


Abstract
Replicative polymerases of eukaryotes, prokaryotes and archaea obtain processivity using ring-shaped DNA sliding clamps that are loaded onto DNA by clamp loaders [replication factor C (RFC) in eukaryotes]. In this study, we cloned the two genes for the subunits of the RFC homologue of the euryarchaeon Archaeoglobus fulgidus. The proteins were expressed and purified from Escherichia coli both individually and as a complex. The afRFC subunits form a heteropentameric complex consisting of one copy of the large subunit and four copies of the small subunits. To analyse the functionality of afRFC, we also expressed the A.fulgidus PCNA homologue and a type B polymerase (PolB1) in E.coli. In primer extension assays, afRFC stimulated the processivity of afPolB1 in afPCNA-dependent reactions. Although the afRFC complex showed significant DNA-dependent ATPase activity, which could be further stimulated by afPCNA, neither of the isolated afRFC subunits showed this activity. However, both the large and small afRFC subunits showed interaction with afPCNA. Furthermore, we demonstrate that ATP binding, but not hydrolysis, is needed to stimulate interactions of the afRFC complex with afPCNA and DNA.
INTRODUCTION
Replicative polymerases from eukaryotes to prokaryotes obtain processivity using ring-shaped DNA sliding clamps that are loaded onto DNA by clamp loader proteins in ATP-dependent reactions. Although the amino acid sequences and the protein complex compositions differ between the various systems, both the overall structure of the clamp/clamp loader proteins and the molecular mechanisms of the clamp-loading process appear to be conserved (1). The Escherichia coli γ complex, whose in vivo composition is γ3δδ′χψ (2), is the most thoroughly analysed clamp loader. A heteropentamer consisting only of γ3δδ′ has been shown to be able to load the dimeric β sliding clamp onto DNA (3). Crystal structures of the γ3δδ′ complex and of the δ subunit in a complex with a mutant monomeric form of β have been published (4,5). Together with earlier biochemical data (6,7), these crystal structures showed that the δ subunit contacts the β clamp and traps β in a conformation where one of the two clamp interfaces is open. ATP binding, but not hydrolysis, is essential for the E.coli clamp loader to open the sliding clamp and to load it onto a primer–template junction of DNA (7). Previous biochemical studies showed that this is also true for the eukaryotic clamp loader, replication factor C (RFC) (8). Unlike the E.coli clamp loader, eukaryotic RFC consists of five different subunits (RFC1–RFC5) (9). However, like the minimal E.coli clamp loader (γ3δδ′), the composition of RFC is heteropentameric (10).
With sequence identities of 30–40%, the clamp loaders of Archaea, the third domain of life, seem to be a closely related, yet simplified, version of their pro- and eukaryotic counterparts. Thus, in contrast to the eukaryotic clamp loader, where each of the five different RFC subunits has been found to be individually essential (9), only two RFC-like subunits are found in each of the completely sequenced archaeal genomes (11). The conserved RFC boxes II–VIII (9) can be found in both the large and the small subunits that constitute archaeal clamp loaders (11). The conserved RFC box I, found only in the large subunit of eukaryotic RFCs (RFC1), is absent in archaeal clamp loaders. However, this motif has been shown to be non-essential for clamp loading (12,13). So far, archaeal clamp loaders from Sulfolobus solfataricus, Methano bacterium thermoautotrophicum and Pyrococcus furiosus have been isolated and shown to stimulate the processivity of their replicative polymerases in PCNA-dependent reactions (14–16). The two subunits of both the crenarchaeal S.solfataricus and the euryarchaeal P.furiosus RFC appear to form complexes with 1:4 (large to small) stoichiometric ratios as shown by gel densitometry and native gel filtration (14,16). In contrast, gel densitometric, as well as combined gel filtration and glycerol gradient centrifugation, data indicate a 2:4 (large to small) subunit composition of the M.thermoautotrophicum RFC (15).
In this study, we isolated and analysed the biochemical properties of the RFC from Archaeoglobus fulgidus, a hyperthermophilic euryarchaeote with an optimal growth temperature of 83°C (17). The results show that the A.fulgidus RFC (afRFC) is composed of two different subunits that form a heteropentamer consisting of one copy of the large subunit and four copies of the small subunit. In common with the E.coli, eukaryotic and other archaeal clamp loaders (7,13–16), afRFC possesses DNA-dependent ATPase activity. The afRFC ATPase activity is stimulated further by A.fulgidus PCNA (afPCNA) in the presence of appropriate polynucleotide substrates. Furthermore, in the presence of afPCNA, afRFC significantly stimulates the processivity of A.fulgidus polymerase B1. In agreement with the E.coli and eukaryotic clamp loaders (7,8), ATP binding is sufficient to stimulate the interactions of afRFC with afPCNA and polynucleotides.
