


Abstract
The salivarian trypanosome Trypanosoma brucei infects mammals and is transmitted by tsetse flies. The mammalian ‘bloodstream form’ trypanosome has a variant surface glycoprotein coat and relies on glycolysis while the procyclic form from tsetse flies has EP protein on the surface and has a more developed mitochondrion. We show here that the mRNA for the procyclic-specific cytosolic phosphoglycerate kinase PGKB, like that for EP proteins, contains a regulatory AU-rich element (ARE) that destabilises the mRNA in bloodstream forms. The human HuR protein binds to, and stabilises, mammalian mRNAs containing AREs. Expression of HuR in bloodstream-form trypanosomes resulted in growth arrest and in stabilisation of the EP, PGKB and pyruvate, phosphate dikinase mRNAs, while three bloodstream-specific mRNAs were reduced in abundance. The synthesis and abundance of unregulated mRNAs and proteins were unaffected. Our results suggest that regulation of mRNA stability by AREs arose early in eukaryotic evolution.
INTRODUCTION
Protist parasites of the order Kinetoplastida cause medically and agriculturally important diseases of man, animals and plants. Many kinetoplastid species rely on an arthropod vectors for transmission between hosts, and adaptation to the different host and vector environments depends on substantial alterations in surface molecules and metabolism. The salivarian trypanosome Trypanosoma brucei, which causes sleeping sickness in humans and Nagana in cattle, is transmitted by tsetse flies. In mammalian blood, the long slender bloodstream trypomastigote form of the parasite relies exclusively on glycolysis for ATP synthesis, and the organisms protect themselves from the host humoral immune system by means of a variant surface glycoprotein (VSG) coat (1). Differentiation starts in the mammal, when a fraction of the parasites change to the non-dividing stumpy bloodstream trypomastigote form (reviewed in 2). Stumpy parasites have more elaborate mitochondria than the long slender bloodstream forms (3) and may be a differentiation intermediate (2,4). Upon entry into the midgut of the tsetse fly, the parasites change to procyclic trypomastigotes, which have extensive mitochondrial function. Procyclic trypanosomes completely lack VSG. Instead the surface is covered with glutamate– proline rich proteins encoded by a small family of two GPEET and six to eight EP genes (5,6).
The kinetoplastids branched very early in eukaryotic evolution so exhibit several remarkable deviations from standard eukaryotic paradigms. Genome organisation is strange in that many open reading frames succeed each other in a head-to-tail arrangement (7). Genes exhibiting completely different regulation may be only 100–200 bp apart, there is no evidence for specific or regulated initiation by RNA polymerase II, and most open reading frames investigated so far seem to be constitutively transcribed (8). Individual mRNAs are excised from polycistronic primary transcripts by trans splicing, so that all mRNAs bear the same trans-spliced leader at the 5′ end. Polyadenylation of each transcript is mechanistically, spatially and temporally coupled to the nearest downstream trans-splicing reaction (8–10).
Given this extraordinary arrangement, post-transcriptional regulation of gene expression is of paramount importance for parasite survival and transmission. Usually, the sequences responsible for regulation of transcript abundance and translation are found in the 3′-untranslated region (UTR) of the mRNA, and transcript abundance is determined by mRNA stability (8). For example, the PGKB gene, which encodes a procyclic-specific phosphoglycerate kinase, is directly upstream of, and co-transcribed with, the PGKC gene, which encodes a bloodstream-specific phosphoglycerate kinase. PGKB is located in the cytosol and PGKC is targeted to a microbody, the glycosome, and inappropriate expression of PGKB in bloodstream forms inhibits growth (11). The regulation of PGK expression lies in the 3′-UTRs of the PGK genes, which give stage-specific expression when appended to a chloramphenicol acetyltransferase (CAT) gene (12); developmental regulation of trans splicing is apparently not involved (13).
Both the VSG genes and the EP/GPEET gene clusters are transcribed by RNA polymerase I (14). VSG gene transcription is shut off in procyclic forms (15 and references therein), whereas EP/GPEET gene transcription is down-regulated just 10-fold in bloodstream forms (16). The EP and GPEET proteins are undetectable in bloodstream forms because the mRNAs are unstable and poorly translated (17,18). The 3′-UTRs of all EP and GPEET mRNAs contain a U-rich 26mer with interspersed A residues which is in an unstable or single-stranded conformation (19). This element is required for RNA instability and repressed translation in bloodstream forms (17,20), and its activity is impaired by mutation of the U residues (17). Degradation of a reporter gene transcript bearing the EP1 3′-UTR is initiated by complete destruction of the 3′-UTR, either via an endonuclease cleavage in the U-rich element or by extremely rapid deadenylation and 3′→5′ exonuclease action (18). In contrast, the mechanism of degradation of unregulated mRNAs in trypanosomes resembles the major mechanism described for yeast and mammals: CAT genes bearing the 3′-UTR from the constitutively expressed actin (ACT) gene or a mutant version of the EP1 3′-UTR lacking the instability element (ep1Δ26) are gradually deadenylated, then are degraded by both 3′→5′ and 5′→3′ exonucleases (18).
The U-rich 26mer instability element, and a similar regulatory element from the mucin genes of the related parasite Trypanosoma cruzi (21), resemble AU-rich elements (AREs) that are found in the 3′-UTRs of transiently expressed proto-oncogenes, lymphokines and cytokines in mammalian cells (22,23). The mechanism of ARE-mediated decay in mammals is not entirely clear (23) and it may be that several processes are influenced by AREs. There is very clear evidence that AREs can stimulate deadenylation (22) and 3′→5′ exonucleolytic degradation (24,25), but it is also possible that they may be attacked by an endonuclease (26–28). The human HuR protein, which is a 36 kDa polypeptide that contains three classical RNA recognition motifs (RRMs) (29,30), binds AREs and U-rich sequences that are in a single-stranded conformation (31–33). Overexpression of HuR in mammalian cells, or inclusion of HuR in a mammalian in vitro degradation assay, lead to stabilisation of reporter mRNAs carrying AREs in their 3′-UTRs (27,28,34).
Recently, ARE-mediated mRNA decay has also been reported in Saccharomyces cerevisiae. The yeast AREs stabilise transcripts in glucose-containing media and the decay is influenced by a HuR-like yeast protein, Pub1p (35), which inhibits deadenylation and subsequent mRNA decapping.
In this study, we show that an ARE in the 3′-UTR of the PGKB mRNA, like that in the EP1 mRNA, stimulates mRNA degradation in bloodstream forms. To investigate the relationship between ARE-mediated degradation in trypanosomes and mammals, we investigated the effects of HuR expression on the abundance of ARE-containing and control mRNAs in trypanosomes.
MATERIALS AND METHODS
Trypanosomes and transfection
Bloodstream and procyclic forms of T.brucei were cultured and transfected as previously described (36,37). The two strains used, BF427-449 and BF927-853, express the bacterial Tet repressor from plasmids pHD449 or pHD853 integrated at the tubulin locus (36,37). Line BF427-449 is not capable of full differentiation into growing procyclic forms. The BF927-853 line is differentiation competent, but the level of Tet repressor will be low in procyclic forms (36). Bloodstream-form 427 trypanosomes expressing phage T7 polymerase (BF427-328) were also used (38).
Plasmid constructs
All plasmids for trypanosomatid expression used are listed in Table 1, and reconstructed sequences are available from the authors. Details of the trypanosomatid expression vectors can be found at http://www.zmbh.uni-heidelberg.de/Clayton/CC_pl.html. Cloning and mutagenesis were all done by PCR using oligonucleotides with appropriate restriction sites, with either plasmid clones or genomic DNA as templates.
