., 3, 4–6. [Google Scholar]
- 36.Zerges W. (2000) Translation in chloroplasts. Biochimie, 82, 583–601. [DOI] [PubMed] [Google Scholar]
- 37.Allison L.A. (2000) The role of sigma factors in plastid transcription. Biochimie, 82, 537–548. [DOI] [PubMed] [Google Scholar]
- 38.Deng X.-W., Tonkyn,J.C., Peter,G.F., Thornber,J.P. and Gruissem,W. (1989) Post-transcriptional control of plastid mRNA accumulation during adaptation of chloroplasts to different light quality environments. Plant Cell, 1, 645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mullet J.E. (1993) Dynamic regulation of chloroplast transcription. Plant Physiol., 103, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Memon A.R., Meng,B. and Mullet,J.E. (1996) RNA-binding proteins of 37/38 kDa bind specifically to the barley chloroplast psbA 3′-end untranslated RNA. Plant Mol. Biol., 30, 1195–1205. [DOI] [PubMed] [Google Scholar]