MATERIALS AND METHODS
Materials
The buffers used and their compositions were: buffer A, 50 mM Tris–HCl (pH 7.5), 1 mM DTT and 1 mM EDTA; buffer B, 20 mM potassium phosphate (pH 7.5) and 500 mM NaCl; buffer C, 50 mM Tris–HCl (pH 8.0), 1 mM DTT and 1 mM EDTA; buffer D, 50 mM MES (pH 5.8), 1 mM DTT and 1 mM EDTA; buffer E, 50 mM Tris–HCl (pH 8.5), 1 mM DTT and 1 mM EDTA; buffer F, 50 mM HEPES–KOH (pH 7.5), 20 mM magnesium chloride and 25 mM potassium chloride. Unless stated otherwise, all chemicals were supplied by Sigma.
Cloning of A.fulgidus genes
afRFC large subunit, afRFC small subunit, afPCNA and afPolB1 genes (AF1195, AF2060, AF0335 and AF0497, respectively; 17) were amplified by polymerase chain reaction from A.fulgidus genomic DNA. The primers for the amplification of the afRFC large subunit (AF1195fw 5′-TCGCGAGCATATGCTGTGGGTTGAGAAGTATC-3′ and AF1195rv 5′-CGCTACAGAGCGGCCGCTTACTAGGAAAAGAAGGAATCGAGAGTT-3′) contained NdeI and NotI restriction sites which were used for insertion into pET28a (Novagen). Expression of this construct resulted in 20 additional N-terminal amino acids, which are not present in the native AF1195. Among these amino acids are six consecutive histidines and residues forming a thrombin cleavage site. The primers for the amplification of the afRFC small subunit (AF2060fw 5′-TGGAGCGCATATGGAGAACTTCGAAATCTGG-3′ and AF2060rv 5′-AAACATTATGCGGCCGCTTACTACTTCTTGGCCAAAGTGGAGAG-3′) contained NdeI and NotI restriction sites which were used for insertion into pET22b (Novagen). The primers for the amplification of afPCNA (AF0335fw 5′-CCGATCGACATATGATAGACGTCATAATGACC-3′ and AF0335rv 5′-CAGCAAGCTTCTATTACTCCGACTCTATGCGCGGAGC-3′) contained NdeI and HindIII restriction sites which were used for insertion into pET22b (Novagen). The primers for the amplification of afPolB1 (AF0497fw 5′-CATGCCATGGAAAGAGTTGAGGGCTGGC-3′ and AF0497rv 5′-AAGAATCGGCCGTTACTAGCTGAAGAATGAATCCAGGCTC-3′) contained NcoI and EagI restriction sites which were used for insertion into pET28a (Novagen).
Cloning of Thermotoga maritima pyruvate kinase and lactate dehydrogenase genes
For the determination of the afRFC ATPase activity in a continous spectrophometric assay, heat-stable pyruvate kinase and lactate dehydrogenase had to be prepared. The genes encoding the pyruvate kinase (TM0208) and lactate dehydrogenase (TM1867) from the thermostable eubacterium T.maritima MSB8 (optimum growth temperature of 80°C) were amplified by polymerase chain reaction from T.maritima genomic DNA. The primers for the amplification of the pyruvate kinase (TM0208fw 5′-GGAATTCCATATGCGAAGTACAAAGATCGTATG-3′ and TM0208rv 5′-CCCAAGCTTACTAATCCACCTTCAACACCCTTATGG-3′) and of the lactate dehydrogenase (TM1867fw 5′-GGAATTCCATATGAAAATAGGTATCGTAGG-3′ and TM1867rv 5′-CCCAAGCTTACTAACCGCTGGTGTTCTGGTGC-3′) contained NdeI and HindIII restriction sites which were in each case used for insertion into pET22b (Novagen).
