


Abstract
Telomeres are essential for genomic stability and cell viability. Telomerase, the enzyme responsible for telomere maintenance, is composed of a reverse transcriptase protein subunit and an integral RNA component which contains the templating domain. In human telomerase, the template region consists of 11 nt (3′-rCAAUCCCAAUC-5′) and comprises an alignment domain (italicised) plus a template sequence encoding the telomeric repeat d(GGT TAG). In this study, the alignment domain of human telomerase was systematically reduced from the 3′ end and the resultant recombinant enzyme activity was evaluated in vitro. Deletion or substitution of one or two residues from the 3′ end of the alignment domain caused only a slight reduction in overall catalytic activity and did not alter the processivity of the enzyme. Deletion or substitution of three or more residues from the 3′ end of the alignment domain resulted in total loss of catalytic activity. These results suggest that the two most 3′ terminal RNA residues are relevant but not essential for overall activity and that the minimal length requirement of the alignment domain is 3 nt. Furthermore, base pairing between the 3′ end of the primer substrate and the first two residues of the alignment domain is also not an absolute requirement for processive synthesis.
INTRODUCTION
Telomeres are found at the termini of eukaryotic chromosomes (1,2) and consist of dGT-rich DNA sequences and their associated proteins (3,4). Telomerase is the cellular reverse transcriptase responsible for the synthesis and maintenance of the chromosomal ends (1). Telomerase compensates for the loss of telomeric DNA that occurs through incomplete replication, thereby protecting the chromosome ends against recombination, chromosome fusion or exonuclease degradation (5,6). Telomerase has also been implicated in the control of cellular lifespan (7,8). In particular, a number of studies have indicated that telomerase expression is associated with cell immortalisation and tumourigenesis, highlighting the enzyme as a potentially promising target for the development of anti-cancer therapeutics (9–11).
Telomerase is a ribonucleoprotein minimally composed of an RNA component (TR) and a catalytic subunit (TERT), which has been identified as a reverse transcriptase in Euplotes (12), Saccharomyces cerevisiae (13), Schizosaccharomyces pombe (14) and human (14). Using biochemical and genetic tools, several telomerase-associated proteins have also been identified to be part of the holoenzyme complex in Tetrahymena and mammals (15–17). Their function remains to be unambiguously determined, but various studies indicate they may participate in telomerase assembly, function or regulation (15,16).
Telomerase elongation and substrate specificity have been characterised in vitro for a wide range of organisms, including protozoa, yeasts and human, by use of a single-stranded DNA primer extension assay (1,18,19). The likely mechanism for telomerase, first proposed by Blackburn and co-workers (20), is distinct from other classes of DNA polymerases (Fig. 1) (21). Telomerase uses a small region of its integral RNA component, called the template domain, to direct the synthesis of the dGT-rich strand of the telomeres whilst reverse transcription is mediated by the catalytic protein subunit of telomerase (TERT) (18,22). The proposed mechanism involves several defined events which include a primer recognition step (Fig. 1B) and an extension step (Fig. 1C) (20). A distinct feature of telomerase is the ability to reiteratively add telomeric repeats to a DNA primer without dissociation of enzyme and substrate. According to the current model, processive repeat addition requires the newly synthesised 3′ end of the primer to re-align on the RNA template after each round of addition (Fig. 1D). Studies on telomerase from different species strongly suggest that telomerase can interact with oligonucleotide primers in a bipartite manner. It has been proposed that the 3′ end of the DNA primer is aligned by hybridisation with the RNA template, whilst the 5′ region of the primer is bound to a second site (the anchor site) which contributes to high affinity substrate binding and processive elongation (23–25). Following a repeat addition (Fig. 1C), the 5′ region of the primer remains bound to the anchor site allowing the newly synthesised 3′ end of the DNA to translocate back to the beginning of the template, without dissociation from the enzyme, for additional repeat synthe sis (Fig. 1D). Alternatively, if both sites are released simultaneously the DNA substrate dissociates from the enzyme with termination of synthesis.
Figure 1.

Telomeric DNA synthesis catalysed by human telomerase. (A) Telomerase catalytic protein (hTERT) is shown with the template sequence spanning nucleotides +46 to +56 of the RNA subunit (hTR) and the anchor site. (B) Primer annealing: the 3′ end of the primer base pairs with the template region of telomerase RNA. (C) Primer extension by reverse transcription until the 5′ end of the template is reached. Newly synthesised DNA is indicated in bold. (D) Translocation and re-alignment of the primer terminus to the 3′ end of the template for a further round of extension. For non-natural substrates, primer annealing during first round synthesis is dictated by sequence complementarity between the primer 3′ end and the template region. In subsequent rounds of synthesis, re-alignment of the primer 3′ end is as described above (D).
Vertebrate telomerase RNA, including those of mammals, birds, fish, reptile and amphibians, vary significantly in length and sequence (26). However, phylogenetic analysis of these RNA sequences from distantly related telomerases show highly conserved structural domains and a remarkably similar core structure despite low primary sequence conservation (26). In particular, 8 nt in the catalytically important template domain are universally conserved throughout the vertebrate kingdom with the consensus sequence 3′-UCCCAAUC-5′ (Fig. 2A). The template region of telomerase may be functionally divided into two distinct domains known as the alignment (italicised) and elongation domains. The elongation domain is invariant for all vertebrates and consists of the 6 nt sequence 3′-CCAAUC-5′, which encodes the telomeric repeat d(GGTTAG) (Fig. 2A). The alignment domain of vertebrates is less conserved and has evolved to have lengths ranging from 2 to 5 nt (Fig. 2A). The sequence conservation of these residues is shown in Figure 2B.
Figure 2.

Vertebrate telomerase RNAs [adapted from Chen et al. (26)]. (A) Alignment and consensus sequence of the template region of telomerase RNA from a variety of vertebrates. The sequences shown are from Homo sapiens (human, hTR), Gallus gallus (chicken, cTR), Rattus norvegicus (rat, rTR) and Mus musculus (mouse, mTR). The template sequence is shown in bold, the alignment domain in bold italic. (B) The consensus sequence. The level of sequence conservation in the template region of vertebrate telomerase RNAs is shown (compiled from 35 sequences of distantly related vertebrates).
The template sequence of human telomerase is 11 residues long (3′-CAAUCCCAAUC-5′), nearly twice the length of a telomeric repeat. Phylogenetic analysis suggests a minimum of 8 nt may be sufficient for function in vertebrates. It has been postulated that the alignment domain functions to re-position the primer substrate by base pairing during processive synthesis (Fig. 1D) (20,27). This translocation step is unique to telomerase amongst the polymerases and represents one of the most intriguing features to understand at the molecular level. However, there is much to be elucidated about factors that influence translocation efficiency and enzyme processivity. Based on the current model, re-alignment of DNA at the alignment domain may be the key for efficient repeat addition. Interestingly, rodent telomerases which can only allow up to 2 bp to form during re-alignment are typically non-processive (28,29) whilst human telomerase which potentially uses 5 bp is significantly more processive (18). Does the shorter alignment domain account for the synthesis of shorter products? The functionally important determinants within the telomerase RNA template have been characterised in ciliates (30–33) but little has been reported for human telomerase. In this paper, we report studies on the effect of systematic reduction in the length of the alignment domain of human telomerase on activity with a particular emphasis on processivity. These studies also define a minimal requirement in the alignment domain for the processive catalytic activity of human telomerase.