Table 1. Plasmids employed in this study.
| pHD no. | Vector plasmid | Promoter | Coding region | 3′-UTR |
|---|---|---|---|---|
| pHD1099 | pHD615 | PEP1Ti | HuR | VSG |
| pHD1138 | pHD617 | PEP1Ti | HuR | ACT |
| pHD869 | pHD774 | PT7 | CAT | PGKB |
| pHD1044 | pHD774 | PT7 | CAT | pgkbΔ(I+II) |
| pHD1139 | pHD774 | PT7 | CAT | pgkbΔII |
| pHD1147 | pHD1034 | PRRNA | CAT | PGKB |
| pHD1148 | pHD1034 | PRRNA | CAT | pgkbΔ(I+II) |
| pHD1149 | pHD1034 | PRRNA | CAT | EP1 |
| pHD1150 | pHD1034 | PRRNA | CAT | ep1Δ26mer |
| pHD1317 | pHD1034 | PRRNA | CAT | PPDK |
| pHD1320 | pHD1034 | PRRNA | CAT | ppdkΔARE700 |
| pHD1318 | pHD1034 | PRRNA | CAT | ppdk528-1002 |
| pHD1319 | pHD1034 | PRRNA | CAT | ppdk528-1002ΔARE700 |
PEP1Ti, EP1 gene promoter with two Tet operator sequences; PRRNA, ribosomal RNA promoter; PT7, T7 polymerase promoter; I and II, two different single-stranded RNA structures predicted in the 3′-UTR of the PGKB gene.
For inducible expression of the HuR protein we used trypanosome expression vectors bearing an EP1 promoter with two tet operators (37). For expression in bloodstream forms, the HuR gene was followed by a VSG 3′-UTR (pHD1099); for procyclic forms, the ACT 3′-UTR was used (37). The coding region of HuR was amplified from a plasmid kindly provided by H. Furneaux (30). The reporter gene constructs for analysis of the PGKB, PPDK and EP1 3′-UTRs were constructed either using plasmid pHD1034, which has a ribosomal RNA promoter, a CAT cassette and a puromycin resistance cassette (C.Hartmann and C.Clayton, unpublished results) or plasmid pHD774, which is similar but has a hygromycin resistance cassette and a T7 promoter (18).
Northern blots and RNA analyses
Trypanosoma brucei RNA was isolated with peqGOLD TriFast™ (peqlab, GmbH). Five micrograms per lane of total RNA were resolved in 1% agarose–formaldehyde gels, transferred to nylon membranes (Nytran-N; Schleicher & Schuell, GmbH) using a Turboblotter™ apparatus (Schleicher & Schuell, GmbH) and hybridised with specific probes. Quantification was done by scanning densitometry or by PhosphorImager. Polyadenylation sites were mapped by RT–PCR (39). Transcription was stopped with 10 µg/ml actinomycin D. RNA folding predictions were generated by the MFOLD programme (Madison, WI) using the default parameters.
Bandshift assay and footprinting
For bandshifts, the EP1 and ep1Δ26 3′-UTRs (17) were amplified from plasmids pHD1 and pHD664 using primers cz841 and cz840 (19). The forward primer, cz841, includes a T7 RNA promoter sequence. The 3′-UTR of the c-fos gene and its ARE-deleted version were prepared from plasmid DNAs, named pAUF and pAUF1/2, provided by H. Furneaux (40). PCR products and linearised plasmids were used for in vitro transcription with [32P]UTP (800 Ci/mmol) (Amersham) using a MAXIscript™ kit (Ambion, Inc.). Full-length transcripts were gel purified. The analyses were done using purified GST-HuR, as described (30), except that after electrophoresis the gel was transferred to a nylon membrane as for northern blots, and exposed to film.
For the footprinting, a single nucleotide ladder was obtained by hydrolysing purified EP1 3′-UTR RNA (19) in 5 vol of formamide/0.5 mM Mg2+ at 100°C for 10 min. The G ladder (T1d in Fig. 3) was obtained by partial digestion of RNA with T1 ribonuclease at 0.1 U/µl under semi-denaturing conditions (buffer 10 mM sodium citrate, 3.5 M urea) at 56°C for 20 min (19). One microlitre of binding buffer (300 mM NaCl, 100 mM Tris pH 7.8, 0.4 µg/µl yeast tRNA) and 1 µl of GST-HuR [in increasing concentrations with 0.6 µg/µl BSA (Sigma)] were added to 1 µl of labelled EP1 RNA (19), mixed and incubated for 10 min at 37°C. After that, 3 µl of T2 (end concentration 0.1 U/µl) were added and incubated for 10 min at room temperature. RNA was extracted and separated on 10% polyacrylamide/7.5 M urea gels at 35 W.
Figure 3.

The EP1 U-rich element binds HuR. (A) Band-shift. An RNA containing a classical ARE (AUF), a version with the ARE deleted (AUF 1/2), EP1 and ep1Δ26 probes were incubated with increasing amounts of GST-HuR and complexes analysed by PAGE. The control lane (labelled C) shows incubation with GST alone (500 nM). Amounts of GST-HuR were, for the AUF panels, 0, 10, 50, 200 and 400 nM; for the EP1 lanes, 0, 2, 10, 50, 100, 200 and 400 nM. (B) Sequence of the EP1 3′-UTR. The nucleotides corresponding to the 26mer are boxed. (C) Footprint of GST-HuR on the EP1 3′-UTR. A 5′-end-labelled EP1 3′-UTR probe was incubated with increasing amounts of GST-HuR and subjected to T2 RNase degradation. After incubation, the resulting fragments were resolved in an acrylamide gel for analysis. Lanes 1, 2, 3, 4, 5 correspond to 0, 2.6, 26.6, 66.6 and 133.3 nM of GST-HuR, digested with T2 ribonuclease; lane C, control reaction without T2 ribonuclease; lane Ld, formamide ladder; and lane T1, limited T1 ribonuclease digestion under semi-denaturing conditions.
Protein analysis
For western blotting, proteins from 2 × 106 cells were fractionated by SDS–PAGE and transferred to a nitrocellulose membrane (Optitran BA-S85; Schleicher & Schuell). Proteins were detected with mouse monoclonal anti-human HuR antibody 19F12 (1/5000 dilution) (gift of H. Furneaux); mouse monoclonal anti-T.brucei procyclin (1/2000 dilution) (CLP001A; Cedarlane); polyclonal rabbit anti-T.cruzi dihydrolipoamide dehydrogenase (DHLADH) (1/1000 dilution, from L. Krauth-Siegel); rabbit polyclonal anti-T.brucei PGK (1/1000 dilution, from P. Michels); rabbit polyclonal anti-T.brucei aldolase (1/1000 dilution); and rabbit anti-Hsp60 (Stressgen, Victoria, Canada, product no. SPA-804), and detected by ECL™ (Amersham). Quantification was performed by scanning, followed by the use of Imagequant software.
Metabolic labelling was done at 5 × 107 cells/ml at 37°C for 2 h in MEM with 10 mM glucose, 1 mM cysteine, and 14 µl/l 2-mercaptoethanol, and [35S]methionine (>1000 Ci/mmol) at 250 µCi/ml. Total cell lysates were electrophoresed on SDS–PAGE gels, the gels were washed three times in methanol/acetic acid then soaked in Entensify® (NEN™), dried, and exposed to film.
