


Abstract
Previous studies have shown that human dihydrofolate reductase (DHFR) acts as an RNA-binding protein, in which it binds to its own mRNA and, in so doing, results in translational repression. In this study, we used RNA gel mobility shift and nitrocellulose filter-binding assays to further investigate the specificity of the interaction between human DHFR protein and human DHFR mRNA. Site-directed mutagenesis was used to identify the critical amino acid residues on DHFR protein required for RNA recognition. Human His-Tag DHFR protein specifically binds to human DHFR mRNA, while unrelated proteins including thymidylate synthase, p53 and glutathione-S-transferase were unable to form a ribonucleoprotein complex with DHFR mRNA. The Cys6 residue is essential for RNA recognition, as mutation at this amino acid with either an alanine (C6A) or serine (C6S) residue almost completely abrogated RNA-binding activity. Neither one of the cysteine mutant proteins was able to repress the in vitro translation of human DHFR mRNA. Mutations at amino acids Ile7, Arg28 and Phe34, significantly reduced RNA-binding activity. An RNA footprinting analysis identified three different RNA sequences, bound to DHFR protein, ranging in size from 16 to 45 nt, while a UV cross-linking analysis isolated an ∼16 nt RNA sequence bound to DHFR. These studies begin to identify the critical amino acid residues on human DHFR that mediate RNA binding either through forming direct contact points with RNA or through maintaining the protein in an optimal structure that allows for the critical RNA-binding domain to be accessible.
INTRODUCTION
The enzyme dihydrofolate reductase (DHFR) (EC 1.5.1.3) catalyzes the NADPH-dependent reduction of dihydrofolate to tetrahydrofolate. This reaction provides the key intermediate in one-carbon transfer reactions. DHFR plays a critical role in folate homeostasis, and it is required for the de novo synthesis of purines, thymidylate and certain amino acids (1,2). For this reason, DHFR has served as a critical target in cancer chemotherapy for well over 40 years.
The intracellular levels of DHFR are regulated by a number of factors including the concentration of serum growth factors, changes in cyclic AMP levels, viral infection and exposure to cytotoxic stresses such as methotrexate (MTX) and the fluoropyrimidines (3–8). Several in vitro and in vivo experimental model systems have shown that exposure of malignant cells to MTX is accompanied by acute increases in DHFR enzyme activity and DHFR protein level (9–13). Recent work from our laboratory observed a significant time- and dose-dependent induction of DHFR protein levels in response to exposure to MTX in the human colon cancer RKO cell line (14). Levels of DHFR were maximally induced by 10–12-fold after treatment with 10 nM MTX for 24 h, while levels of DHFR mRNA remained unchanged in control and MTX-treated cells. These findings suggested that the expression of DHFR in response to cytotoxic agents is, in part, controlled by a translational regulatory event.
Using a rabbit reticulocyte lysate in vitro translation system, our laboratory showed that the addition of pure human recombinant DHFR protein specifically repressed the translation of human DHFR mRNA in a dose-dependent manner (15). An RNA electrophoretic gel shift assay system confirmed that human DHFR protein interacted with its target DHFR mRNA in a direct and specific manner (15,16). Based on these studies, a model of DHFR translational autoregulation was proposed. This regulatory mechanism would appear to have biological relevance in that it provides an efficient mechanism for the levels of DHFR to be tightly controlled within a given cell. Disruption of this normal regulatory process may provide an efficient mechanism for malignant cells to protect themselves following exposure to cytotoxic stress and, in so doing, may lead to the rapid development of cellular drug resistance.
In the present study, we investigated the molecular elements underlying the interaction between human DHFR protein and its target DHFR mRNA. Specifically, our studies focused on characterizing the critical amino acid residues on the protein that are required for RNA recognition. We show that the cysteine positioned at amino acid residue 6 as well as Ile7, Arg28 and Phe34 are critical for RNA binding.
MATERIALS AND METHODS
Synthesis of plasmid constructs
The full-length, 700-nt, human DHFR cDNA (Fig. 1) was isolated from human breast cancer MCF-7 cells and cloned into pGEM-7Z (Promega, Madison, WI) as previously described by Chu et al. (15) to give the recombinant plasmid pGEM-7Z:DHFR. The coding region of human DHFR cDNA (564 nt) was amplified from pGEM-7Z:DHFR by PCR amplification using the sense primer: 5′-ATCGCATATGATGGTTGGTTCGCTAAACTGC-3′ and the antisense primer: 5′-ATCGGGATTCTTAATCATTCTTCTCATA-3′. The underlined bases represent the NdeI and BamHI restriction sites, respectively. The PCR product was analyzed by non-denaturing gel electrophoresis, and the DNA corresponding to the protein-coding region of the DHFR cDNA (nucleotides 1–564) was digested with NdeI and BamHI and cloned into the NdeI and BamHI restriction sites of pET28a(+) (Novagen, Madison, WI) to give pET28a(+):DHFR. The sequence was confirmed by sequencing analysis as performed by the Keck Facility of the Yale University School of Medicine.
Figure 1.

Map of the human DHFR cDNA. The 700-nt DHFR cDNA includes 59 nt of the 5′-UTR, the coding region (564 nt) and 77 nt of the 3′-UTR.