MATERIALS AND METHODS
hTERT and hTR plasmid constructs
The hTERT expression plasmid was generated by sub-cloning the wild-type hTERT coding region from the pCl-Neo-hTERT plasmid (34) (obtained from Dr R. Weinberg) using the forward primer 5′-CATATGCCGCGCGCTCCCCGCTG-3′ and the reverse primer 5′-GAATTCTCAGTCCAGGATGGTCTTGA-3′ containing a NdeI and EcoRI restriction site, respectively. The PCR fragments were digested with NdeI and EcoRI and subsequently cloned into plasmid pET28a digested with the same enzymes. This resulted in the pET28a-hTERT expression vector in which the hTERT insert is flanked 5′ by a T7 RNA polymerase promoter.
The full-length (451 nt) RNA gene of human telomerase (hTR) was amplified from HeLa genomic DNA (Sigma) (26). A subsequent PCR amplification was carried out using the forward primer 5′-GGGGAAGCTTTAATACGACTCACTATAGGGTTGCGGAGGGTGGGCCTG-3′, containing a HindIII site followed by the promoter for the T7 RNA polymerase, and the reverse primer 5′-CCCCGGATCCTGCGCATGTGTGAGCCGAGTCCTGG-3′, bearing a BamHI restriction site. The PCR product was cloned into pUC18 for in vitro transcription, resulting in the pUC18-hTR+1 plasmid encoding for the wild-type RNA subunit of telomerase.
Site-directed mutagenesis
Mutations in hTR were performed on the pUC18-hTR+1 plasmid using the QuikChange site-directed mutagenesis kit according to the manufacturer’s recommendations (Stratagene). Primers 5′-CCATTTTTTGTCTAACCCTATGAGAAGGGCGTAGGCG-3′ (sense) and 5′-CGCCTACGCCCTTCTCATAGGGTTAGACAAAAAATGG-3′ (antisense) were used to amplify plasmid pUC18-hTR+1 and gave plasmid pUC18-hTR-Δ2 bearing deletions in the first two residues of the alignment domain (56CA55). The primer pair 5′-CCATTTTTTGTCTAACCCTATGTGAGAAGGGCGTAGGC-3′ (sense) and 5′-GCCTACGCCCTTCTCACATAGGGTTAGACAGTGG-3′ (antisense) was used to amplify plasmid pUC18-hTR+1 giving rise to plasmid pUC18-hTR-sub2 with substitutions in the first two residues of the alignment domain of telomerase RNA. Similar primer sets containing the corresponding modifications were used to generate the other hTR variants described in this paper. Thermocycling was for 30 s at 95°C, followed by 15 cycles of 30 s at 95°C, 1 min at 55°C and 7 min at 68°C. All mutations were checked by sequencing of the corresponding plasmids.
In vitro transcription and reconstitution of human telomerase activity
Plasmids containing the gene encoding the wild-type and mutant versions of the human telomerase RNA were linearised with BamHI prior to in vitro transcription. Plasmid pET28a-hTERT was digested using NotI. The transcription reaction (100 µl) contained 40 mM Tris–HCl, pH 8.0, 2 mM spermidine, 8 mM MgCl2, 50 mM NaCl, 30 mM dithiothreitol, 1 mM each NTP, 200 U RNAguard, 2 µg of linearized template DNA and 100 UT7 RNA polymerase (Stratagene). After 2 h incubation at 37°C, the DNA template was digested with 2.5 U/µg DNA of RNase-free DNase I (Amersham Pharmacia Biotech) for 10 min at 37°C. Free NTPs were removed by size exclusion chromatography (NAP-5 column; Amersham Pharmacia Biotech). Transcripts were precipitated with ethanol and dissolved in water prior to use. The integrity and size of the RNA transcripts were determined by staining with ethidium bromide. Telomerase activity was reconstituted using the nuclease-treated rabbit reticulocyte lysate system (RRL) (Promega) at 70% total concentration complemented with purified hTERT mRNA transcripts (25 ng/µl lysate) and wild-type or hTR variants (0.1 µg/µl lysate). Other components of the transcription mix were as recommended by the RRL manufacturer. The reaction mixtures were incubated for 90 min at 30°C and either assayed immediately for activity or quick frozen and kept at –78°C until use.
Primer extension assays of human telomerase activity
The TRAP assay (Intergen) was performed in 50 µl total volume containing 1× CHAPS buffer (20 mM Tris–HCl pH 8.3, 63 mM KCl, 1.5 mM MgCl2, 0.05% v/v Tween-20, 1 mM EGTA), 50 µM dNTPs, 2 µl of rabbit reticulocyte lysate unless otherwise mentioned and 0.1 µg of either 32P-radiolabeled TS primer (5′-AATCCGTCGAGCAGAGTT-3′) or 32P-radiolabeled primers identical in sequence to the original TS oligonucleotide other than the terminal 3′ nucleotides dGTT, which were substituted by either dGGT or dGGG. Each reaction mixture was incubated for 45 min at 30°C. The telomerase products were phenol/chloroform extracted and ethanol precipitated or alternatively purified using the QIAquick Nucleotide Removal Kit (Qiagen) and dissolved in PCR-grade water prior to amplification. PCR amplification was also carried out in 50 µl total volume including 1× CHAPS buffer, 2 U Taq polymerase (Amersham Pharmacia Biotech), 0.1 µg reverse primer [5′-GCGCGG(CTTACC)3CTAACC-3′], 0.01 amol internal control (IC) primer (5′-AATCCGTCGAGCAGAGTTAAAAGGCCGAGAAGCGAT-3′) and 0.1 µg IC specific reverse primer (5′-ATCGCTTCTCGGCCTTTT-3′). Each reaction mixture was placed in a pre-heated GENEAMP 2400 thermocycler block for 28 cycles at 94°C for 30 s, 59°C for 30 s and 72°C for 30 s, followed by 8 min at 72°C. The PCR products were separated on a 12.5% non-denaturing polyacrylamide gel. The gels were dried and exposed for autoradiography for 12–24 h. The reaction products were visualised and quantitated using a phosphorimager (Molecular Dynamic) and the densitometric analysis was performed with the ImageQuant v.3.0 software. The signal intensity in the RRL control [lysate (–hTR, +hTERT)] was subtracted as background. All the results given in this paper are the average from at least three independent experiments unless otherwise mentioned.