RESULTS
The PGKB 3′-UTR contains a regulatory ARE-like instability element
The cytosolic phosphoglycerate kinase PGKB is expressed exclusively in procyclic forms. The 3′-UTR of the PGKB mRNA is ∼160 nt long (Fig. 1A) with several alternative polyadenylation sites (determined by RT–PCR, data not shown). The 3′-UTR includes a 46 nt region with a high U content (positions 88–133) (Fig. 1A). Flanking several stretches of Us there are A residues, in one case giving the typical AUUUA pentamer described for AREs. Secondary-structure predictions for the PGKB 3′-UTR (Fig. 1B) suggest that the PGKB ARE is likely to be single stranded, forming two domains separated by a short stretch of four to five paired nucleotides. To test whether this element influences mRNA stability in bloodstream forms, we first cloned the intergenic region (from the stop codon of the PGKB gene to the initiation codon of the PGKC gene) downstream of a CAT reporter gene that was driven by a T7 promoter (Fig. 1C). The construct was transfected into bloodstream-form trypanosomes which constitutively express T7 polymerase. To analyse the role of the putative ARE we deleted either the whole ARE or region II (Fig. 1C, top). The steady-state levels of the reporter mRNAs were assessed by northern blotting and the CAT activity was measured (Fig. 1C). Both deletions resulted in an increased abundance of the transcripts and increased CAT activity, relative to the construct containing the intact PGKB 3′-UTR. All constructs were transcribed from the T7 promoter and contained identical 5′ trans splicing and polyadenylation signals, so neither transcription nor processing should be affected by the deletions. The results therefore indicate that the ARE-like sequence in the PGKB 3′-UTR is required for destabilisation of the mRNA. Thus, the PGKB ARE resembles the 26mer in the 3′-UTR of the EP1 gene both structurally and functionally.
Figure 1.

Analysis of an mRNA-instability element present in the 3′-UTR of the PGKB gene. (A) Sequence of the 3′-UTR of the PGKB mRNA. Polyadenylation sites are indicated by arrows. Regions I and II, which contain poly-U tracts, are boxed. (B) Detail of a representative secondary structure predicted for a relevant region of the 3′-UTR of PGKB. The single-stranded regions actually extend beyond regions I and II. (C) Role of the PGKB ARE in determining mRNA levels. Total RNA was taken from parasites expressing a CAT reporter gene with different versions of the 3′-UTRs of PGKB and EP1 (illustrated at the top). The ‘PGKB’ blot shows mRNAs bearing the complete 3′-UTR (lane 3′UTR), or with the complete ARE or region II deleted as indicated. Sample RT–PCRs of transcripts with the deleted ARE were polyadenylated at C(160) and the ΔII transcripts were polyadenylated after G(164) [see (A)]. The EP1 blot shows mRNA levels of reporter transcripts bearing the complete EP1 3′-UTR (lane 3′UTR) or deleted for the 26mer ARE (lane Δ26mer). Lane (I+II) shows RNA with a hybrid 3′-UTR, where regions I and II of PGKB replaced the 26mer in the 3′-UTR of EP1. Sample loading was assessed with a tubulin probe (TUB). Bottom, CAT activities of cell extracts prepared from the different cell lines, mean and standard deviation of at least three measurements.
To see if the PGKB ARE-like sequence could function in another context, we inserted it into a version of the EP1 3′-UTR lacking the 26mer ARE, the ep1Δ26 3′-UTR. The resulting 3′-UTR had the PGKB ARE instead of the EP1 ARE. Three different permanent transformed cell lines were examined and all gave similar results; a typical example is shown. The PGKB ARE was able to reduce CAT mRNA and activity by approximately one-third in bloodstream trypanosomes when placed in the context of the EP1 3′-UTR (Fig. 1C) but it was not as effective as the EP1 26mer, suggesting that the context of the ARE is important. Similar results were previously obtained with the EP1 3′-UTR: a construct in which the EP1 26mer was placed between the CAT gene and the actin (ACT) 3′-UTR gave the same amount of CAT as a construct with the ACT 3′-UTR alone (17).
ARE-mediated PGKB mRNA degradation
mRNAs bearing an ACT 3′-UTR or a version of the EP1 3′-UTR lacking the ARE (ep1Δ26) are subject to deadenylation followed by degradation by 3′→5′ and 5′→3′ exonucleases, with a half-life of 15–20 min. In contrast, a reporter mRNA bearing the EP1 3′-UTR has a half-life of ∼5 min in bloodstream forms and its degradation is initiated by the rapid disappearance of the 3′-UTR (18). To confirm that the PGKB ARE was stimulating mRNA degradation, we used the bloodstream trypanosomes expressing CAT reporter genes from the T7 promoter, because these express sufficient mRNA to allow half-life determinations. The cells were incubated with actinomycin D, RNA was purified and the level of CAT mRNA was determined (18), using the stable SRP RNA (signal recognition particle RNA) as a loading control (41). In five independent experiments, the CAT mRNA bearing the pgkbΔARE 3′-UTR had a half-life of ∼15 min and the CAT mRNA with the PGKB 3′-UTR had a half-life of ∼7 min (Fig. 2 shows the results of two experiments). The level of CAT-pgkbΔARE mRNA showed a clear increase in abundance in the first 5–10 min after actinomycin D addition. The reason for this increase, which has previously been documented for endogenous actin RNA and CAT RNAs bearing EP1 and actin 3′-UTRs (17,18), is not known. The CAT-PGK mRNA was destroyed without any initial rise in abundance, similarly to CAT-EP1 mRNA (17,18). Preliminary results suggested that the mechanism of degradation was also very similar (C.Ben-Dov and H.Irmer, data not shown), but further experiments will be required to confirm this.
Figure 2.

Degradation mediated by the PGKB 3′-UTR. RNA was purified from cells expressing either CAT-PGKB (filled symbols) or CAT-pgkbΔARE (open symbols) at the indicated times after actinomycin D addition. CAT and SRP RNAs were detected by hybridisation with 32P-labelled probes. The blot result is reproduced from X-ray film, while the quantification shown of CAT mRNA relative to the SRP standard was done for two independent experiments (circles and triangles) using a PhosphorImager. The half-life of the CAT-pgkbΔARE mRNA was calculated from the descending part of the curve.
Human HuR interacts with the EP1 ARE
Overexpression of the mammalian protein HuR results in stabilisation of ARE-containing mRNAs in mammalian cells. Given the resemblance between the EP1 and PGKB instability elements and the ARE consensus from mammals, both at the primary and (predicted) secondary structure levels, we decided to test the effect of HuR in trypanosomes. The binding of HuR to U-rich sequences without a classical AUUUA element, like the EP1 26mer, is well documented (31,32). Nevertheless we felt it necessary to first confirm that HuR can bind to the EP1 3′-UTR by bandshift assays. In vitro-transcribed probes containing either the complete EP1 3′-UTR or the derivative without the 26mer, ep1Δ26, were used. The controls were the 3′-UTR of the human proto-oncogene c-fos, ‘AUF’, which contains two AREs, and a version lacking both AREs, named AUF1/2 (30). The probes were incubated with increasing amounts of purified recombinant GST-HuR. As expected, a shift in the migration of the AUF probe, but not AUF1/2, was seen at low and medium input of HuR (Fig. 3A). The EP1 3′-UTR probe, which contains the ARE-like element, also gave a shift, although the binding was not as strong as that mediated by the two AREs in the c-fos positive control. The ep1Δ26 sequence behaved similarly to the AUF1/2 negative control, suggesting that binding of HuR by EP1 mRNA was 26mer dependent. At very high input levels of HuR, non-specific binding was also observed.
To confirm that HuR interacted mainly with the EP1 ARE we mapped the binding site by RNase protection. Results are shown in Figure 3C. The ARE is indicated by the vertical line in Figure 3C and is boxed in Figure 3B. As can be seen from Figure 3C, lanes 1–5, the whole sequence corresponding to the 26mer was increasingly protected from T2 degradation as the amount of added GST-HuR was raised. Other neighbouring sequences were simultaneously more exposed to the enzyme. This confirmed that human HuR selectively bound to the EP1 ARE-like instability element.