Point mutations were created using the QuikChange™ Site-Directed Mutagenesis Kit (Clontech, Palo Alto, CA). In brief, each reaction (total volume, 50 µl) contained 30 ng of plasmid pET28a(+):DHFR, 125 ng of sense oligonucleotide mutant primer and 125 ng of antisense oligonucleotide mutant primer 1 µl of dNTP mix and 2.5 U Pfu Turbo DNA polymerase. Each reaction was overlaid with 30 µl of mineral oil. PCRs were performed as follows: initial incubation at 95°C for 30 s, denaturation at 95°C for 30 s, annealing at 55°C for 1 min and extension at 68°C for 12 min for a total of 16 cycles. Following PCR cycling, reactions were cooled to room temperature. The parent plasmid was removed by digestion with 1 µl of the DpnI restriction enzyme (10 U/µl) at 37°C for 30 min. The mutant plasmid was then transformed into Epicurian Coli XL1-Blue supercompetent cells. To verify the presence of the respective point mutations, each construct was sequenced by the Keck Facility, Yale University School of Medicine. All primers used for mutagenesis are listed as follows (the bold letters represent the mutant sequences), and the resulting mutant proteins are listed in Table 1: C6A, sense primer, 5′-ATGGTTGGTTCGCTAAACGCTATCGTCGCTGTGTCC-3′; C6A, antisense primer, 5′-GGACACAGCGACGATAGCGTTTAGCGAACCAACCAT-3′; C6S, sense primer, 5′-ATGGTTGGTTCGCTAAACTCTATCGTCGCTGTGTCC-3′; C6S, antisense primer, 5′-GGACACAGCGACGATAGAGTTTAGCGAACCAACCAT-3′; I7A, sense primer, 5′-GGTTCGCTAAACTGCGCTGTCGCTGTGTCCCAGAAC-3′; I7A, antisense primer, 5′-GTTCTGGGACACAGCGACAGCGCAGTTTAGCGAACC-3′; I7F, sense primer, 5′-GGTTCGCTAAACTGCTTTGTCGCTGTGTCCCAGAAC-3′; I7F, antisense primer, 5′-GTTCTGGGACACAGCGACAAAGCAGTTTAGCGAACC-3′; L22F, sense primer, 5′-GGCAAGAACGGGGACTTTCCCTGGCCACCGCTC-3′; L22F, antisense primer, 5′-GAGCGGTGGCCAGGGAAAGTCCCCGTTCTTGCC-3′; L22R, sense primer, 5′-GGCAAGAACGGGGACAGACCCTGGCCACCGCTC-3′; L22R, antisense primer, 5′-GAGCGGTGGCCAGGGTCTGTCCCCGTTCTTGCC-3′; R28A, sense primer, 5′-CCCTGGCCACCGCTCGCTAATGAATTCAGATATTTC-3′; R28A, antisense primer, 5′-GAAATATCTGAATTCATTAGCGAGCGGTGGCCAGGG-3′; E30A, sense primer, 5′-CCACCGCTCAGGAATGCTTTCAGATATTTCCAGAG-3′; E30A, antisense primer, 5′-CTCTGGAAATATCTGAAAGCATTCCTGAGCGGTGG-3′; E30Q, sense primer, 5′-CCACCGCTCAGGAATCAGTTCAGATATTTCCAGAG-3′; E30Q, antisense primer, 5′-CTCTGGAAATATCTGAACTGATTCCTGAGCGGTGG-3′; F31A, sense primer, 5′-CCGCTCAGGAATGAAGCTAGATATTTCCAGAGATG-3′; F31A, antisense primer, 5′-CATTCTCTGGAAATATCTAGCTTCATTCCTGAGCGG-3′; F31G, sense primer, 5′-CCGCTCAGGAATGAAGGTAGATATTTCCAGAGATG-3′; F31G, antisense primer, 5′-CATTCTCTGGAAATATCTACCTTCATTCCTGAGCGG-3′; R32A, sense primer, 5′-CTCAGGAATGAATTCGCTTATTTCCAGAGAATG-3′; R32A, antisense primer, 5′-CATTCTCTGGAAATAAGCGAATTCATTCCTGAG-3′; F34A, sense primer, 5′-AATGAATTCAGATATGCTCAGAGAATGACCACA-3′; F34A, antisense primer, 5′-TGTGGTCATTCTCTGAGCATATCTGAATTCATT-3′; F34S, sense primer, 5′-AATGAATTCAGATATTCTCAGAGAATGACCACA-3′; F34S, antisense primer, 5′-TGTGGTCATTCTCTGAGAATATCTGAATTCATT-3′.
Table 1. Relative binding affinity of DHFR and control RNA transcripts.
| RNA |
IC50 (nM) |
| DHFR:1–700 | 3.16 ± 0.18 |
| DHFR:1–564 | 3.64 ± 0.23 |
| DHFR:5′-UTR | >200 |
| DHFR:3′-UTR | >200 |
| TS-30 | >200 |
| Yeast tRNA | >200 |
Binding of human recombinant His-Tag DHFR protein to full length, human DHFR mRNA and unrelated RNAs was determined by comparing the concentration of unlabeled RNA that inhibited specific binding of 32P-radiolabeled human DHFR mRNA coding region by 50%. The competition experiments were performed as described in the Materials and Methods. Each value represents the mean ± SD of at least three experiments.
Escherichia coli DHFR cDNA was amplified with two primers (sense primer, ATCG GGA TCC ATG ATC AGT CTG ATT GCG GCG; antisense primer, ATCG AAG CTT TTA CCG CCG CTC CAG AAT CTC; the underlined nucleotides represent the restriction enzyme sites of BamHI and HindIII, respectively) and cloned into pET28a(+) (digested with BamHI and HindIII) to give pET28a(+): DHFR(Ecoli). The sequence was confirmed by sequencing analysis as performed by the Keck Facility of the Yale University School of Medicine.
In vitro RNA transcription
A 32P-radiolabeled, human DHFR mRNA probe (DHFR: 1–564) was synthesized in vitro with T7 RNA polymerase (Promega) after linearization of pET28a(+):DHFR with HindIII. Unlabeled, full-length DHFR mRNA (DHFR: 1–700), which included 59 nt of the 5′-untranslated region (UTR), the entire coding region and 77 nt of the 3′-UTR, was synthesized in vitro with the MEGAscript SP6 kit (Ambion, Austin, TX) after linearization of pGEM-7Z:DHFR with BamHI. β-Actin antisense RNA (245 nt), GAPDH mRNA (316 nt) and 18S rRNA (80 nt) were synthesized in vitro using T7 polymerase and pTRI-actin-mouse (Ambion), pTRI-GAPDH-Human (Ambion) and pTRI-RNA-18S (Ambion) templates, respectively. TS30 RNA, which represents the 5′-upstream binding site on TS mRNA and whose sequence is 5′-CCG CCC GCC GCG CCA UGC CUG UGG CCG GCU-3′, was chemically synthesized by the Keck Facility, Yale University School of Medicine. Sequences corresponding to the 5′- (59 nt) and 3′-UTRs (77 nt) of human DHFR mRNA were synthesized in vitro using PCR-generated DNAs and the T7-MEGAshortscript™ kit (Ambion). The primers used in the PCR are listed as follows, and the underlined nucleotides represent the T7 promoter/primer sequence: 5′-UTR, sense primer, 5′-TAATACGACTCACTATAGGGCGCCAAACTTGACCGCGC-3′; 5′-UTR, antisense primer, 5′-GACAGCAGCGGGAGGACCTC-3′; 3′-UTR, sense primer, 5′-TAA TACGACTCACTATAGGGTATGAAGGTGTTTTCTAGT-3′; 3′-UTR, antisense primer, 5′-ACCTTTTCTAATGTAAAAATACATA-3′. All RNA transcripts were resolved on a 15% polyacrylamide–8 M urea gel to confirm their integrity and sizes, and all RNA transcripts were subsequently excised and gel purified. The concentration of unlabeled RNA was determined by UV spectrophotometry. Labeled transcripts were made by inclusion of [α-32P]CTP at 800 Ci/mmol (NEN Dupont, Boston, MA), and the concentration of radioactively labeled RNA was determined from the specific activity of 32P incorporation.