Expression and purification of recombinant proteins
afRFC, afRFCsm, afRFCla, afPCNA, afPolB1, tmPK and tmLDH were overexpressed as follows: 2 l of E.coli cells, harboring the different plasmids, were grown at 37°C in Luria–Bertani (LB) medium in the presence of the appropriate antibiotics. afRFC large and small subunits were both co-expressed and expressed separately in BL21-CodonPlus (DE3)-RIL cells (Stratagene). afPCNA and afPolB1 were overexpressed in B834(DE3)pLysS cells (Novagen) carrying a plasmid (pSJS1240, gift from S. J. Sandler) to express low abundance tRNAs (18). tmPK and tmLDH were over expressed in BL21(DE3) cells (Novagen) carrying the pSJS1240 plasmid. When the cultures reached optical densities at 600 nm of 0.6, protein expression was induced by incubation in the presence of 1 mM IPTG for 3 h after which time the cells were harvested. Bacterial lysates were prepared by sonication (five 30-s pulses using a Sonifier 250, Branson) in 30 ml of buffer A containing 200 mM NaCl, 20% (w/v) sucrose, 1 mM 4-(2-aminoethyl)benzenesulphonylfluoride (Calbiochem), 140 ng/ml Pepstatin and 400 ng/ml Leupeptin. Cell debris was removed by centrifugation at 20 000 g for 30 min, and the supernatants were heated at 65°C for 15 min. This heat step was left out for the preparation of the afRFC large subunit, since the isolated large subunit proved to be heat sensitive. After centrifugation at 20 000 g for 30 min to partially remove denatured E.coli proteins, ammonium sulfate was added to the supernatants to 80% saturation and the precipitates were collected by centrifugation. For the purification of the afRFC complex and the afRFC large subunit, the precipitates were resuspended in buffer B containing 50 mM imidazole and loaded in three batches onto a 5-ml HiTrap Chelating HP column (Amersham Biosciences) previously charged with NiCl. After washing with 30 ml of buffer B containing 50 mM imidazole, bound proteins were eluted with buffer B containing 500 mM imidazole. Ammonium sulfate was added to the eluates to 80% saturation and the precipitates were collected by centrifugation. The precipitates were resuspended in 2 ml buffer A containing 500 mM NaCl and applied to a Superdex 200 gel filtration column (Amersham Biosciences) equilibrated with buffer A containing 500 mM NaCl. For the purification of afRFCsm, the precipitate was resuspended in buffer A to obtain a conductivity approximately corresponding to 100 mM NaCl and loaded in two batches onto a 5-ml HiTrap Heparin HP column (Amersham Biosciences). The column was washed with 30 ml of buffer A containing 100 mM NaCl and developed with a 50-ml linear gradient of NaCl from 100 mM to 1 M in buffer A. The pooled protein peak, which eluted at ∼250 mM NaCl, was diluted 2-fold with buffer A and loaded in two batches onto a Mono Q HR 10/10 column (Amersham Biosciences) equilibrated with buffer A containing 100 mM NaCl. The column was washed with 50 ml of buffer A containing 100 mM NaCl and developed with a 64-ml linear gradient of NaCl from 100 to 500 mM in buffer A. The pooled protein peak, which eluted at ∼300 mM NaCl, was concentrated to 2 ml using a Centricon YM-10 device (Millipore) and applied to a Superdex 200 gel filtration column (Amersham Biosciences) equilibrated with buffer A containing 500 mM NaCl. For the purification of afPCNA, the precipitate was resuspended in buffer C to obtain a conductivity approximately corresponding to 100 mM NaCl and loaded onto a 15-ml SOURCE 30 Q column (Amersham Biosciences) equilibrated with buffer C. The column was washed with 90 ml of buffer C and developed with a 160-ml linear gradient of NaCl from 0 to 500 mM in buffer C. The protein peak, which eluted at ∼300 mM NaCl, was pooled, adjusted to 1 M ammonium sulfate, and applied to a 10-ml SOURCE 15 ISO column (Amersham Biosciences) equilibrated with buffer C containing 1 M ammonium sulfate. After washing with 60 ml of buffer C containing 1 M ammonium sulfate, the column was developed with a 100-ml linear gradient of ammonium sulfate from 1 to 0 M in buffer C. The pooled protein peak was diluted with buffer C to a conductivity corresponding approximately to 200 mM NaCl and loaded onto a Mono Q HR 10/10 column (Amersham Biosciences) equilibrated with buffer C. The column was washed with 50 ml of buffer C, developed with a 64-ml linear gradient of NaCl from 0 to 500 mM in buffer C, and the protein peak, which eluted at ∼300 mM NaCl, was pooled. For the purification of afPolB1, the precipitate was resuspended in buffer D to obtain a conductivity corresponding