Expression of human HuR inhibits growth of T.brucei
To express HuR in T.brucei, we cloned the HuR gene into an expression vector containing a tetracycline-inducible promoter, and transfected the construct into trypanosomes expressing the Tn10 tet repressor. Two lines were made, based either on the monomorphic 427 strain or the more pleomorphic (differentiation competent) 927 strain (36) (see Materials and Methods). Results are shown for the 427 line; the 927 line behaved similarly. The permanent transformants expressed low levels of HuR in the absence of tetracycline and high levels (>10-fold more) if tetracycline was added to the culture medium (Fig. 4A). A cross-reacting trypanosome protein of similar mobility and unknown identity was also seen in all cells (*) but the intensity was low and variable.
Figure 4.

Expression of HuR in T.brucei. (A) Bloodstream-form or procyclic-form trypanosomes containing an inducible HuR gene were incubated with the indicated concentrations of tetracycline for 18 h. Total protein extracts were analysed by western blot with the anti-HuR monoclonal 19F12 antibody to assess HuR induction. WT, cells without an inducible HuR gene. The cross-reacting trypanosome protein band is marked by an arrow and asterisk. (B) Effect of HuR on cell growth. Tetracycline (50 ng/ml) was added at time 0. Open squares, the control, –Tet; filled squares, +Tet. Every day cells were diluted to 2 × 105 cells/ml as required.
The cells growing in the absence of tetracycline exhibited normal growth, with a division time similar to many other trypanosome clones in our laboratory, whereas high levels of HuR caused rapid growth arrest and eventual cell death (Fig. 4B). These effects are specific to HuR; studies by many laboratories have shown that the addition of tetracycline, and the expression of several other non-trypanosome proteins such as reporters and selectable markers (references in 42), have no effect whatsoever on trypanosome growth, even when the very active T7 promoter is used. Immunofluorescent staining with the antibody against HuR in the absence of tetracycline gave a faint cytoplasmic fluorescence, which was considerably brighter 24 h after tetracycline addition (data not shown); we could see no evidence of any HuR in the nucleus.
We also analysed the effects of inducible HuR expression in procyclic trypanosomes, by constructing an appropriate cell line. The level of HuR expression 18 h after tetracycline addition was indistinguishable from that seen in bloodstream forms (Fig. 4A) but an inhibition of cell growth only became apparent on the second day (Fig. 4B). Thus, HuR had more rapid effects on bloodstream forms than on procyclics.
In trypanosomes, different cell cycle stages can be distinguished readily as the kinetoplast divides before the nucleus (43). Cells with one nucleus and one kinetoplast (1K1N) are in G1 or S phase, cells with 2K1N are in G2/M, and 2K2N are in M and cytokinesis (43). The bloodstream-form cell line containing the uninduced HuR gene exhibited a slight decrease in 1K1N relative to the parent cell line and a corresponding minor increase in the numbers of 2K1N, 2K2N and abnormal forms (data not shown). In the cells after 24 h of HuR induction, the main difference from the controls was an increase in the number of abnormal forms. However, none of the differences were very great and an arrest at a particular cell cycle stage could not be documented.
Effects of HuR on developmentally regulated trypanosome mRNAs
To analyse the effects of HuR on mRNA levels we concentrated on a few regulated and control mRNAs (Fig. 5A). All experiments were repeated several times for both cell lines, and quantified for at least three independent experiments; just one example is shown. EP and PGKB mRNAs occur in procyclic forms and are almost undetectable in bloodstream forms. Conversely, the PGKC, VSG and ALD (aldolase) messengers are abundant in bloodstream forms and strongly down-regulated in procyclics. The control mRNAs, ACT, TUB (data not shown) and PGKA, show no marked developmental regulation. The first two lanes of Figure 5A illustrate that the expression patterns in established bloodstream forms and procyclic forms were as anticipated. The other lanes show the effects of HuR expression in bloodstream forms. The ACT, TUB (data not shown) and PGKA mRNAs, which are not developmentally regulated, were unaffected or very slightly reduced by HuR expression. In contrast, HuR had pronounced effects on the expression of regulated mRNAs. The PGKB mRNA was up-regulated by the induction of HuR expression. Quantification of several blots revealed, on average, a 6-fold increase 24 h after tetracycline addition, as against the 10-fold increase in established procyclic forms. For EP, accumulation was noticeable 6 h after induction and the level at 24 h was 4–5-fold higher than in the normal bloodstream cells. Even without tetracycline, the 427 (and 927; data not shown) cell lines containing the integrated inducible copy of HuR exhibited an increase of 10–20% in the level of EP and PGKB mRNAs, most likely because of low-level HuR expression. These effects were specific to the HuR protein, as the expression of other foreign proteins such as selectable markers and reporters has no effect on the levels of developmentally regulated and constitutive mRNAs (some examples are listed in 42).
Figure 5.

ARE-containing mRNAs are up-regulated by induction of HuR in T.brucei bloodstream forms. (A) RNA was extracted from bloodstream-form parasites at the indicated times (hours) after tetracycline addition. Total RNA was blotted and hybridised with probes for the indicated mRNAs. Non-transfected bloodstream forms and procyclics (B and P, respectively) were included as controls (wt). (B) Effect of HuR expression on the abundance of reporter gene (CAT) mRNAs with different 3′-UTRs. The constructs are illustrated below; the arrow represents the RRNA promoter. RNA was purified before (– lanes) and 18 h after (+ lanes) Tet addition. (C) Effect of aphidicolin on mRNA levels. Bloodstream forms were incubated in the presence of aphidicolin (60 µM) for the indicated times (hours). Control cells (lane C) were cultured only with solvent (ethanol); P is procyclic total RNA. (D) Cells expressing CAT genes driven by a T7 promoter, with the indicated 3′-UTRs (EP1 or PGK), were incubated with aphidicolin (+) for 18–20 h and RNA detected with the probes indicated. The RRNA panel is a methylene blue stain.
The effects of HuR expression on EP and PGKB expression would be consistent with a direct stabilising effect mediated by interaction of HuR with the AREs. To our surprise, however, the expression of HuR also resulted in down-regulation of mRNAs specific to bloodstream forms, VSG, ALD and PGKC (Fig. 5A). The reductions were 2-fold for VSG, 2.5-fold for ALD and 5-fold for PGKC. In normal procyclics the corresponding reductions in mRNA abundance were >100×, 4× and 20×. These mRNAs do not contain ARE-like sequences. Note that the PGKC and PGKB genes are only ∼100 bp apart on the same transcription unit, confirming that the effects seen must occur post-transcriptionally.
To confirm that the effects of HuR expression on the EP and PGKB transcripts required the presence of the AREs, and were transcription independent, we made constructs in which CAT genes bearing 3′-UTRs of the EP1 and PGKB genes, along with deleted versions lacking the AREs, were placed downstream of a constitutive rRNA promoter. These were transfected into the cell line that inducibly expressed HuR and stable cell lines were generated. The reporter mRNAs containing ARE-like sequences (Fig. 5B, EP1, PGKB) were, as expected, at low levels (EP1) or undetectable in cells without tetracycline (Fig. 5B, – lanes), and were reproducibly much more abundant when HuR was induced (+ lanes). The controls lacking AREs (Fig. 5B, ep1Δ26, pgkbΔARE) were expressed at a higher level than those with the ARE, and the mRNA levels were unaffected by HuR induction. This result showed that the effect of HuR induction on the EP and PGKB mRNAs was dependent on the presence of ARE-like regulatory sequences.