Expression and purification of wild-type and mutant recombinant DHFR proteins
Escherichia coli BL21 (DE3) competent cells (Invitrogen, Carlsbad, CA) were transformed with recombinant wild-type or mutant human DHFR cDNA plasmids and grown overnight at 37°C in Luria–Bertani medium containing 50 µg/ml kanamycin sulfate. The cells were placed in fresh medium at a dilution of 1:10, grown to an absorbance of 0.6–0.8 at 600 nm, and then induced with 1 mM isopropyl-β-d thiogalactopyranoside (American Bioanalytical, Natick, MA) for 4 h at 37°C. Cells were pelleted at 8000 r.p.m. at 4°C and then freeze–thawed three times. Cells were resuspended in binding buffer (100 mM NaH2PO4, 10 mM Tris–HCl pH 8.0 and 5 mM imidazole) and incubated on ice for 30 min after adding lysozyme to a final concentration of 1 mg/ml. After sonication on ice for 15 min, the lysates were centrifuged at 20 000 r.p.m. at 4°C for 20 min to remove the insoluble material. His-Tag recombinant proteins were purified with a 1-ml polypropylene column (Qiagen, Valencia, CA) packed with TALON metal affinity resin (Clontech) and eluted with buffer containing 100 mM NaH2PO4, 10 mM Tris–HCl pH 8.0 and 150 mM imidazole. The integrity and purity of the recombinant His-Tag proteins were analyzed by resolving SDS–polyacrylamide gel electrophoresis (SDS–PAGE) (12.5% acrylamide) followed by Coomassie blue staining. His-Tag E.coli DHFR protein was prepared using the same method as His-Tag human DHFR protein. Glutathione-S-transferase (GST) and GST-p53 were prepared according to the method described by Albor et al. (17).
The catalytic activity of recombinant wild-type and mutant His-Tag DHFR proteins was determined using a spectrophotometric assay as previously described (18).
RNA gel mobility shift assay
The RNA electrophoretic gel mobility shift assay was performed using a modification of previously described methods (19–21). Each reaction (total volume, 20 µl) contained 18 mM HEPES, pH 7.5, 0.9 mM MgCl2, 18 mM KCl, 100 mM 2-mercaptoethanol (2-ME) (Sigma, St Louis, MO), 3% glycerol, 40 U Prime RNase Inhibitors (Eppendorf-5 Prime, Boulder, CO), 20 µg/ml bovine serum albumin (BSA), 25 µg/ml yeast tRNA, 32P-radiolabeled DHFR mRNA (100 000 c.p.m.; 3.8 fmol) and DHFR protein. The initial incubation was performed for 15 min at room temperature. RNase T1 (15 U; Ambion) was then added for 10 min, followed by incubation with heparin (5 mg/ml; Sigma) for an additional 10 min at room temperature. The entire reaction sample was resolved on a 4% non-denaturing polyacrylamide [acrylamide/methylenebis (acrylamide)/60:1] gel (gel dimensions, 15 × 17 cm) for ∼30 min at 500 V. Gels were dried and then exposed to Kodak XMR film (Eastman Kodak Company, Rochester, NY) and visualized by autoradiography.
Competition experiments were performed with human recombinant DHFR protein (42.6 pmol) and 32P-radiolabeled DHFR mRNA (3.8 fmol; 100 000 c.p.m.). These conditions were selected based on control experiments using a fixed amount of radiolabeled DHFR mRNA with varying concentrations of DHFR protein to determine the linearity of binding.
Nitrocellulose filter-binding assay
The nitrocellulose filter-binding assay was performed using a modification of previously described methods (22). Each reaction (total volume, 20 µl) contained 18 mM HEPES pH 7.5, 0.9 mM MgCl2, 18 mM KCl, 100 mM 2-ME, 40 U Prime RNase Inhibitor, 20 µg/ml BSA, 25 µg/ml yeast tRNA, 32P-radiolabeled DHFR mRNA and DHFR protein. The initial reaction was performed for 15 min at room temperature, after which 15 U RNase T1 were added for 15 min, followed by incubation with heparin (5 mg/ml) for an additional 10 min at room temperature. The mixtures were filtered through a nitrocellulose 0.22 µm filter (Millipore, Bedford, MA) supported in a Millipore glass-fritted 25-mm filter apparatus. After washing the nitrocellulose filters twice with 30 mM HEPES (pH 7.5), the bound radiolabeled RNA retained on the filter was counted in a scintillation counter in 10 ml of Bio-Safe II™ scintillant (Research Product International Corp., Mt. Prospect, IL).
In vitro translation
In vitro translation reactions were performed using a rabbit reticulocyte lysate system (Promega) as previously described (21). In brief, rabbit lysates were incubated with human DHFR mRNA in the presence or absence of recombinant DHFR protein along with 16 µCi [35S]methionine (NEN Dupont) and 0.4 U Prime RNase Inhibitor (Eppendorf-5 Prime). Reactions (total volume, 25 µl) were incubated for 1 h at 30°C, after which two volumes of Laemmli Sample Buffer (Bio-Rad Laboratories, Hercules, CA) were added to the reaction mix, followed by a 10-min incubation at 65°C. The reaction samples were resolved on 12.5% SDS–PAGE according to the method of Laemmli (23), and the gels were processed as previously described. After drying for 1 h, the in vitro translation products were visualized by autoradiography.
UV cross-linking analysis
32P-radioabeled DHFR RNA (3.8 fmol) and human His-Tag DHFR protein (42.6 pmol) were incubated at room temperature for 15 min in an RNA-binding reaction as described above. The DHFR ribonucleoprotein (RNP) complexes were UV cross-linked for 15 min at 254 nm (Stratalinker 1800 UV crosslinker; Stratagene, La Jolla, CA) and then digested with RNase A (0.1 µg/µl; Qiagen) for 15 min at 37°C. The complexes were resolved on an SDS–15% PAGE. The gel was dried and subjected to autoradiography.