The modified version of the TRAP was performed as described by Aradi et al. (35) using 2 µl of RRL. The DNA products of telomerase extension were purified prior to amplification and analysed as described above. For a quantitative comparison of processivity, the processivity of a particular repeat i was defined as Pi = ∑(Ti + 1 + Ti + 2 + … + Tn)/∑(Ti + Ti + 1 + … + Tn), where Ti designated the signal of position Pi determined using a phosphorimager and n designated the highest number of repeats discernable on the telomerase ladder (35).
Competitive inhibition of the wild-type hTR–protein complex formation
Reconstitution of telomerase activity was performed as described above in the absence or presence of increasing amounts of competitor RNA (up to 50-fold molar excess). The competitor RNAs (Δ4 or Δ5) or non-specific human transfer RNA (tRNA from Sigma) were added to the RRL in conjunction with wild-type hTR and before translation of hTERT. The total amount of RNA within each reaction mix was kept constant at a concentration of 0.1 µg/µl lysate used. Activity was assayed immediately after incubation using 2 µl of lysate and quantified as described above.
RESULTS
Human telomerase elongates DNA substrates with tandemly repeated telomeric sequences d(GGTTAG) generating a characteristic ladder with a periodicity of 6 nt (18). The bands on the ladder arise from pausing and/or dissociation events during the extension process (18,36). In vitro co-expression of the RNA subunit (TR) and protein reverse transcriptase (TERT) component of telomerase in RRL suffices to reconstitute wild-type-like telomerase activity (37,38). Telomerase activity was reconstituted in vitro with a fixed amount (0.1 µg/µl lysate) of either wild-type or mutated RNAs using the RRL expression system. To study the alignment domain requirements, the effects of both deletions and substitutions at the 3′ end of the alignment domain on telomerase activity were evaluated (Table 1). An important aspect of the experimental design is that systematic alterations from the 3′ end simply reduce the length of the alignment sequence, but without causing internal mismatches or discontinuity between alignment and templating sequences, which may also interfere with telomerase function. Assays were performed by either the standard TRAP or an adapted protocol using recombinant telomerase (see Materials and Methods) (35,39). Products were separated by gel electrophoresis and quantified by integrating the radioactive signal of the ladder in each lane divided by the signal from the respective internal control as described by Kim and Wu (39). The activity for all the mutants was background corrected (see Materials and Methods) and normalised relative to telomerase activity reconstituted by wild-type hTR, which represents 100% activity.
Table 1. Nomenclature, modifications and activity of the human telomerase RNA mutants.
| hTR mutant | Specific nucleotide change | Alignment domain | Telomerase activity (%) (±SD) |
|---|---|---|---|
| Wild-type | None | 56CAAUC52 | 100 |
| Δ1 | Δ56C | AAUC52 | 84 ± 8 |
| Δ2 | Δ56CA | AUC52 | 43 ± 9 |
| Δ3 | Δ56CAA | UC52 | 0 |
| Δ4 | Δ56CAAU | C52 | 0 |
| Δ5 | Δ56CAAUC | None | 0 |
| Sub1 | 56C→G | AAUC52 | 77 ± 3 |
| Sub2 | 56CA→GT | AUC52 | 37 ± 7 |
| Sub3 | 56CAA→GTT | UC52 | 0 |
| Sub4 | 56CAAU→GTTA | C52 | 0 |
| Sub5 | 56CAAUC→GTTAG | None | 0 |
| RRL control | 0 |
Δ, deletion; Sub, substitution. The sequence of the alignment domain is written 3′→5′. The values for activity are background corrected with respect to the RRL control (which showed 3 ± 1% activity) and normalised with respect to the wild-type (taken as 100%). The values for activity represent the mean of at least three independent experiments.
3′-Terminal deletions in the alignment domain of human telomerase
The pentanucleotide alignment sequence 3′-56CAAUC52-5′ of human telomerase was systematically shortened by the deletion of residues from the 3′ end. The numbering of hTR nucleotides is based on the sequence described by Feng et al. (40). All the mutants constructed and discussed in this paper are given in Table 1.
Deletion of the first nucleotide 56C (mutant hereafter referred to as Δ1) decreased the catalytic activity to 84 ± 8% as measured by TRAP assay (Table 1 and Fig. 3, lane 6). Deletion of the dinucleotide sequence 3′-56CA55-5′ (Δ2) decreased activity to 43 ± 9% (Fig. 3, lane 7). Further shortening of the alignment domain by deletion of the tri- and tetranucleotide sequences 3′-56CAA54-5′ (Δ3) and 3′-56CAAU53-5′ (Δ4) respectively or deletion of all five residues 3′-56CAAUC52-5′ (Δ5) resulted in no detectable activity above background (Table 1 and Fig. 3, lanes 8–10, respectively). A low level of background activity (3%) endogeneous to the RRL was detectable (Fig. 3, lane 12) and all data were corrected accordingly (Table 1). Deletion of residue 56C does not significantly impair telomerase activity, suggesting that this residue makes a minor contribution to overall activity. Similarly, the dinucleotide sequence 56CA55 is required but not essential for overall activity. The progressive decrease in overall activity with reduced alignment domain reaches a critical cut-off point at 3 nt, corresponding to the remaining sequence 3′-54AUC52-5′, where further length reduction leads to complete loss of activity.
Figure 3.
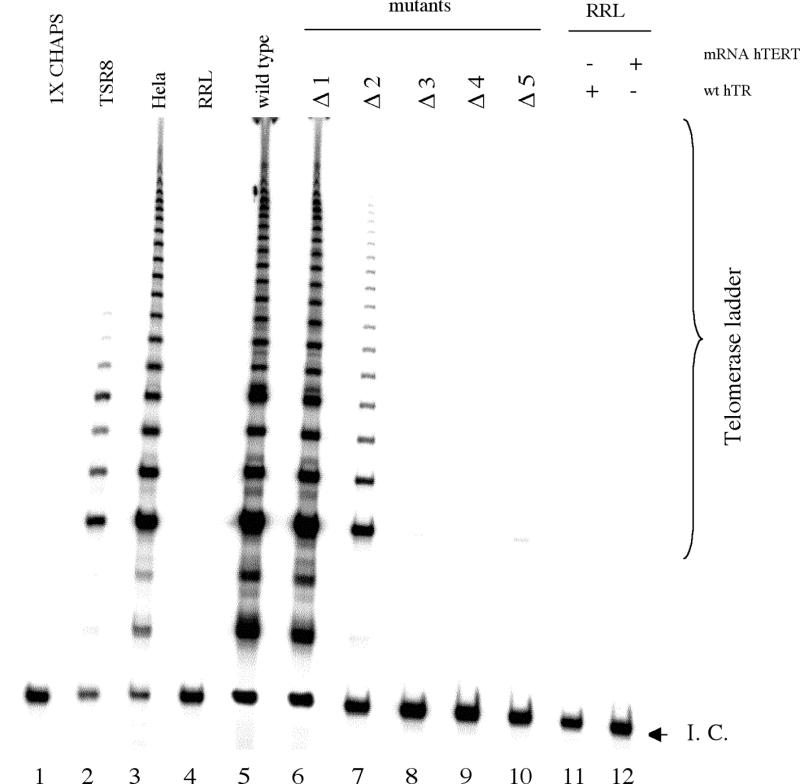
Effect of deleting residues 56C–52C from the 3′ end of the alignment domain on telomerase activity by TRAP assay. Lane 1, 1× CHAPS buffer; lane 2, TSR8 template amplification control; lane 3, wild-type telomerase from HeLa cell extracts; lane 4, RRL control (–hTR, –hTERT mRNA); lane 5, telomerase activity reconstituted from wild-type hTR; lanes 6–10, recombinant mutants Δ1, Δ2, Δ3, Δ4 and Δ5, respectively; lane 11, RRL control (+hTR, –hTERT mRNA); lane 12, RRL (–hTR, +hTERT mRNA). The internal PCR control band (I.C.) for the assay is indicated.