The experiments so far had shown that expression of HuR causes rapid growth arrest in bloodstream trypanosomes, and more gradual growth inhibition in procyclic trypanosomes. The growth arrest in bloodstream trypanosomes was accompanied by down-regulation of three bloodstream-form-specific RNAs, and up-regulation of two ARE-containing, procyclic-form-specific mRNAs. Although it was clear that the up-regulation of the two procyclic-specific mRNAs was ARE dependent, it was not proven that this up-regulation depended on an in vivo interaction of these sequences with HuR. Another possibility was that the regulation was a consequence of growth arrest. This seemed especially likely because the non-dividing ‘stumpy’ bloodstream-form trypanosome is thought to be an intermediate prepared for procyclic differentiation. To find out whether growth arrest alone might cause mRNA changes, we incubated bloodstream trypanosomes with the inhibitor aphidicolin, which causes immediate cell cycle arrest in S-phase (44). We observed EP mRNA up-regulation, which was similar to that seen upon HuR induction (Fig. 5C). Both PGKC and TUB mRNAs were reproducibly somewhat reduced (Fig. 5C). Notably, however, no up-regulation of PGKB was detected in several experiments. The PGKB mRNA remained undetectable using the PGK coding-region probe, which hybridises with all three PGK isoform mRNAs (Fig. 5A).
Our failure to detect an increase in PGKB mRNA could have been due to its low abundance. To circumvent this problem, and to check whether the effects of aphidicolin were dependent on the transcribing polymerase or the promoter, we added aphidicolin to cells expressing CAT-PGKB and CAT-EP1 from the T7 promoter (Fig. 5D). As before, the treatment with aphidicolin did not enable us to detect the PGKB transcript from the endogenous locus. In addition, neither of the T7 transcripts, CAT-EP1 and CAT-PGKB, was increased in abundance (Fig. 5D). T7-produced CAT-pgkbΔARE mRNA behaved just like CAT-PGKB mRNA (data not shown), and we have also confirmed (using the cell lines shown in Fig. 5B) that neither CAT-EP1 nor CAT-PGKB transcripts are affected by aphidicolin when transcribed from the RRNA promoter (data not shown). It appears, therefore, that the accumulation of EP mRNA after aphidicolin treatment is promoter dependent. The EP promoter, unlike RRNA and T7 promoters, shows a 10-fold up-regulation during bloodstream-to-procyclic differentiation. It is possible that similar effects might be induced by cell cycle arrest, but this was irrelevant to the project so was not analysed further.
HuR expression does not inhibit protein synthesis
The experiments so far had demonstrated that ARE-containing mRNAs were stabilised by the expression of HuR, and that the effect was not a side effect of growth arrest. The inhibition of protein synthesis has also been shown to cause an increase in EP and some other mRNAs (20,45–47). To make sure that the effects of HuR on mRNAs were not a side effect of protein synthesis inhibition, cells of both the HuR-expressing lines were metabolically labelled for 2 h with [35S]methionine, starting 4, 8 and 18 h after HuR induction. Newly synthesised proteins were detected by one-dimensional gel electrophoresis and autoradiography. Results for one line are shown; the pattern for the other was indistinguishable. The pattern of Coomassie-stain-detectable proteins was unaltered by HuR induction (data not shown). In addition, no gross alterations in the level or pattern of newly synthesised proteins were seen (Fig. 6A). Notable were a rise in the synthesis of a protein with the expected size of HuR (* in Fig. 6A) and decreases in the synthesis of proteins that migrated similarly to aldolase (black triangle) and VSG (open triangle). Synthesis of one protein was transiently activated ∼4 h after tetracycline addition in both cell lines (filled circle). Clearly, HuR did not have a generalised toxic effect on protein synthesis. Interestingly, induction of HuR did not induce detectable EP or PGKB expression, despite the increases in mRNA, suggesting that either the controls on translation continued to operate or the protein could not be processed and was degraded. The levels of aldolase and PGKC were reduced, reflecting the decreases in mRNA (Fig. 6B). Dihydrolipoamide dehydrogenase (DHALDH) is a marker that is induced in stumpy and procyclic forms (3). HuR expression initially caused a decrease of DHALDH, but the level then recovered to marginally above the initial level (Fig. 6B). Expression of mitochondrial HSP60 followed a similar pattern but the differences were less marked (data not shown). Aphidicolin treatment, in contrast, had no reproducible effect on expression of either DHALDH or the other proteins (data not shown).
Figure 6.
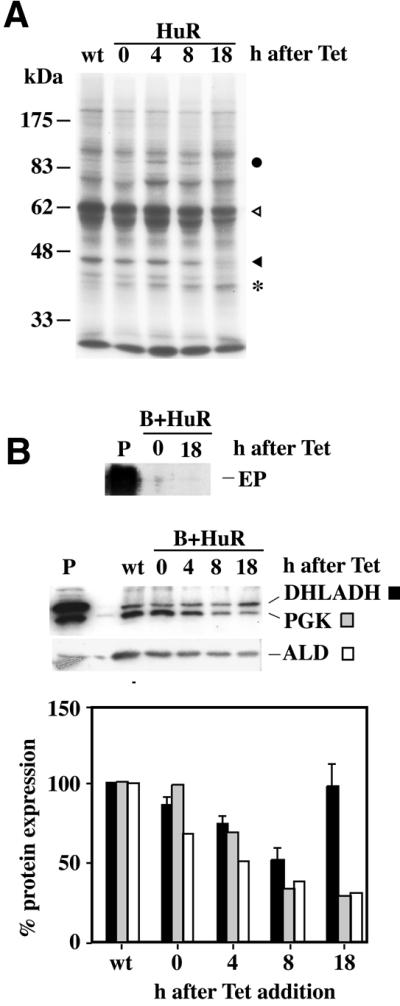
Effect of HuR expression on protein synthesis. (A) Effect on total protein synthesis. Bloodstream cells were pulsed with [35S]methionine at the indicated times (hours) after HuR induction. Several proteins underwent a decrease in their synthesis rate (triangles), one (possibly HuR) showed a steady increase (asterisk), and at least one was transiently activated (filled circle). (B) Effect on the abundance of specific proteins 18 h after tetracycline addition. Western blots (top) and quantification (bottom), where the level of each protein was calculated relative to the level of the same protein in cells not containing the HuR gene (wt). The bars for DHLADH show the mean and standard deviation for three independent experiments, while the PGK and ALD were quantified for one representative blot.