Isolation of bound RNAs from the DHFR RNP complex
32P-radiolabeled DHFR mRNA probe corresponding to DHFR:1–564 (3 000 000 c.p.m.; 114 fmol) was incubated with His-Tag DHFR protein (128 pmol) in a standard RNA-binding reaction as previously described (19–21). After incubation at room temperature for 15 min, the reaction mixture was incubated with 300 U RNase T1 and 50 ng of RNase A for 15 min, after which heparin was added for an additional 10 min. The mixtures were filtered through a 0.22 µm nitrocellulose filter (Millipore). After washing the filters with 5 ml of 30 mM HEPES pH 7.5, the radiolabeled RNA retained on the filter was eluted with an RNA elution buffer (0.6 M sodium acetate, 1 mM EDTA and 0.1% SDS) at 37°C overnight. The supernatant was then extracted with phenol/chloroform (1:1), chloroform/isoamyl alcohol (24:1) and precipitated with 2.5 volumes of ethanol. The pelleted RNAs were resuspended in molecular biology grade water (Eppendorf-5 Prime) and resolved on a 15% polyacrylamide–8 M urea gel at 500 V for 1.5 h. The gel was then subjected to wet autoradiography.
RESULTS
Characterization of the interaction between human recombinant DHFR protein and human DHFR mRNA
Previous work from this laboratory had shown that human recombinant DHFR protein specifically interacted with its own DHFR mRNA (15). The DHFR protein that had been used in this original series of experiments had been purified to homogeneity by isoelectric focusing. Unfortunately, this source of human recombinant DHFR protein was no longer available for further studies, and it was necessary to identify an alternate source of protein. As a result, we purified wild-type, human recombinant DHFR protein that expressed a His-Tag sequence located immediately upstream of the DHFR coding sequence. This human His-Tag DHFR was purified by affinity chromatography using the TALON metal affinity resin. The purity of this recombinant protein was shown to be >95% pure by Coomassie blue staining.
A 32P-radiolabeled, human DHFR mRNA (564 nt), corresponding to the protein-coding sequence, was then used in RNA gel shift and nitrocellulose filter-binding assays. As seen in Figure 2A, wild-type, human His-Tag DHFR protein formed a complex with the radiolabeled DHFR mRNA with a delayed migration on the non-denaturing gel (Fig. 2A, lanes 2–4). An identical complex was observed when the radiolabeled DHFR mRNA probe was incubated with wild-type human recombinant DHFR protein (data not shown), and we have previously published these results (15). In contrast, no complex formation was observed when radiolabeled human DHFR mRNA was incubated in the presence of other proteins including His-Tag E.coli DHFR, His-Tag human TS, GST and GST-p53 (Fig. 2A, lanes 5–8). Competition experiments revealed that the addition of unlabeled human DHFR RNA corresponding to the coding region (DHFR:1–564) inhibited complex formation in a dose-dependent manner (Fig. 2B, lanes 3–5). However, the addition of unrelated RNAs, including yeast tRNA, GAPDH mRNA, 18S rRNA, TS30 and β-actin antisense RNA was unable to reduce the level of complex formation (Fig. 2B, lanes 6–10).
Figure 2.


(A) Specificity of human DHFR protein binding to human DHFR mRNA. 32P-radiolabeled human DHFR RNA (100 000 c.p.m.; 3.8 fmol) was incubated in the absence (lane 1) or presence of 21.3 pmol (lane 2), 42.6 pmol (lane 3), 85.2 pmol (lane 4) of His-Tag recombinant human DHFR protein as described in the Materials and Methods. Radiolabeled DHFR mRNA was also incubated in the presence of 238 pmol of His-Tag E.coli DHFR (lane 5), 139 pmol of His-Tag human TS (lane 6), 192 pmol of GST (lane 7) and 63.3 pmol of GST-p53 (lane 8). (B) RNA gel-shift competition experiment to show specificity of binding of DHFR protein to DHFR mRNA. A radiolabeled DHFR mRNA probe was incubated in the absence (lane 1) or presence (lanes 2–10) of His-Tag DHFR protein (42.6 pmol) as described in the Materials and Methods. Competition studies were performed with a 25- (lane 3), 125- (lane 4) and 250-fold (lane 5) molar excess of unlabeled DHFR mRNA and a 250-fold molar excess of yeast tRNA (lane 6), GAPDH mRNA (lane 7), 18S rRNA (lane 8), TS30 (lane 9) or β-actin antisense RNA (lane 10).
To confirm the results observed with the RNA gel shift assay, we employed the nitrocellulose filter-binding assay. Using this method, we were able to more precisely quantify the relative binding affinity of the various competitor RNAs. As seen in Table 1, the addition of cold, unlabeled DHFR: 1–700 RNA, a sequence corresponding to the full-length DHFR mRNA, significantly reduced the level of binding of the radiolabeled probe to the DHFR protein with an IC50 of 3.16 nM. An RNA sequence corresponding to the 564-nt coding region of DHFR mRNA (DHFR:1–564) displayed a nearly identical relative binding affinity as the full-length RNA sequence (IC50 = 3.64 nM). In contrast, sequences corresponding to the respective 5′- and 3′-UTRs of DHFR mRNA had markedly reduced binding affinities, with IC50 values >200 nM. Unrelated RNAs, including a 30-nt RNA corresponding to nucleotides 80–109 of the TS mRNA (TS30) and yeast tRNA were unable to compete for binding. These studies provided further support for the specific nature of the interaction of human DHFR protein and its own human DHFR mRNA.
We also tested the effect of substrate binding on the ability of DHFR protein to interact with its own DHFR mRNA. For these studies, we employed the filter-binding assay, and incubated DHFR protein with varying concentrations of MTX. As seen in Table 2, incubation of DHFR with MTX effectively abrogated the RNA binding of DHFR in a dose-dependent manner, and an IC50 value of 0.29 µM was obtained. In contrast, incubation with the fluoropyrimidine 5-FU (IC50 > 5000 µM) did not affect the RNA-binding activity of DHFR and other antifolate analogs such as ZD1694 (IC50 = 6.6 nM), ZD9331 (IC50 = 16.8 nM) or 1843U89 (IC50 = 48.5 nM) were significantly less effective at abrogating the RNA-binding activity of DHFR when compared with MTX.