Analysis of the elongation products provides information on telomerase processivity. The TRAP assay conditions include a large excess of primer, such that repeats added to any given primer will result from a single primer-binding event, with synthesis effectively terminating for a given primer upon dissociation from the enzyme (39). Although the original distribution of DNA products from the telomerase extension is subject to scrambling (owing to multiple reverse primer-binding sites during the PCR amplification), the overall method occurs without further lengthening of the telomerase products (39). The TRAP may therefore provide a qualitative measure of processivity by counting the longest repeats on the ladder (39). Gel electrophoresis analysis of the telomerase ladder from wild-type telomerase and the Δ1 and Δ2 mutant enzymes revealed a comparable pattern of product elongation (Fig. 4, lanes 5–7, respectively). In particular, the longest products generated by the Δ1 and Δ2 mutants are comparable in size to those generated by the wild-type enzyme (approximately 20 added repeats), indicative of similar processivity. Therefore, deletion of residue 56C or of the dinucleotide 56CA55 does not significantly alter the processivity of human telomerase. Further confirmation of these results was obtained using a modified version of the TRAP specially developed by Szatmari and Aradi to analyse telomerase processivity (35). The assay includes a step which caps the 3′ ends of the telomerase extension products prior to PCR amplification. The PCR amplification uses a reverse primer complementary to the unique capping sequence and thus preserves both the initial size and length distribution of the telomerase extension products (35). Using this method, qualitative analysis of the products ladder indicated that the longest products generated by the Δ1 and Δ2 mutants were comparable to those generated by the wild-type enzyme (Fig. 4A, lanes 3, 4 and 6, respectively), consistent with the data obtained with the conventional TRAP assay (Fig. 3).
Figure 4.


(A) Activity and processivity of human telomerase mutants by a modified TRAP assay. Lane 1, 1× CHAPS control; lane 2, RRL without hTR; lane 3, recombinant telomerase activity reconstituted from wild-type hTR; lanes 4–13, recombinant telomerase activity reconstituted from variant telomerase RNAs, Δ1–Δ5 and Sub1–Sub5. For the standard assay conditions see Materials and Methods. The product bands selected for quantitative processivity analysis (Materials and Methods) are indicated by arrows. (B) Processivity of wild-type and mutants telomerases. P1 represents the processivity of wild-type and mutated telomerases at the first visible telomeric repeat; P2 is the processivity at the second repeat and so forth. See Materials and Methods for a description of quantitation and calculations performed. Reactions were performed in duplicate and the average processivity and standard deviation for each position were determined and plotted alongside the wild-type values.
A quantitative analysis of processivity was also carried out (as described in Materials and Methods) (35). The first and second visible repeat of a given ladder were chosen as representative of the short products and repeats 9 and 10 were chosen as the long products (Fig. 4A, arrows). The data are summarised in Figure 5B, where Pi designates the processivity of telomerase at a particular repeat i of the ladder. The processivity values P1, P2, P9 and P10 for the mutant enzymes Δ1 and Δ2 and wild-type recombinant telomerase were identical within experimental error (Fig. 4B). Taken together, these results further confirm that shortening of the alignment domain from the 3′ end by one (56C) or two (56CA55) residues does not alter the processivity of human telomerase.
Figure 5.

Effect of deleting residues 56C–52C from the 3′ end of the alignment domain on telomerase activity by TRAP assay using the alternative primer TS-GGG. Lane 1, telomerase activity reconstituted with wild-type hTR (wt hTR); lanes 2–6, recombinant mutants Δ1, Δ2, Δ3, Δ4 and Δ5, respectively. The percentage of telomerase activity reconstituted in each reaction is indicated relative to wild-type hTR under the respective lanes. The internal PCR control band (I.C.) for the assay is indicated.
PCR amplification of the telomerase products by the standard TRAP assay is restricted to detecting extension products that contain at least four telomeric repeats (39). Therefore, a poorly processive enzyme activity cannot be precluded for mutants Δ3–Δ5. In an attempt to detect low levels of poor processive activity for mutant enzymes Δ3, Δ4 and Δ5, we have either used 10-fold more enzyme in the assay to enhance detection sensitivity (data not shown) or employed the modified TRAP capable of amplifying a primer extended by as few as two telomeric repeats (Fig. 4A, lanes 8, 10 and 12, respectively) (35). Neither attempt resulted in detectable level of telomerase product for Δ3, Δ4 and Δ5 variants, thus reflecting a serious impairment of activity and/or processive synthesis for these mutant enzymes. In addition, wild-type and mutant telomerases (Δ1–Δ5) were also assayed with alternative primers (TS-GGT and TS-GGG) identical in sequence to the original TS except that the terminal 3′ nucleotides dGTT were substituted by the trinucleotides dGGT and dGGG, respectively. These primers are designed to base pair with the elongation domain only, which remained unchanged, and thus normalise the first round synthesis in all mutants (18,25). The levels and patterns of reconstituted catalytic activity observed were comparable to those described for the TS primer (Fig. 5), indicating that the effects of mutations are general to all three DNA substrates.
3′-Terminal substitutions of residues in the alignment domain of human telomerase
It is possible that the deletion of residues from the alignment domain of hTR may inadvertently disrupt telomerase activity by possibly frame-shifting the RNA template with respect to the active site of telomerase protein. To rule out this possibility, we generated and studied a series of RNA variants in which residues in the alignment domain have been substituted from the 3′ end by analogy with the deletions. For the substitution mutants, purines were replaced by pyrimidines and pyrimidines by purines (Table 1). The activity of reconstituted telomerase mutants was measured as described previously. Substitution of residue 56C by G (Sub1) decreased activity to 77 ± 3% (Table 1 and Fig. 6, lane 6). Substitution of 56CA55 by GT (Sub2) resulted in 37 ± 7% activity compared to wild-type (Fig. 6, lane 7). Further substitutions of the sequences 56CAA54 by GTT (Sub3), 56CAAU53 by GTTA (Sub4) and 56CAAUC52 by GTTAG (Sub5) in the alignment domain resulted in no telomerase activity detectable above background levels (Table 1 and Fig. 6, lanes 8–10). These results are identical, within experimental error, to those obtained for the corresponding deletion mutants and support the hypothesis that observed changes in activity result from shortening of the alignment sequence per se rather than non-specific effects due a change in register between the template and the active site of hTERT.