Regulation of PPDK expression
The fact that both EP and PGKB mRNAs contained regulatory AREs suggested that there might be a whole class of procyclic-specific, ARE-containing mRNAs. To identify further members of this class we took advantage of recently developed T.brucei genomic arrays (48). RNA was prepared from bloodstream trypanosomes containing an inducible HuR gene 24 h after addition of tetracycline, and fluorescently labelled cDNA was hybridised to arrays containing approximately 20 000 random genomic fragments of ∼2 kb. Labelled cDNA corresponding to RNA from wild-type trypanosomes served as a control. Preliminary results indicated that a small subset of procyclic-specific clones was up-regulated by HuR. One of these, encoding pyruvate, phosphate dikinase (PPDK) was chosen for further analysis because it was functionally characterised and the polyadenylation site had already been mapped (49). It was already known that the PPDK mRNA is well expressed in procyclic forms and barely detectable in bloodstream forms (49). In our array analysis, the ratio of fluorescent probe hybridisation indicated 9-fold regulation between bloodstream and procyclic forms, and a 4-fold increase in PPDK mRNA in bloodstream forms upon induction of HuR expression, which could be confirmed by northern blot analysis (data not shown). The PPDK 3′-UTR is 1.2 kb long and contains several U-rich tracts. To identify potential AREs we examined the potential secondary structure as predicted by MFOLD (Fig. 7A). Just one U-rich region, around nucleotide 700, was predicted to be single stranded in all 10 of the lowest-energy predicted structures (Fig. 7B). As for the PGKB 3′-UTR, we have not yet confirmed this structure experimentally. Nevertheless, to find out if this played any role in regulating the level of PPDK mRNA, we made permanent bloodstream trypanosome lines expressing CAT with either the PPDK 3′-UTR or various deleted versions, using the RRNA promoter for expression (Fig. 7C). The levels of CAT activity were enhanced ∼4-fold, and mRNA levels ∼2-fold, by deletion of the putative ARE region from the PPDK 3′-UTR (Fig. 7D); variations between independent cloned cell lines containing the same construct are presumably due to the fact that the construct is integrated into the spacers of different ribosomal RNA arrays (16). The effects of HuR expression on the mRNAs from these constructs are shown in Figure 7E. The CAT mRNA with the PPDK 3′-UTR was increased in abundance by expression of HuR, whereas the level of CAT-PPDK-ΔARE mRNA was only slightly affected by HuR expression. Similar results were obtained from a truncated version of the 3′-UTR (ppdk528-1002) consisting of the predicted branched stem including the ARE.
Figure 7.

Analysis of an mRNA-instability element present in the 3′-UTR of the PPDK gene. (A) Representative predicted structure of the complete PPDK 3′-UTR. Positions 528 and 1002 are indicated, and the region enlarged in (B) is boxed. (B) Detail of the secondary structure predicted for the region containing the possible ARE around nucleotide 700. (C) Maps of CAT mRNAs produced from constructs designed to determine the role of the PPDK ARE in determining mRNA levels and CAT activity. Transcription was from the RRNA promoter and constructs were targeted to RRNA spacer regions. 3′-UTRs were either the whole UTR (PPDK), the whole UTR with a 23 nt deletion (from nucleotides 686 to 710 inclusive, ppdkΔARE), or a truncated version from nucleotides 528 to 1002 (ppdk528-1002) and the corresponding deletion mutant (ppdk528-1002ΔARE). The truncated sequence has a slightly different predicted folding pattern than the complete 3′-UTR but the ARE sequence is predicted to remain single stranded. (D) CAT activities of cell extracts prepared from different cell lines expressing the CAT reporter gene with different versions of the PPDK 3′-UTR [shown in (C)], mean and standard deviation of six (PPDK) or nine (PPDKΔARE) independent cell lines measured in duplicate, with a representative northern blot beneath. (E) Total RNA was taken from parasites expressing the CAT reporter gene with different 3′-UTRs. At least three different cell lines were examined for each 3′-UTR variant. The northern blots show representative results with (16 h) and without tetracycline induction of HuR.
Overall the results from PPDK suggest that structural predictions combined with the expression of RNA-binding proteins can greatly assist in the identification of regulatory motifs.
DISCUSSION
The results described in this paper show that the 3′-UTRs of three similarly controlled, but unrelated, T.brucei mRNAs, EP, PGK and PPDK, share similar regulatory motifs consisting of a U-rich region, which may include the consensus AUUUA motif (ARE) found in unstable cytokine and oncogene mRNAs in mammals. The U-rich regions were all predicted to be single stranded. This structure was previously confirmed for EP RNA by chemical and enzymatic analysis (19). Each of the mRNAs has low abundance in bloodstream trypanosomes, and the abundance could be increased by the expression of HuR, a protein that specifically binds to single-stranded U-rich RNA (31,32) and can stabilise ARE-containing RNAs in mammalian cells and in vitro extracts (see Introduction). Correspondingly, deletion of the U-rich elements from the affected mRNAs resulted in an increase in mRNA abundance, and eliminated the response to HuR expression.
Trypanosomes depend almost exclusively on post- transcriptional regulation, and different sets of mRNAs and proteins accumulate at different times in the life cycle. It is therefore attractive to suppose that mRNAs that show similar regulation will all share common regulatory motifs. Indeed, an RNA element of 450 nt has recently been found in a large set of mRNAs that share a common developmental regulation pattern in Leishmania (50); in this particular case, the regulation is mostly at the level of translation. The U-rich sequence described here is completely unrelated to the Leishmania element, but very similar indeed to an ARE-like element found in a developmentally regulated mucin mRNA in T.cruzi (21), suggesting that ARE-mediated degradation may be generally important in trypanosome differentiation.
The effects of HuR in increasing mRNA levels in bloodstream-form trypanosomes were specific for mRNAs that are normally present at low abundance and contain destabilising U-rich sequences in the 3′-UTRs. The increases were also completely independent of the promoter–polymerase combination, as they were seen for RNAs transcribed by polymerase II and polymerase I. Both the sequence specificity of the effects and the absence of any evidence for accumulation of precursor RNAs make it very unlikely that HuR generally inhibits RNA processing. Polyadenylation of each transcript is mechanistically linked to trans splicing of the downstream RNA; however, the linked PGKA, PGKB and PGKC genes were all affected in different ways. Although treatment of the cells with Aphidicolin to stop the cell cycle induced EP RNA, the effect was restricted to the mRNA transcribed from the EP1 promoter and was not seen for RNAs transcribed by polymerase II or from the RRNA or T7 promoters. Thus, the results are consistent with the hypothesis that the effects of HuR in enhancing mRNA abundance were due to alterations in mRNA stability through binding of HuR to the destabilising AREs. This is entirely consistent with the effects of HuR in mammalian cells. Since HuR is not a trypanosome protein, the most likely mechanism would be to shield or hide the AREs from recognition by the trypanosome degradation machinery.
In addition to the specific increase in known ARE-containing developmentally regulated mRNAs in bloodstream forms, HuR expression had more general effects: a reduction in the abundance of bloodstream-form-specific mRNAs in the bloodstream forms, and inhibition of the growth of procyclic forms (where so far we have found no effect on a very limited number of mRNAs). In bloodstream forms, growth inhibition could be due to metabolic imbalances caused by stabilisation of ARE-containing developmentally regulated RNAs. The fact that growth of procyclic forms was also gradually affected could either be due to binding of HuR to additional unstable mRNAs whose destruction is required for cell cycle progression in both life cycle stages, or it may be that prolonged expression of HuR results in less specific effects. In mammalian cells also, there are different classes of ARE-containing mRNAs (33,51).