Table 2. Effect of antifolate drugs on RNA-binding activity of His-Tag recombinant DHFR protein.
| Anti-folate drug | IC50 (µM) ± SD |
|---|---|
| MTX | 0.29 ± 0.140 |
| TDX | 6.60 ± 2.25 |
| ZD9331 | 16.8 ± 4.13 |
| ZD1843 | 48.5 ± 2.50 |
| 5-FU | >5000 |
Effect of inhibitor compounds on binding of human recombinant His-Tag DHFR protein to full length, human DHFR mRNA was determined by comparing the concentration of anti-folate drug that inhibited specific binding of 32P-radiolabeled human DHFR mRNA by 50%. The experiments were performed as described in the Materials and Methods. Each value represents the mean ± SD of at least three experiments.
Identification of amino acid residues involved in RNA recognition
Previous studies identified several point mutations within the active site of human DHFR protein as playing a major role in the development of a cellular drug resistant to the antifolate analog MTX and the related compound trimetrexate. Of note, several mutant proteins have been expressed that display significantly reduced binding affinity to these respective antifolate analogs, but as yet maintain intact catalytic activity. Such mutant proteins include I7F, L22Y, F34S and R70K (24–27). Recently, mutant proteins with substitutions at Leu22 and/or Phe31 were shown to retain RNA-binding activity (28). In the present report, we focused our initial attention on investigating the potential RNA-binding activity of these well characterized mutant proteins. In addition, we expressed and purified proteins with mutations at the sole cysteine amino acid residue at position 6 of the protein, as cysteine residues have been documented to play a critical role in RNA recognition (29–34).
For this series of experiments, we used site-directed mutagenesis to express and purify 14 different His-Tag DHFR mutant proteins. These mutant proteins are listed in the Materials and Methods. As seen in Figure 3A, mutation at the Cys6 residue with either an alanine (C6A) or serine (C6S) residue almost completely abrogated the RNA-binding activity. Of note, the catalytic activity of each of these cysteine mutant proteins was reduced by only 20–25% (Fig. 3B). Three other mutant proteins, I7A, R28A and F34S, were significantly impaired in their ability to bind DHFR mRNA. In contrast, mutant proteins E30A, E30Q and F31A showed a significant increase in their respective RNA-binding properties.
Figure 3.


(A) RNA-binding activity of wild-type and mutant DHFR proteins using the nitrocellulose filter-binding assay. 32P-radiolabeled DHFR mRNA (100 000 c.p.m.; 3.8 fmol) was incubated with 42.6 pmol of wild-type or mutant His-Tag recombinant DHFR proteins using the nitrocellulose filter-binding assay as described in the Materials and Methods. Each value represents the mean ± SD of at least three experiments. (B) Enzyme activity of wild-type and mutant human DHFR proteins. The enzyme activity of wild-type and mutant DHFR proteins was determined using the spectrophotometric assay, as outlined in the Materials and Methods. Each value represents the mean ± SD of at least three experiments.
To begin to determine the potential functional significance of these mutations, the effect of these mutant proteins on DHFR mRNA translation was investigated by means of the in vitro rabbit reticulocyte lysate translation system. When human DHFR mRNA was included in the rabbit lysate, a band corresponding to the His-Tag DHFR protein was observed, resolving at a molecular weight of ∼24 kDa (Fig. 4, lane 1). Translation of DHFR mRNA was completely repressed upon the addition of human His-Tag DHFR protein (Fig. 4, lane 2). In contrast, the addition of mutant proteins C6A, C6S, I7A, R28A and F34S did not result in translational inhibition (Fig. 4, lanes 3–7). Incubation of DHFR mRNA with mutant proteins E30Q, F31A and I7F resulted in a significant degree of translational repression, to nearly the same degree as observed with the wild-type DHFR protein (Fig. 4, lanes 8–10). Finally, the addition of a DHFR protein isolated from a different species, namely E.coli DHFR, was unable to inhibit translation of human DHFR mRNA (Fig. 4, lane 11).
Figure 4.

Effect of wild-type and mutant human DHFR proteins on translation of human DHFR mRNA. Human DHFR mRNA (0.24 nmol) was incubated in the absence (lane 1) or presence of wild-type or mutant DHFR proteins (85.2 pmol) as described in the Materials and Methods. Lane 1 contains only DHFR mRNA; lane 2, wild-type human DHFR protein; lane 3, C6A; lane 4, C6S; lane 5, I7A; lane 6, R28A; lane 7, F34S; lane 8, E30Q; lane 9, F31A; lane 10, I7F; lane 11, His-Tag E.coli DHFR protein.
UV cross-linking analysis
A UV cross-linking analysis was performed to determine the approximate molecular weight of the DHFR RNP complex. As seen in Figure 5, a complex resolving at a molecular weight of ∼29 kDa was observed when the 564-nt radiolabeled probe was incubated with wild-type, His-Tag DHFR protein (Fig. 5, lane 3). UV cross-linking was absolutely essential for visualization of this complex, as an identical reaction mixture not subjected to UV cross-linking did not reveal such a complex (Fig. 5, lane 2). A complex was also observed when the radiolabeled probe was incubated with the mutant E30A mutant protein (Fig. 5, lane 5). In contrast, the C6A mutant DHFR protein and a GST protein were unable to form a complex with the radiolabeled probe (Fig. 5, lanes 4 and 6). As the His-Tag DHFR protein resolves at a molecular weight of ∼24 kDa, these findings suggest that the main RNA sequence protected from RNase digestion is ∼16 nt in length.
Figure 5.

UV cross-linking analysis. 32P-radiolabeled human DHFR mRNA (100 000 c.p.m.; 3.8 fmol) was incubated with 42.6 pmol of wild-type or mutant human His-Tag DHFR protein as described in the Materials and Methods. After UV cross-linking, the reaction mixture was incubated with RNase A to digest the unprotected RNAs. The UV cross-linked complexes were then resolved on SDS–12.5% PAGE. Lane 1, probe only; lane 2, His-Tag DHFR protein without UV cross-linking; lane 3, His-Tag DHFR protein; lane 4, C6A; lane 5, E30A; lane 6, GST.
Isolation of bound RNAs from the DHFR RNP complex
We next performed a footprinting analysis to determine the approximate size of the DHFR RNA sequence bound by the DHFR protein. For this set of experiments, we used the nitrocellulose filter-binding assay. A standard RNA-binding reaction was performed, and the RNAs bound to the human DHFR protein were eluted from the nitrocellulose filter and subsequently resolved on a denaturing 15% polyacrylamide– 8 M urea gel. As seen in Figure 6, three distinct RNA species were identified, ranging in size from 16 to 45 nt.