Figure 6.

Effect of substitutions of residues 56C–52C on telomerase activity as assayed by TRAP. Lane 1, 1× CHAPS buffer; lane 2, TSR8 template amplification; lane 3, wild-type telomerase from HeLa cell extracts; lane 4, RRL (–hTR, –hTERT mRNA); lane 5, telomerase activity reconstituted from wild-type hTR (wt hTR); lanes 6–10, Sub1, Sub2, Sub3, Sub4 and Sub5, respectively; lane 11, RRL (+hTR, –hTERT mRNA); lane 12, –hTR, +hTERT mRNA. The internal control (I. C.) for the assay is indicated.
We also assayed the mutant enzymes Sub3, Sub4 and Sub5 with the modified version of the TRAP (Fig. 4A, lanes 9, 11 and 13, respectively). In good agreement with the deletion experiments, we found that the Sub3, Sub4 and Sub5 mutants did not generate any detectable level of products above background, further indicating that the trinucleotide sequence 54AUC52 is apparently absolutely critical and represents the minimum length requirement of the alignment domain of human telomerase for efficient extension. Qualitative analysis of the telomerase ladder generated by the mutants Sub1 and Sub2 confirmed that these mutants and wild-type telomerase generated identical product profiles (Fig. 4A, lanes 3, 5 and 7). This was further supported by our quantitation data of processivity, whereby P1, P2, P9 and P10 of both mutants Sub1 and Sub2 were identical within experimental error to wild-type telomerase, further demonstrating that the processivity of telomerase is not sensitive to modifications of the most 3′ residues 56C and 56CA55 of the alignment domain (Fig. 4B).
Specific inhibition of human telomerase ribonucleoprotein formation
To preclude the possibility that changes in activity may result from a failure of mutant hTR to bind hTERT protein, we carried out a competition assay to determine the ability of the RNA variants to interact with hTERT (41,42). Specifically, we investigated whether the most drastically altered RNA variant Δ5 could compete with wild-type hTR for the formation of the RNA–hTERT complex, by testing the level of activity reconstituted in the absence and presence of competitor RNA (Fig. 7). Standard competition assays were performed with a fixed amount of total RNA (0.1 µg/µl lysate) in the presence of increasing amounts of the competitor RNA Δ5 (1-, 10-, 20- and 50-fold molar excess). Both RNAs were added simultaneously to the RRL prior to hTERT translation and the reconstituted telomerase activity measured by TRAP as described above. Telomerase activity reconstituted in the presence of Δ5 was significantly reduced in a concentration-dependent manner, indicating a specific competition with wild-type hTR for protein binding (Fig. 7, lanes 2–6). In particular, telomerase reconstituted in the presence of a 20-fold molar excess of competitor Δ5 was very weakly active relative to telomerase reconstituted with wild-type hTR alone at the corresponding concentration (0.005 µg/µl lysate) (Fig. 7, compare lanes 5 and 8, respectively). The variant RNA Δ4 was equally able to specifically compete with wild-type hTR (Fig. 7, lanes 9 and 10), whilst human tRNA (20-fold molar excess) had no effect on the telomerase RNA–protein complex formation (Fig. 7, lane 11). These results indicate that the deletion of four or five residues from the alignment sequence 56CAAUC52 does not preclude the ability of the RNA mutants to associate with hTERT. Therefore, the observation that certain hTR variants do not support catalytic activity in RRL are unlikely to be simply due to the inability of the RNA to bind the protein component of human telomerase.
Figure 7.

Specific inhibition of human telomerase activity by competitor RNAs Δ4 and Δ5. Standard competition assays (see Materials and Methods) were performed with increasing amounts of the indicated competitor RNA. Lane 1, RRL control (–hTR, –hTERT mRNA); lane 2, wild-type hTR incubated without competitor RNA (wt hTR); lanes 3–6, wild-type hTR incubated with increasing amounts (1-, 10-, 20- and 50-fold molar excess) of mutant RNA Δ5, 1- and 20-fold molar excess of variant Δ4 (lanes 9 and 10) and 20-fold molar excess of non-specific human tRNA (lane 11); lane 7, Δ5 mutant added alone (0.1 µg/µl lysate); lane 8, as lane 5 but wild-type hTR (0.005 µg/µl lysate) was incubated without competitor RNA. The competitor and wild-type hTR were added simultaneously to the lysate before synthesis of hTERT. The percentage of telomerase activity reconstituted in each reaction is indicated under the respective lanes. The internal control (I.C.) for the TRAP assay is indicated.
DISCUSSION
Telomerase shares significant structural (12,43) and mechanistic (44) traits with retroviral reverse transcriptases (RT) such as HIV-1 RT (45), but there are features that clearly distinguish it from conventional RTs (46). Most notably, telomerase carries an intrinsic RNA component which, in addition to structural functions, contains the template sequence that directs telomere repeat synthesis (Fig. 1) (40). The template region of human telomerase consists of an 11 nt sequence 3′-CAAUCCCAAUC-5′ complementary to nearly two telomeric repeats d(GGTTAG). The template sequences of other vertebrates vary in length, ranging from 11 to 8 nt, suggesting that 8 nt maybe sufficient for telomerase function (Fig. 2A) (26,47). Of these eight residues, six form a universally conserved (100% sequence identity) (26) elongation domain (3′-CCAAUC-5′) which codes for a single telomeric repeat (Fig. 2A). The alignment domain, contiguous to the 3′ end of the elongation domain, consists of additional nucleotides complementary to the telomeric sequence (Fig. 2A). Unlike the elongation domain, the alignment domain of telomerase is not completely conserved throughout vertebrates and varies in length from 2 nt (3′-UC-5′) for rodents to a maximum of 5 nt (3′-CAAUC-5′) for humans (Fig. 2A). This strongly suggests that not all nucleotides in the alignment domain of human telomerase are required for function.