A global analysis of trypanosome gene expression using genomic arrays that cover most of the genome revealed approximately 200 mRNAs that show significant differences in level between bloodstream and procyclic forms and are regulated at the post-transcriptional level (48). We do not yet know how many of these contain U-rich regions in the 3′-UTRs, or how many contain other conserved elements, as the vast majority of the polyadenylation sites have not been mapped. Our first results from the pilot array experiment with HuR, however, suggest a general method to find both regulatory regions and the proteins which bind to them. Most of the T.brucei genome sequence is now available at least as a single-pass sequence, but so far no direct homologues of HuR are present and there are no predicted proteins with three RRMs. There are, however, many predicted proteins with one or two RNA-binding domains, some of which are flanked by glutamine-rich regions. These proteins are reminiscent of the mammalian AUF1 protein which destabilises ARE-containing mRNAs (52). One of the T.brucei genes encodes a protein which is strongly related to the T.cruzi protein TcUBP1 (53): moderate overexpression of TcUBP1 in T.cruzi resulted in destabilisation of an ARE-containing mRNA (53). Preliminary results indicate that expression of either TcUBP1 or the T.brucei homologue in T.brucei inhibits cell growth and alters the abundance of several mRNAs; notably no effects on EP1 mRNA were detected (C.Hartmann and G.Erdmann, unpublished results). Results from other laboratories have tentatively implicated additional classes of proteins in mRNA stabilisation or destabilisation (54,55). Overexpression of these proteins in bloodstream and procyclic trypanosomes, combined with array analysis, should reveal which proteins are regulatory and enable us to identify both the likely mRNA targets and the consensus sequences required for their regulation.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Henry Furneaux (Memorial Sloan Kettering Cancer Center, New York, NY) for HuR and c-fos plasmids, and for the anti-HuR monoclonal antibody. We thank Steve Beverley (University of Washington, MO, USA) and Keith Matthews (Manchester University, UK) for very useful criticism and suggestions, Shula Michaeli (Bar-Ilan University, Israel) for the SRP RNA clone and Paul Michels (ICP, Brussels) for anti-PGK. We thank other members of the laboratory, particularly A. Estévez, for discussions, and L. Krauth-Siegel, BZH, Heidelberg, for the DHLADH antibody. We thank Gitta Erdmann for contributions made during an undergraduate laboratory rotation. L.Q. was supported by an EMBO fellowship, and C.G. by a DAAD scholarship. C.B.-D. contributed during a short-term visit funded by the DAAD. The rest of the support came from the Deutsche Forschungsgemeinschaft.
REFERENCES
- 1.Cross G.A.M. (1997) Antigenic variation in trypanosomes: secrets surface slowly. Bioessays, 18, 283–291. [DOI] [PubMed] [Google Scholar]
- 2.Matthews K.R. (1999) Developments in the differentiation of Trypanosoma brucei. Parasitol. Today, 15, 76–80. [DOI] [PubMed] [Google Scholar]
- 3.Tyler K.M., Matthews,K.R. and Gull,K. (1997) The bloodstream differentiation-division of Trypanosoma brucei studied using mitochondrial markers. Proc. R. Soc. London, 264, 1481–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tasker M., Wilson,J., Sarkar,M., Hendriks,E. and Matthews,K. (2000) A novel selection regime for differentiation defects demonstrates an essential role for the stumpy form in the life cycle of African trypanosomes. Mol. Biol. Cell, 11, 1905–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roditi I., Furger,A., Ruepp,S., Schurch,N. and Butikofer,P. (1998) Unravelling the procyclin coat of Trypanosoma brucei. Mol. Biochem. Parasitol., 91, 117–130. [DOI] [PubMed] [Google Scholar]
- 6.Roditi I. and Clayton,C.E. (1999) An unambiguous nomenclature for the major surface glycoprotein genes of the procyclic form of Trypanosoma brucei. Mol. Biochem. Parasitol., 103, 99–100. [DOI] [PubMed] [Google Scholar]
- 7.Myler P.J., Audleman,L., deVos,T., Hixson,G., Kiser,P., Lemley,C., Magness,C., Rickel,E., Sisk,E., Sunkin,S., Swartzell,S., Westlake,T., Bastien,P., Fu,G., Ivens,A. and Stuart,K. (1999) Leishmania major Friedlin chromosome 1 has an unusual distribution of protein-coding genes. Proc. Natl Acad. Sci. USA, 96, 2902–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clayton C.E. (2002) Developmental regulation without transcriptional control? From fly to man and back again. EMBO J., 21, 1881–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ullu E., Matthews,K.R. and Tschudi,C. (1993) Temporal order of RNA-processing reactions in trypanosomes: rapid trans splicing precedes polyadenylation of newly synthesized tubulin transcripts. Mol. Cell. Biol., 13, 720–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matthews K.R., Tschudi,C. and Ullu,E. (1994) A common pyrimidine-rich motif governs trans-splicing and polyadenylation of tubulin polycistronic pre-mRNA in trypanosomes. Genes Dev., 8, 491–501. [DOI] [PubMed] [Google Scholar]
- 11.Blattner J., Helfert,S., Michels,P. and Clayton,C.E. (1998) Compartmentation of phosphoglycerate kinase in Trypanosoma brucei plays a critical role in parasite energy metabolism. Proc. Natl Acad. Sci. USA, 95, 11596–11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blattner J. and Clayton,C.E. (1995) The 3′-untranslated regions from the Trypanosoma brucei phosphoglycerate kinase genes mediate developmental regulation. Gene, 162, 153–156. [DOI] [PubMed] [Google Scholar]
- 13.Kapotas N. and Bellofatto,V. (1993) Differential response to RNA trans-splicing signals within the phosphoglycerate kinase gene cluster in Trypanosoma brucei. Nucleic Acids Res., 21, 4067–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laufer G. and Günzl,A. (2001) In-vitro competition analysis of procyclin gene and variant surface glycoprotein gene expression site transcription in Trypanosoma brucei. Mol. Biochem. Parasitol., 113, 55–65. [DOI] [PubMed] [Google Scholar]
- 15.Horn D. and Cross,G.A.M. (1997) Position-dependent and promoter-specific regulation of gene expression in Trypanosoma brucei. EMBO J., 16, 7422–7431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biebinger S., Rettenmaier,S., Flaspohler,J., Hartmann,C., Peña-Diaz,J., Wirtz,L.E., Hotz,H.R., Barry,J.D. and Clayton,C.E. (1996) The PARP promoter of Trypanosoma brucei is developmentally regulated in a chromosomal context. Nucleic Acids Res., 24, 1202–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hotz H.-R., Hartmann,C., Huober,K., Hug,M. and Clayton,C.E. (1997) Mechanisms of developmental regulation in Trypanosoma brucei: a polypyrimidine tract in the 3′-untranslated region of a trypanosome surface protein mRNA affects RNA abundance and translation. Nucleic Acids Res., 25, 3017–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Irmer H. and Clayton,C.E. (2001) Degradation of the EP1 mRNA in Trypanosoma brucei is initiated by destruction of the 3′-untranslated region. Nucleic Acids Res., 29, 4707–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drozdz M. and Clayton,C.E. (1999) Structure of a regulatory 3′-untranslated region from Trypanosoma brucei. RNA, 5, 1632–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schürch N., Furger,A., Kurath,U. and Roditi,I. (1997) Contribution of the procyclin 3′ untranslated region and coding region to the regulation of expression in bloodstream forms of Trypanosoma brucei. Mol. Biochem. Parasitol., 89, 109–121. [DOI] [PubMed] [Google Scholar]