Figure 6.
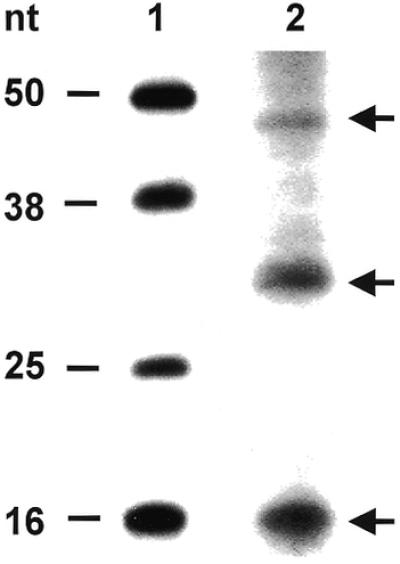
Isolation of bound RNAs from DHFR RNP complexes. 32P-radiolabeled human DHFR mRNA (3 000 000 c.p.m.; 114 fmol) was incubated with wild-type, His-Tag human DHFR protein (127.8 pmol), followed by digestion with RNase T1 and RNase A, and the addition of heparin. RNAs bound to the DHFR protein were eluted and resolved on a 15% polyacrylamide–8 M urea gel as described in the Materials and Methods. Lane 1, RNA markers; lane 2, RNAs isolated from the DHFR RNP complex.
DISCUSSION
In this study, we present evidence that demonstrates a specific interaction between human His-Tag DHFR protein and its cognate human DHFR mRNA. This interaction was confirmed by using two different experimental approaches, an RNA gel mobility shift assay and a nitrocellulose filter-binding method. The human DHFR initially studied by our laboratory was a recombinant protein that had been purified to homogeneity by isoelectric focusing (15), and was therefore completely substrate-free. In contrast, the DHFR protein employed by Erickan-Abali et al. (16) was only ∼20% pure. Nevertheless, the findings described herein are entirely consistent with those previously observed by our group and by Ercikan-Abali et al. (16). Our studies with the recombinant His-Tag DHFR protein suggest that the presence of the His-Tag does not adversely affect the RNA-binding activity of DHFR. McPherson et al. (28) also observed that a human His-Tag DHFR fusion protein retained the ability to bind to its target DHFR mRNA. These findings suggest that the recombinant His-Tag DHFR fusion protein can be used as an appropriate source of human DHFR in future studies to identify the specific RNA-binding domain(s) on the protein. In addition, the original His-Tag DHFR cDNA construct used to express this recombinant protein can be used as a template for site-directed mutagenesis studies to generate mutant DHFR proteins.
To begin to identify the amino acid residues on DHFR that are required for RNA recognition, we used a site-directed mutagenesis strategy to express and purify mutant DHFR proteins. In contrast to wild-type protein, mutant proteins I7A, R28A and F34S did not bind to the target DHFR mRNA and, as a result, were unable to inhibit DHFR mRNA translation. However, three mutant proteins E30Q, F31A and I7F, which all bound with increased relative affinity to DHFR mRNA, repressed translation to nearly the same extent as the wild-type protein. These studies identify Ile7, Arg28 and Phe34 as being important amino acid residues involved in RNA binding and suggest that the folate-binding site may represent an important RNA-binding domain on DHFR protein. Moreover, the fact that catalytically inactive mutant proteins retain RNA-binding activity indicates that the functions of RNA binding and enzyme catalysis are not mediated by the same domain(s) on the protein. The specific mechanism(s) by which the folate-binding region mediates RNA binding remains unclear. It is conceivable that this site interacts directly with the target RNA and that these three amino acids function as critical contact points. However, an alternative possibility is that these amino acids maintain the protein in a certain conformational state that then allows for optimal RNA binding.
Several studies have focused on characterizing the molecular elements that mediate the interaction between RNA-binding proteins and their target RNAs. The R17 bacteriophage coat protein (29,30), aminoacyl-tRNA synthetase (31), and iron-responsive element factor (32,33) represent three well characterized RNA-binding proteins. In each case, a free cysteine sulfhydryl group(s) on the RNA-binding protein forms a covalent Michael adduct with the C-6 position on the corresponding uracil ring of the target RNA. With this in mind, we designed recombinant DHFR proteins with a mutation at the sole cysteine residue located at amino acid position 6. Substitutions at Cys6 with either an alanine or serine residue resulted in nearly complete abrogation of RNA-binding activity. Of note, the catalytic activity of each of these mutant proteins was almost completely preserved. These findings suggest then that the sulfhydryl residue at Cys6 is critical for RNA binding. While the precise mechanism by which this occurs remains unclear, there are, at least, three possibilities to explain its role. First, the cysteine sulfhydryl may form a direct Michael adduct with the C-6 position of a uracil ring on DHFR mRNA. Secondly, occupation of the cysteine moiety may result in altered RNA binding through a steric hindrance mechanism. Finally, the cysteine residue may be critical in maintaining the protein in a certain conformational structure that then allows the true binding domain on DHFR to be readily accessible to the target RNA.
One issue that we wished to determine was the approximate size of the DHFR RNA sequence bound by DHFR protein. For this work, two different strategies were taken. The first approach was a UV cross-linking assay to isolate and determine the molecular weight of the RNA–protein complex, while the second one incorporated an RNA footprinting analysis. In the UV cross-linking analysis, an RNP complex was indeed observed when wild-type DHFR protein was incubated with a radiolabeled DHFR mRNA probe and then subjected to UV photochemical cross-linking. This complex resolved at a molecular weight of ∼29 kDa. This finding is in sharp contrast to the 80 kDa RNP complex identified previously by Ercikan-Abali et al. (16). One potential explanation for this rather sharp difference is that the DHFR protein used in our experiments was nearly homogeneously pure whereas the DHFR protein used by Ericikan-Abali et al. (16) was only 20% pure. The use of only partially pure DHFR protein, in their case, raises concerns for the specificity of this RNP complex and suggests binding of a cellular protein other than DHFR to the DHFR mRNA. Another possibility is that a significantly longer sequence of RNA (200 nt) was bound to the human recombinant DHFR protein purified by Erickan-Abali et al. (16) as a result of incomplete and/or insufficient RNase digestion. In the present report, we were able to document the specificity of the DHFR RNP complex by the inability of mutant DHFR protein-C6A and an unrelated GST protein to form such a complex with the same radiolabeled DHFR mRNA probe.