Altering the sequence of the alignment domain of human telomerase at the first two positions from the 3′ end (56C and 56CA55, respectively) influences some aspects of telomerase activity. Deletion of the first 3′ residue (56C) of the alignment domain marginally reduced overall activity to 84 ± 8% whilst deletion of the sequence 56CA55 (mutant Δ2) had a more pronounced effect, but the decrease in overall activity was still of only ∼50% (Table 1 and Fig. 3). These observations demonstrate that the most 3′ terminal residues 56C and 55A of the alignment domain of human telomerase do contribute to the optimal function of telomerase but are not essential for catalytic activity in vitro. Each mutant was assayed with three alternative primers differing just in their three most 3′ terminal nucleotides. Two of these primers (TS-GGT and TS-GGG) were designed to base pair only with the elongation domain of human telomerase, which remained unaltered for all mutants (18,25). The initial primer recognition was therefore normalised during first round synthesis for all the mutants, which helps preclude changes in activity that would result purely from initiation. Comparable levels of activity and processivity profiles were generated with all three primers (Fig. 5), indicating that the variations in telomerase activity between the mutants reflect intrinsic biochemical properties of the core enzymes. The substitution mutants Sub1 and Sub2 show activities comparable to the corresponding deletions, which supports the hypothesis that the changes in activity relate to shortening of the alignment sequence, rather than a potential change in register between the RNA template and the catalytic core of the protein which might arise upon nucleotide deletion. This also indicates a tolerance in the hTR structure towards slight modifications in the region between the template and the upstream conserved pseudoknot structure (spanning nucleotides 57–64). This is consistent with the fact that the length of this particular region is not conserved in vertebrate telomerase RNA (26).
Interestingly, deletion or substitution of the first 1 or 2 nt 56C and 55A of the alignment sequence did not alter the processivity of the enzyme as compared to wild-type telomerase (Fig. 4). If repeat addition processivity is dependent on the re-alignment efficiency, one might have expected that a longer alignment domain, providing more base pairs, would favour repeat addition synthesis. The finding that processivity was insensitive to a loss of base-pairing potential from the first 2 nt, out of a total of 5 nt, of the alignment domain challenges this model. If DNA primers are re-aligned exclusively by base-pairing interaction during processive synthesis then loss of base pairing to both 56C and 55A would result in an estimated loss in free energy (ΔΔG°) of +1.9 kcal/mol (at 37°C) (48). Our data do not support that having a 4 or 5 nt long alignment domain is beneficial for processive synthesis of human telomerase, but rather that the 3 nt sequence 3′-54AUC52-5′ is sufficient for repeat addition with an efficiency comparable to wild-type telomerase. Non-base-pairing interactions between human telomerase and the DNA substrate may exist near to the alignment domain, as shown in yeast and Tetrahymena (49,50), cooperating with base pairing to position the 3′ end of the primer at the catalytic site. It is possible that such interactions could compensate for the difference in binding energy between the 3 and 5 bp hybrid duplex, thus allowing mutants Δ1, Δ2, Sub1 and Sub2 to display wild-type-like processivity.
Shortening of the alignment sequence beyond residue 55A had marked qualitative and quantitative effects on catalytic activity. In particular, the specific deletion or substitution of the trinucleotide 56CAA54 completely abolished enzymatic activity, clearly indicating that the minimal length of the alignment domain required to sustain human telomerase activity in vitro is three residues spanning nucleotides 52C–54A. Subsequently, mutants Δ4, Δ5, Sub4 and Sub5 were all judged inactive by both the conventional and modified TRAP assays. Despite the fact that none of the modified RNA residues were in a domain predicted to be involved in protein binding (36,51), we performed a competition assay to show that even the most significantly altered RNA variants, Δ4 and Δ5, bind specifically to telomerase protein with an affinity comparable to wild-type hTR (Fig. 7, lanes 2–10). Therefore, it is very unlikely that the defects in reconstituting telomerase activity with some of the RNA mutants was due to the inability of these RNA variants to associate with hTERT. In addition, increasing the quantity of enzyme used in the assays by 10-fold still showed no activity, suggesting a drop in activity of >2 orders of magnitude for mutants Δ4, Δ5, Sub4 and Sub5. Activity loss was independent of the three DNA primers used as substrate for telomerase extension, suggesting that the effect cannot be simply due to initial primer recognition. The modified TRAP, capable of detecting as few as two repeats addition (Fig. 4B, lanes 8–13), also failed to show activity. Taken together, the fact that an initiation problem during first round synthesis is unlikely and the absence of even two repeat addition products strongly suggests that human telomerase mutants with an alignment domain shorter than the trinucleotide sequence 3′-54AUC52-5′ is unable to carry out post-first round synthesis. Studies on Tetrahymena thermophila have shown that shortening the alignment domain from 3 (wild-type) to 2 and 1 nt greatly impair multiple repeat synthesis while sustaining catalytic activity (31). Although the sequence of the T.thermophila telomerase alignment domain is not totally conserved with that of human telomerase, both enzymes are known to be normally very processive, suggesting that a 3 nt alignment domain may be a general minimum requirement of telomerases for processive synthesis. This is also consistent with the observation that most rodent telomerases, which have only a 2 nt alignment domain, are typically non-processive in vitro (29). The dinucleotide sequence 3′-54UC52-5′ of the rodent telomerase alignment domain is the shortest known alignment sequence, which may be the minimum alignment domain for basic catalytic activity of telomerase.
The alignment domain and the anchor site are both important sites for primer interaction with the telomerase ribonucleoprotein (25). Interactions of the telomerase product with these binding sites are likely to determine whether re-alignment and further elongation or dissociation is most likely. Anchor site interactions may be especially important for telomerase function in ciliates, since the development of macronuclei and chromosome healing events frequently require the extension of DNA substrates whose 3′ termini are not complementary to the template sequence (22,24,52). However, little is known about the relative contribution of the anchor site and alignment domain interactions with the primers for human telomerase. Our results suggest that at least 3 bp of the DNA substrate with the RNA template is required for efficient re-alignment and processive synthesis. These results imply that deletion of three or more residues from the alignment domain may destabilise the product DNA-alignment domain heteroduplex favouring product release from the enzyme. In light of our results, it would appear that for processivity control in human telomerase, the anchor site might only play a role secondary to the alignment domain. This is in good agreement with observations previously made that a strong interaction of human telomerase at the anchor site is insufficient to overcome a limited base-pairing interaction between the primer 3′ end and the template (25). By gradually deleting residues from the 3′ end of the DNA primer, Morin demonstrated that as few as two base-pairing interactions in the last 4 nt suffice for initiation and elongation by telomerase. While those primer modifications only affected first round synthesis (i.e. primer recognition), our study complements these findings and suggest that at least three base-pairing interactions are required for efficient synthesis in subsequent rounds of primer extension.