- 21.Di Noia J.M., D’Orso,I., Sánchez,D.O. and Frasch,A.C.C. (2000) AU-rich elements in the 3′-untranslated region of a new mucin-type gene family of Trypanosoma cruzi confers mRNA instability and modulates translation efficiency. J. Biol. Chem., 275, 10218–10227. [DOI] [PubMed] [Google Scholar]
- 22.Chen C.Y. and Shyu,A.B. (1995) AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci., 20, 465–470. [DOI] [PubMed] [Google Scholar]
- 23.Mitchell P. and Tollervey,D. (2000) mRNA stability in eucaryotes. Curr. Opin. Genet. Dev., 10, 193–198. [DOI] [PubMed] [Google Scholar]
- 24.Chen C.Y., Gherzi,R., Ong,S.E., Chan,E.L., Raijmakers,R., Pruijn,G.J., Stoecklin,G., Moroni,C., Mann,M. and Karin,M. (2001) AU binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell, 107, 451–464. [DOI] [PubMed] [Google Scholar]
- 25.Mukherjee D., Gao,M., O’Connor,J.P., Raijmakers,R., Pruijn,G., Lutz,C.S. and Wilusz,J. (2002) The mammalian exosome mediates the efficient degradation of mRNAs that contain AU-rich elements. EMBO J., 21, 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Z., Chang,F.-C. and Furneaux,H.M. (2000) The identification of an endonuclease that cleaves within an HuR binding site in mRNA. Nucleic Acids Res., 28, 2695–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fan X.C. and Steitz,J.A. (1998) Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J., 17, 3448–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng S.S., Chen,C.Y., Xu,N. and Shyu,A.B. (1998) RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J., 17, 3461–3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Myer V.E., Fan,X. and Steitz,J.A. (1997) Identification of HuR as a protein implicated in AUUUA-mediated mRNA decay. EMBO J., 16, 2130–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma W.J., Cheng,S., Campbell,C., Wright,A. and Furneaux,H. (1996) Cloning and characterization of HuR, a ubiquitously expressed Elav-like protein. J. Biol. Chem., 271, 8144–8151. [DOI] [PubMed] [Google Scholar]
- 31.Antson A.A. (2000) Single stranded RNA binding proteins. Curr. Opin. Genet. Dev., 10, 87–94. [DOI] [PubMed] [Google Scholar]
- 32.Wang X. and Tanaka Hall,T.M. (2001) Structural basis for the recognition of AU-rich element RNA by the HuD protein. Nature Struct. Biol., 8, 141–145. [DOI] [PubMed] [Google Scholar]
- 33.Peng S.S., Chen,C.Y. and Shyu,A.B. (1996) Functional characterization of a non-AUUUA AU-rich element from the c-jun proto-oncogene mRNA: evidence for a novel class of AU-rich elements. Mol. Cell. Biol., 16, 1490–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ford L.P., Watson,J., Keene,J.D. and Wilusz,J. (1999) ELAV proteins stabilize deadenylated intermediates in a novel in vitro mRNA deadenylation/degradation system. Genes Dev., 13, 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vasudevan S. and Peltz,S.W. (2001) Regulated ARE-mediated mRNA decay in Saccharomyces cerevisiae. Mol. Cell, 7, 1191–1200. [DOI] [PubMed] [Google Scholar]
- 36.van Deursen F.J., Shahi,S.H., Turner,C.M.R., Hartmann,C., Guerra-Giraldez,C., Matthews,K.R. and Clayton,C.E. (2001) Characterisation of the growth and differentiation in vivo and in vitro of bloodstream-form Trypanosoma brucei strain TREU 927. Mol. Biochem. Parasitol., 112, 163–172. [DOI] [PubMed] [Google Scholar]
- 37.Biebinger S., Wirtz,L.E. and Clayton,C.E. (1997) Vectors for inducible over-expression of potentially toxic gene products in bloodstream and procyclic Trypanosoma brucei. Mol. Biochem. Parasitol., 85, 99–112. [DOI] [PubMed] [Google Scholar]
- 38.Wirtz L.E., Hartmann,C. and Clayton,C.E. (1994) Gene expression mediated by bacteriophage T3 and T7 RNA polymerases in transgenic trypanosomes. Nucleic Acids Res., 22, 3887–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartmann C., Hotz,H.-R., McAndrew,M. and Clayton,C. (1998) Effect of multiple downstream splice sites on polyadenylation in Trypanosoma brucei. Mol. Biochem. Parasitol., 93, 149–152. [DOI] [PubMed] [Google Scholar]
- 40.Chung S., Jiang,L., Cheng,S. and Furneaux,H. (1996) Purification and properties of HuD, a neuronal RNA-binding protein. J. Biol. Chem., 271, 11518–11524. [DOI] [PubMed] [Google Scholar]
- 41.Michaeli S., Podell,D., Agabian,N. and Ullu,E. (1992) The 7SL RNA homologue of Trypanosoma brucei is closely related to mammalian 7SL RNA. Mol. Biochem. Parasitol., 51, 55–64. [DOI] [PubMed] [Google Scholar]
- 42.Clayton C.E. (1999) Genetic manipulation of Kinetoplastida. Parasitol. Today, 15, 372–378. [DOI] [PubMed] [Google Scholar]
- 43.Sherwin T. and Gull,K. (1989) The cell division cycle of Trypanosoma brucei brucei: timing of event markers and cytoskeletal modulations. Phil. Trans. R. Soc. London, 323, 573–588. [DOI] [PubMed] [Google Scholar]
- 44.Mutomba M.C. and Wang,C.C. (1996) Effects of aphidodocolin and hydroxyurea on the cell cycle and differentiation of Trypanosoma brucei bloodstream forms. Mol. Biochem. Parasitol., 80, 89–102. [DOI] [PubMed] [Google Scholar]
- 45.Dorn P.L., Aman,R.A. and Boothroyd,J.C. (1991) Inhibition of protein synthesis results in super-induction of procyclin (PARP) RNA levels. Mol. Biochem. Parasitol., 44, 133–140. [DOI] [PubMed] [Google Scholar]
- 46.Graham S.V. and Barry,J.D. (1996) Polysomal, procyclin mRNAs accumulate in bloodstream forms of monomorphic and pleomorphic trypanosomes treated with protein synthesis inhibitors. Mol. Biochem. Parasitol., 80, 179–192. [DOI] [PubMed] [Google Scholar]
- 47.Vanhamme L., Postiaux,S., Poelvoorde,P. and Pays,E. (1999) Differential regulation of ESAG transcripts in Trypanosoma brucei. Mol. Biochem. Parasitol., 102, 35–42. [DOI] [PubMed] [Google Scholar]
- 48.Diehl S., Diehl,F., El-Sayed,N.M., Clayton,C.E. and Hoheisel,J.D. (2002) Analysis of stage-specific gene expression in the bloodstream and the procyclic form of Trypanosoma brucei using a genomic DNA microarray. Mol. Biochem. Parisitol., in press. [DOI] [PubMed] [Google Scholar]
- 49.Bringaud F., Baltz,D. and Baltz,T. (1998) Functional and molecular characterization of a glycosomal PPi-dependent enzyme in trypanosomatids: pyruvate phosphate dikinase. J. Biol. Chem., 95, 7963–7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boucher N., Wu,Y., Dumas,C., Dube,M., Sereno,D., Breton,M. and Papadopoulou,B. (2002) A common mechanism of stage-regulated gene expression in Leishmania mediated by a conserved 3′ UTR element. J. Biol. Chem., 277, 19511–19520. [DOI] [PubMed] [Google Scholar]
- 51.Chen C.Y., Xu,N. and Shyu,A.B. (1995) mRNA decay mediated by two distinct AU-rich elements from c-fos and granulocyte-macrophage colony-stimulating factor transcripts: different deadenylation kinetics and uncoupling from translation. Mol. Cell. Biol., 15, 5777–5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.DeMaria C.T. and Brewer,G. (1996) AUF1 binding affinity to A+U-rich elements correlates with rapid mRNA degradation. J. Biol. Chem., 271, 12179–12184. [DOI] [PubMed] [Google Scholar]
- 53.D’Orso I. and Frasch,A.C.C. (2001) TcUBP-1, a developmentally regulated U-rich RNA-binding protein involved in selective mRNA destabilization in trypanosomes. J. Biol. Chem., 276, 34801–34809. [DOI] [PubMed] [Google Scholar]
- 54.Hoek M., Zanders,T. and Cross,G.A.M. (2002) Trypanosoma brucei expression-site-associated-gene 8 protein interacts with a Pumilio family protein. Mol. Biochem. Parasitol., 120, 269–284. [DOI] [PubMed] [Google Scholar]
- 55.Hendriks E.F., Robinson,D.R., Hinkins,M. and Matthews,K.R. (2001) A novel CCCH protein which modulates differentiation of Trypanosoma brucei to its procyclic form. EMBO J., 20, 6700–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]