The molecular weight of the His-Tag DHFR protein, as resolved on a denaturing SDS gel, is 24 kDa. If one assumes a 1:1 binding stoichiometry between DHFR protein and DHFR RNA, the remaining 5000 molecular weight must be contributed by the bound RNA. Based on these calculations, the size of the RNA sequence bound to the protein would be ∼16 nt in length. As a complementary method, we used an RNA footprinting analysis that resolved three main RNA species, 16, 30 and 45 nt in length, with the most intense signal being observed at 16 nt. Taken together, these findings suggest that a core RNA sequence of 16 nt is bound to DHFR protein. However, our results do not rule out the possibility that longer-sized RNA sequences containing this 16-nt core sequence are bound to DHFR protein or that there are, in fact, three different RNA elements with which the DHFR protein specifically interacts. Finally, a nearly 5-fold higher amount of RNase A was included in the UV cross-linking analysis when compared with what was used for the footprinting method. Thus, it is conceivable that a more complete digestion of the unbound RNA was achieved with the cross-linking method, thereby yielding a smaller-sized RNA sequence bound to DHFR protein. Further studies are in progress to more carefully elucidate the RNA sequences bound to DHFR protein in the RNP complexes both in vitro and in vivo.
One final issue relates to whether the ability of DHFR to recognize RNA is conserved in evolution. As presented herein, recombinant E.coli DHFR protein was unable to bind to human DHFR mRNA as well as to its own E.coli DHFR mRNA. This result may not be entirely surprising, as the level of homology between the human and E.coli DHFR is only on the order of 27%. Moreover, when one compares the level of amino acid similarity in the folate-binding region in which the C6, I7, R28 and F34 amino acids reside, there is even less homology (<20%). This finding is in marked contrast to TS in which the RNA-binding activity is maintained across all species studies to date (34). Despite these initial results, further studies are needed to more precisely determine whether the RNA-binding activity of DHFR is conserved amongst the different species.
Further studies are needed to more carefully elucidate the key molecular elements underlying the translational autoregulation of DHFR and the interaction between DHFR protein and its target mRNA. Our laboratory is presently dissecting the critical cis-elements on DHFR mRNA that are required both in vitro and in vivo for RNA recognition. In addition, studies are in progress to determine the precise intracellular localization of the DHFR RNP complex in intact human colon cancer cells. Such work may help to further characterize the biological significance of this particular RNP complex. While four different amino acids have been shown to be important for RNA recognition, it remains unclear as to whether these amino acids form direct contact points with the target DHFR mRNA or whether they maintain the protein in a certain conformational state that then allows for the proper binding domain on the protein to interact with its target mRNA. Studies are now planned to resolve the crystal structure of the DHFR protein–DHFR mRNA complex in an attempt to address this critical issue. Finally, as the process of translational autoregulation is becoming an increasingly recognized mechanism for the control of gene expression, the studies described herein may provide new insights that can be applied to other critical cellular genes.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by grants from the National Cancer Institute (CA16359 and CA82897 to E.C.). The authors also wish to thank the Yale Cancer Center and the VA CT Cancer Center, VA CT Healthcare System for their continued support of this research.
REFERENCES
- 1.Blakely R.L. and Cocco,L. (1984) Dismutation of dihydrofolate by dihydrofolate reductase. Biochemistry, 23, 2377–2383. [DOI] [PubMed] [Google Scholar]
- 2.Chu E. and Allegra,C.J. (1996) Antifolates. In Chabner,B.A. and Longo,D.L. (eds), Cancer Chemotherapy: Principles and Practice. Lippincott, Philadephia, PA, pp. 110–153.
- 3.Frearson P.M., Kit,S. and Dubbs,D.R. (1966) Induction of dihydrofolate reductase activity by SV40 and polyoma virus. Cancer Res., 26, 1653–1660. [PubMed] [Google Scholar]
- 4.Kellems R.E., Morhenn,V.B., Pfendt,E.A., Alt,F.W. and Schimke,R.T. (1979) Polyoma virus and cyclic AMP-mediated control of dihydrofolate reductase mRNA abundance in methotrexate-resistant mouse fibroblasts. J. Biol. Chem., 254, 680–685. [PubMed] [Google Scholar]
- 5.Hendrickson S.L., Wu,J.-S.R. and Johnson,L.F. (1980) Cell cycle regulation of dihydrofolate reductase mRNA metabolism in mouse fibroblasts. Proc. Natl Acad. Sci. USA, 77, 5140–5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gudewicz T.M., Morhenn,V.B. and Kellems,R.E. (1981) The effect of polyoma virus, serum factors, and dibutyryl cyclic AMP on dihydrofolate reductase synthesis and the entry of quiescent cells into S phase. J. Cell Physiol., 108, 1–8. [DOI] [PubMed] [Google Scholar]
- 7.Yoder S.S., Robberson,B.L., Leys,E.J., Hook,A.G., Al-Ubaidi,M., Yeung,C.-Y., Kellems,R.E. and Berget,S.M. (1983) Control of cellular gene expression during adenovirus infection: induction and shut-off of dihydrofolate reductase gene expression by adenovirus type 2. Mol. Cell. Biol., 3, 819–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schuetz J.D., Gorse,K.M., Goldman,I.D. and Westin,E.H. (1988) Evidence for a functional defect in the translocation of the methotrexate transport carrier in a methotrexate-resistant murine L1210 leukemia cell line. J. Biol. Chem., 263, 7708–7712. [PubMed] [Google Scholar]