Detailed functional analysis of the template residues in telomerase have previously been limited largely to ciliates, including T.thermophila and Euplotes crassus (53). This is the first study towards a detailed elucidation of the role of specific template residues in the function of human telomerase. Ideally, the recombinant human telomerase mutants described in this paper will be studied by a direct assay, for which there is no routine methodology yet established, owing to difficulties in expression and reconstitution. Consequently, we cannot absolutely rule out the possibility that substitution or deletion in the alignment domain of human telomerase may affect catalytic activity in ways that do not manifest under the conditions of our assays. There also exists the possibility that certain residues in the alignment domain of human telomerase may contribute to enzyme catalytic activity in other ways besides base pairing, such as the structure of the catalytic pocket. Our results do indicate that the integrity in both length and sequence of the alignment domain of human telomerase contributes to optimal overall activity, but with only a minor contribution from the first two 3′ residues in vitro. On the basis of this study, we conclude that three residues (3′-54AUC52-5′) are the minimum for efficient processive repeat addition by human telomerase. Our results are consistent with the level of sequence conservation in the alignment domain of vertebrate telomerase RNAs (Fig. 3B), with the least conserved (34%) residue of the 11 nt template region 56C not being essential. The next two residues in the alignment domain, 55A and 54A, respectively, are both 90% conserved (Fig. 2B). Interestingly, mutants with modification of the dinucleotide sequence 56CA55 show significant telomerase activity and high processivity, which supports the hypothesis that strongly conserved residue 55A is actually not essential for in vitro activity. However, replacement of both 55A and 54A abolished telomerase activity (Δ3 and Sub3), which is consistent with the conservation of both residues. Reasons for the presence of additional (>3) alignment domain residues of certain vertebrates, including human, are unclear. One possibility is that a more optimal structure in the RNA template for active site function results from the additional residues. Alternatively, there may exist an unknown in vivo function for these additional residues in certain organisms.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Robert Weinberg for plasmid pCl-Neo-hTERT. We also thank Dr Wjatschesslaw Wlassoff and Andrew Whitney for helpful discussions. This work was funded by a grant from the BBSRC and the Cambridge European Trust.
REFERENCES
- 1.Greider C.W. and Blackburn,E.H. (1985) Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell, 43, 405–413. [DOI] [PubMed] [Google Scholar]
- 2.Zakian V.A. (1995) Telomeres: beginning to understand the end. Science, 270, 1601–1607. [DOI] [PubMed] [Google Scholar]
- 3.Blackburn E.H. and Grieder,C.W. (1995) Telomeres. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 4.Blackburn E.H. (1991) Structure and function of telomeres. Nature, 350, 569–573. [DOI] [PubMed] [Google Scholar]
- 5.Gottschling D.E. and Zakian,V.A. (1986) Telomere proteins: specific recognition and protection of the natural termini of Oxytricha macronuclear DNA. Cell, 47, 195–205. [DOI] [PubMed] [Google Scholar]
- 6.Sandell L.L. and Zakian,V.A. (1993) Loss of a yeast telomere: arrest, recovery, and chromosome loss. Cell, 75, 729–739. [DOI] [PubMed] [Google Scholar]
- 7.Bodnar A.G., Ouellette,M., Frolkis,M., Holt,S.E., Chiu,C.P., Morin,G.B., Harley,C.B., Shay,J.W., Lichtsteiner,S. and Wright,W.E. (1998) Extension of life-span by introduction of telomerase into normal human cells. Science, 279, 349–352. [DOI] [PubMed] [Google Scholar]
- 8.Counter C.M., Meyerson,M., Eaton,E.N., Ellisen,L.W., Caddle,S.D., Haber,D.A. and Weinberg,R.A. (1998) Telomerase activity is restored in human cells by ectopic expression of hTERT (hEST2), the catalytic subunit of telomerase. Oncogene, 16, 1217–1222. [DOI] [PubMed] [Google Scholar]
- 9.Holt S.E. and Shay,J.W. (1999) Role of telomerase in cellular proliferation and cancer. J. Cell Physiol., 180, 10–18. [DOI] [PubMed] [Google Scholar]
- 10.Kim N.W., Piatyszek,M.A., Prowse,K.R., Harley,C.B., West,M.D., Ho,P.L., Coviello,G.M., Wright,W.E., Weinrich,S.L. and Shay,J.W. (1994) Specific association of human telomerase activity with immortal cells and cancer. Science, 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- 11.Herbert B., Pitts,A.E., Baker,S.I., Hamilton,S.E., Wright,W.E., Shay,J.W. and Corey,D.R. (1999) Inhibition of human telomerase in immortal human cells leads to progressive telomere shortening and cell death. Proc. Natl Acad. Sci. USA, 96, 14276–14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lingner J., Hughes,T.R., Shevchenko,A., Mann,M., Lundblad,V. and Cech,T.R. (1997) Reverse transcriptase motifs in the catalytic subunit of telomerase. Science, 276, 561–567. [DOI] [PubMed] [Google Scholar]
- 13.Counter C.M., Meyerson,M., Eaton,E.N. and Weinberg,R.A. (1997) The catalytic subunit of yeast telomerase. Proc. Natl Acad. Sci. USA, 94, 9202–9207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakamura T.M., Morin,G.B., Chapman,K.B., Weinrich,S.L., Andrews,W.H., Lingner,J., Harley,C.B. and Cech,T.R. (1997) Telomerase catalytic subunit homologs from fission yeast and human. Science, 277, 955–959. [DOI] [PubMed] [Google Scholar]
- 15.Harrington L., McPhail,T., Mar,V., Zhou,W., Oulton,R., Bass,M.B., Arruda,I. and Robinson,M.O. (1997) A mammalian telomerase-associated protein. Science, 275, 973–977. [DOI] [PubMed] [Google Scholar]
- 16.Holt S.E., Aisner,D.L., Baur,J., Tesmer,V.M., Dy,M., Ouellette,M., Trager,J.B., Morin,G.B., Toft,D.O., Shay,J.W. et al. (1999) Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev., 13, 817–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Collins K., Kobayashi,R. and Greider,C.W. (1995) Purification of Tetrahymena telomerase and cloning of genes encoding the two protein components of the enzyme. Cell, 81, 677–686. [DOI] [PubMed] [Google Scholar]
- 18.Morin G.B. (1989) The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell, 59, 521–529. [DOI] [PubMed] [Google Scholar]
- 19.Cohn M. and Blackburn,E.H. (1995) Telomerase in yeast. Science, 269, 396–400. [DOI] [PubMed] [Google Scholar]
- 20.Shippen-Lentz D. and Blackburn,E.H. (1990) Functional evidence for an RNA template in telomerase. Science, 247, 546–552. [DOI] [PubMed] [Google Scholar]
- 21.Cech T.R. (2000) Life at the end of the chromosome: telomere and telomerase. A review. Angew. Chem. Int. Ed., 39, 35–43. [DOI] [PubMed] [Google Scholar]