- 9.Hillcoat B.L., Swett,V. and Bertino,J.R. (1967) Increase of dihydrofolate reductase activity in cultured mammalian cells after exposure to methotrexate. Proc. Natl Acad. Sci. USA, 58, 1632–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bastow K., Prabhu,R. and Cheng,Y.C. (1984) The intracellular content of dihydrofolate reductase: possibilities for control and implications for chemotherapy. Adv. Enzyme Regul., 22, 15–26. [DOI] [PubMed] [Google Scholar]
- 11.Cowan K.H., Goldsmith,M.E., Ricciardone,M.D., Levine,R., Rubalcaba,E. and Jolivet,J. (1986) Regulation of dihydrofolate reductase in human breast cancer cells and in mutant hamster cells transfected with a human dihydrofolate reductase minigene. Mol. Pharmacol., 30, 69–76. [PubMed] [Google Scholar]
- 12.Domin B.A., Grill,S.P., Bastow,K.F. and Cheng,Y.C. (1982) Effect of methotrexate on dihydrofolate reductase activity in methotrexate-resistant human KB cells. Mol. Pharmacol., 21, 478–482. [PubMed] [Google Scholar]
- 13.Grem J.L., Voeller,D.M., Geoffroy,F., Horak,E., Johnston,P.G. and Allegra,C.J. (1994) Determinants of trimetrexate lethality in human colon cancer cells. Br. J. Cancer, 70, 1075–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gollerkeri A. and Chu,E. (2001) Translational regulation of DHFR expression in human colon cancer RKO cells. Proc. Am. Assoc. Cancer Res., 14, 783a. [Google Scholar]
- 15.Chu E., Takimoto,C.H., Voeller,D.M., Grem,J.L. and Allegra,C.J. (1993) Specific binding of human dihydrofolate redcutase protein to dihydrofolate redcutase messenger RNA in vitro. Biochemistry, 32, 4756–4760. [DOI] [PubMed] [Google Scholar]
- 16.Ercikan-Abali E.A., Banerjee,D., Waltham,M.C., Skacel,N., Scotto,K.W. and Bertino,J.R. (1997) Dihydrofolate reductase protein inhibits its own translation by binding to dihydrofolate reductase mRNA sequences within the coding region. Biochemistry, 36, 12317–12322. [DOI] [PubMed] [Google Scholar]
- 17.Albor A., Kaku,S. and Kulesz-Martin,M. (1998) Wild-type and mutant forms of p53 activate human topoisomerase I: a possible mechanism for gain of function in mutants. Cancer Res., 58, 2091–2094. [PubMed] [Google Scholar]
- 18.Delcamp T.J., Susten,S.S., Blankenship,D.T. and Freisheim,J.H. (1983) Purification and characterization of dihydrofolate reductase from methotrexate-resistant human lymphoblastoid cells. Biochemistry, 22, 633–639. [DOI] [PubMed] [Google Scholar]
- 19.Leibold E.A. and Munro,H.N. (1988) Cytoplasmic protein binds in vitro to a highly conserved sequence in the 5′ untranslated region of ferritin heavy and light subunit mRNAs. Proc. Natl Acad. Sci. USA, 85, 2171–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chu E., Koeller,D.M., Casey,J.L., Drake,J.C., Chabner,B.A., Elwood,P.C., Zinn,S. and Allegra,C.J. (1991) Autoregulation of human thymidylate synthase messenger RNA translation by thymidylate synthase. Proc. Natl Acad. Sci. USA, 88, 8977–8981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chu E., Voeller,D.M., Koeller,D.M., Drake,J.C., Takimoto,C.H., Maley,G.F., Maley,F. and Allegra,C.J. (1993) Identification of an RNA binding site for human thymidylate synthase. Proc. Natl Acad. Sci. USA, 90, 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hall K.B. and Kranz,J.K. (1999) Nitrocellulose filter binding for determination of dissociation constants. In Haynes,S. (ed.), Methods in Molecular Biology: RNA–Protein Interaction Protocols. Humana Press, Totowa, NJ, Vol. 108, pp. 105–114. [DOI] [PubMed]
- 23.Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 24.Petal M., Sleep,S.E.H., Lewis,W.S., Spencer,H.T., Mareya,S.M., Sorrentino,B.P. and Blakely,R.L. (1997) Comparison of the protection of cells from antifolates by transduced human dihydrofolate reductase mutants. Hum. Gene Ther., 8, 2069–2077. [DOI] [PubMed] [Google Scholar]
- 25.Lewis W.R., Cody,V., Galitsky,N., Luft,J.R., Pangborn,W., Chunduru,S.K., Spencer,H.T., Appleman,J.R. and Blakely,R.L. (1995) Methotrexate-resistant variants of human dihydrofolate reductase with substitutions of leucine 22. Kinetics, crystallography, and potential as selectable markers. J. Biol. Chem., 270, 5057–5064. [DOI] [PubMed] [Google Scholar]
- 26.Nakano T., Spencer,H.T., Appleman,J.R. and Blakely,R.L. (1994) Critical role of phenylalanine 34 of human dihydrofolate reductase in substrate and inhibitor binding and in catalysis. Biochemistry, 33, 9945–9952. [DOI] [PubMed] [Google Scholar]
- 27.Thompson P.D. and Freisheim,J.H. (1991) Conversion of arginine to lysine at position 70 of human dihydrofolate reductase: generation of a methotrexate-insensitive mutant enzyme. Biochemistry, 30, 8124–8130. [DOI] [PubMed] [Google Scholar]
- 28.McPherson J.P., Saurbrey,A., Meehl,M., Russo,A., Skacel,N. and Bertino,J.R. (1999) Characterization of RNA binding by mutant variants of human dihydrofolate reductase. Proc. Am. Assoc. Cancer Res., 40, 676a. [Google Scholar]
- 29.Carey J., Cameron,V., deHaseth,P.L. and Uhlenbeck,O.C. (1983) Sequence-specific interaction of R17 coat protein with its ribonucleic acid binding site. Biochemistry, 22, 471–478. [DOI] [PubMed] [Google Scholar]
- 30.Romaniuk P.J. and Uhlenbeck,O.C. (1985) Nucleoside and nucleotide inactivation of the R17 coat protein: evidence for a transient covalent RNA–protein bond. Biochemistry, 24, 4239–4244. [DOI] [PubMed] [Google Scholar]
- 31.Starzyk R.M., Koontz,S.W. and Schimmel,P.R. (1979) A covalent adduct between the uracil ring and the active site of an aminoacyl-tRNA synthetase. Nature, 298, 136–140. [DOI] [PubMed] [Google Scholar]
- 32.Theil E.C. (1990) Regulation of ferritin and transferrin receptor mRNA. J. Biol. Chem., 265, 4771–4774. [PubMed] [Google Scholar]
- 33.Klausner R.D., Rouault,T.A. and Hartford,J.B. (1993) Regulating the fate of mRNA: the control of cellular iron metabolism. Cell, 72, 19–28. [DOI] [PubMed] [Google Scholar]
- 34.Voeller D.M., Changchien,L.M., Maley,G.F., Maley,F., Takechi,T., Turner,R.E., Montfort,W.R., Allegra,C.J. and Chu,E. (1995) Characterization of a specific interaction between Escherichia coli thymidylate synthase and Escherichia coli thymidylate synthase mRNA. Nucleic Acids Res., 23, 869–875. [DOI] [PMC free article] [PubMed] [Google Scholar]