- 22.Greider C.W. and Blackburn,E.H. (1987) The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell, 51, 887–898. [DOI] [PubMed] [Google Scholar]
- 23.Hammond P.W., Lively,T.N. and Cech,T.R. (1997) The anchor site of telomerase from Euplotes aediculatus revealed by photo-cross-linking to single- and double-stranded DNA primers. Mol. Cell. Biol., 17, 296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harrington L.A. and Greider,C.W. (1991) Telomerase primer specificity and chromosome healing. Nature, 353, 451–454. [DOI] [PubMed] [Google Scholar]
- 25.Morin G.B. (1991) Recognition of a chromosome truncation site associated with alpha-thalassaemia by human telomerase. Nature, 353, 454–456. [DOI] [PubMed] [Google Scholar]
- 26.Chen J.L., Blasco,M.A. and Greider,C.W. (2000) Secondary structure of vertebrate telomerase RNA. Cell, 100, 503–514. [DOI] [PubMed] [Google Scholar]
- 27.Autexier C. and Greider,C.W. (1994) Functional reconstitution of wild-type and mutant Tetrahymena telomerase. Genes Dev., 8, 563–575. [DOI] [PubMed] [Google Scholar]
- 28.Maine I.P., Chen,S.F. and Windle,B. (1999) Effect of dGTP concentration on human and CHO telomerase. Biochemistry, 38, 15325–15332. [DOI] [PubMed] [Google Scholar]
- 29.Prowse K.R., Avilion,A.A. and Greider,C.W. (1993) Identification of a nonprocessive telomerase activity from mouse cells. Proc. Natl Acad. Sci. USA, 90, 1493–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gilley D., Lee,M.S. and Blackburn,E.H. (1995) Altering specific telomerase RNA template residues affects active site function. Genes Dev., 9, 2214–2226. [DOI] [PubMed] [Google Scholar]
- 31.Gilley D. and Blackburn,E.H. (1996) Specific RNA residue interactions required for enzymatic functions of Tetrahymena telomerase. Mol. Cell. Biol., 16, 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Autexier C. and Greider,C.W. (1995) Boundary elements of the Tetrahymena telomerase RNA template and alignment domains. Genes Dev., 9, 2227–2239. [DOI] [PubMed] [Google Scholar]
- 33.Autexier C. and Greider,C.W. (1998) Mutational analysis of the Tetrahymena telomerase RNA: identification of residues affecting telomerase activity in vitro. Nucleic Acids Res., 26, 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meyerson M., Counter,C.M., Eaton,E.N., Ellisen,L.W., Steiner,P., Caddle,S.D., Ziaugra,L., Beijersbergen,R.L., Davidoff,M.J., Liu,Q. et al. (1997) hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell, 90, 785–795. [DOI] [PubMed] [Google Scholar]
- 35.Szatmari I. and Aradi,J. (2001) Telomeric repeat amplification, without shortening or lengthening of the telomerase products: a method to analyze the processivity of telomerase enzyme. Nucleic Acids Res., 29, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamilton S.E., Pitts,A.E., Katipally,R.R., Jia,X., Rutter,J.P., Davies,B.A., Shay,J.W., Wright,W.E. and Corey,D.R. (1997) Identification of determinants for inhibitor binding within the RNA active site of human telomerase using PNA scanning. Biochemistry, 36, 11873–11880. [DOI] [PubMed] [Google Scholar]
- 37.Weinrich S.L., Pruzan,R., Ma,L., Ouellette,M., Tesmer,V.M., Holt,S.E., Bodnar,A.G., Lichtsteiner,S., Kim,N.W., Trager,J.B. et al. (1997) Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nature Genet., 17, 498–502. [DOI] [PubMed] [Google Scholar]
- 38.Beattie T.L., Zhou,W., Robinson,M.O. and Harrington,L. (1998) Reconstitution of human telomerase activity in vitro. Curr. Biol., 8, 177–180. [DOI] [PubMed] [Google Scholar]
- 39.Kim N.W. and Wu,F. (1997) Advances in quantification and characterization of telomerase activity by the telomeric repeat amplification protocol (TRAP). Nucleic Acids Res., 25, 2595–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feng J., Funk,W.D., Wang,S.S., Weinrich,S.L., Avilion,A.A., Chiu,C.P., Adams,R.R., Chang,E., Allsopp,R.C., Yu,J. et al. (1995) The RNA component of human telomerase. Science, 269, 1236–1241. [DOI] [PubMed] [Google Scholar]
- 41.Autexier C. and Triki,I. (1999) Tetrahymena telomerase ribonucleoprotein RNA–protein interactions. Nucleic Acids Res., 27, 2227–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bachand F., Triki,I. and Autexier,C. (2001) Human telomerase RNA–protein interactions. Nucleic Acids Res., 29, 3385–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harrington L., Zhou,W., McPhail,T., Oulton,R., Yeung,D.S., Mar,V., Bass,M.B. and Robinson,M.O. (1997) Human telomerase contains evolutionarily conserved catalytic and structural subunits. Genes Dev., 11, 3109–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bosoy D. and Lue,N.F. (2001) Functional analysis of conserved residues in the putative “Finger” domain of telomerase reverse transcriptase. J. Biol. Chem., 276, 46305–46312. [DOI] [PubMed] [Google Scholar]
- 45.Peng Y., Mian,I.S. and Lue,N.F. (2001) Analysis of telomerase processivity: mechanistic similarity to HIV-1 reverse transcriptase and role in telomere maintenance. Mol. Cell, 7, 1201–1211. [DOI] [PubMed] [Google Scholar]
- 46.Beattie T.L., Zhou,W., Robinson,M.O. and Harrington,L. (2000) Polymerization defects within human telomerase are distinct from telomerase RNA and TEP1 binding. Mol. Biol. Cell, 11, 3329–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blasco M.A., Funk,W., Villeponteau,B. and Greider,C.W. (1995) Functional characterization and developmental regulation of mouse telomerase RNA. Science, 269, 1267–1270. [DOI] [PubMed] [Google Scholar]
- 48.Sugimoto N., Nakano,S., Katoh,M., Matsumura,A., Nakamuta,H., Ohmichi,T., Yoneyama,M. and Sasaki,M. (1995) Thermodynamic parameters to predict stability of RNA/DNA hybrid duplexes. Biochemistry, 34, 11211–11216. [DOI] [PubMed] [Google Scholar]
- 49.Collins K. and Greider,C.W. (1995) Utilization of ribonucleotides and RNA primers by Tetrahymena telomerase. EMBO J., 14, 5422–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lue N.F. and Peng,Y. (1998) Negative regulation of yeast telomerase activity through an interaction with an upstream region of the DNA primer. Nucleic Acids Res., 26, 1487–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Antal M., Boros,E., Solymosy,F. and Kiss,T. (2002) Analysis of the structure of human telomerase RNA in vivo. Nucleic Acids Res., 30, 912–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bednenko J., Melek,M., Greene,E.C. and Shippen,D.E. (1997) Developmentally regulated initiation of DNA synthesis by telomerase: evidence for factor-assisted de novo telomere formation. EMBO J., 16, 2507–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Collins K. (1999) Ciliate telomerase biochemistry. Annu. Rev. Biochem., 68, 187–218. [DOI] [PubMed] [Google Scholar]