to 100 mM NaCl and loaded onto a 5-ml HiTrap Heparin HP column (Amersham Biosciences) equilibrated with buffer D containing 100 mM NaCl. The column was washed with 30 ml of buffer D containing 100 mM NaCl and developed with a 50-ml linear gradient of NaCl from 100 mM to 1 M in buffer D. The pooled protein peak, which eluted at ∼500 mM NaCl, was concentrated to 2 ml using a Centricon YM-50 device (Millipore) and applied to a Superdex 75 gel filtration column (Amersham Biosciences) equilibrated with buffer A containing 200 mM NaCl. After purification, 10% (v/v) glycerol was added to the proteins, which were subsequently stored in small aliquots at –80°C and only thawed immediately before use. For the purification of tmPK, the precipitate was resuspended in buffer E to obtain a conductivity corresponding to 50 mM NaCl and loaded onto a 15-ml SOURCE 30 Q column (Amersham Biosciences) equilibrated with buffer E. The column was washed with 90 ml of buffer E and developed with a 200-ml linear gradient of NaCl from 0 to 500 mM in buffer E. The protein peak, which eluted at ∼100 mM NaCl, was pooled and applied to a 30-ml F3GA-blue Sepharose column pre-equilibrated with buffer E containing 100 mM NaCl. The protein was eluted with buffer E containing 1 M NaCl, followed by concentration to 5 mg/ml using an ultrafiltration cell equipped with a YM-30 ultrafiltration membrane (Millipore). For the purification of tmLDH, the precipitate was resuspended in buffer C to a conductivity corresponding to 50 mM NaCl and applied to a 30-ml F3GA-blue Sepharose column pre-equilibrated with buffer C containing 50 mM NaCl. The protein was eluted with buffer C containing 2 mM NADH and 50 mM NaCl, the pH of the eluate was adjusted to 8.5, and the protein was loaded onto a 15-ml SOURCE 30 Q column (Amersham Biosciences) equilibrated with buffer E. The column was washed with 90 ml of buffer E and developed with a 200-ml linear gradient of NaCl from 0 to 500 mM in buffer E. The protein peak, which eluted at ∼200 mM NaCl, was pooled and dialysed against buffer E containing 100 mM NaCl and 1 g/l (w/v) charcoal. Ammonium sulfate was added to the purified tmPK and tmLDH proteins to 80% saturation and the proteins were stored at 4°C. For use in the continous spectrophometric ATPase assay, on the same day of use the tmPK and tmLDH precipitates were collected by centrifugation, resuspended in 50 mM HEPES–KOH (pH 7.5), 7 mM MgCl2 and 100 mM KCl and concentrated using Centricon YM-10 devices (Millipore) to obtain final concentrations of 1.2 µg/µl tmPK and 0.8 µg/µl tmLDH.
Denaturing gel filtration of afRFC
Gel filtration under denaturing conditions was used to determine the afRFC complex stoichiometry. afRFC (2 nmol) was mixed with an equal volume of column buffer containing 50 mM potassium phosphate (pH 7.5), 2 mM DTT and 6 M guanidinium hydrochloride. The protein was applied in a final volume of 200 µl to a Superdex 200 HR10/30 gel filtration column (Amersham Biosciences) equilibrated with column buffer. The column was connected to an ÄKTApurifier system (Amersham Biosciences). Peak quantification of the elution profile obtained at 280 nm was completed according to the supplier’s instructions. The obtained values, 68.71 mAU for the peak representing the afRFC large subunit or 101.06 mAU for the peak representing the afRFC small subunit, were divided by the respective extinction coefficients, 66 570 for afRFCla or 23 140 for afRFCsm, which were calculated using ProtParam software (19). 250 µl of the collected 500 µl column fractions were buffer exchanged against buffer B, concentrated to 10 µl using Microcon YM-10 devices (Millipore) and separated on a 12.5% SDS–PAGE gel.
Analytical ultracentrifugation
Sedimentation equilibrium experiments were carried out in a Beckman XL-A analytical ultracentrifuge (Beckman, Palo Alto, CA) using an AnTi 60 rotor. Samples were exhaustively dialysed against a buffer containing 50 mM Tris–HCl (pH 7.5), 500 mM NaCl and 10% (v/v) glycerol, and loaded into a six-channel centrepiece. Equilibrium was attained at 5000, 7000 and 8500 r.p.m. Buffer density and partial specific volume was calculated according to the method of Laue et al. (20). Data from each channel were excised and analysed using the software provided with the centrifuge.
Elongation of a singly primed M13 DNA template by afPolB1
afPolB1 catalysed elongation of singly primed M13 single-stranded circular DNA was carried out in reaction mixtures (20 µl) containing 40 mM Tris–HCl (pH 7.5), 0.5 mM DTT, 10 µg/ml BSA, 7 mM magnesium chloride, 200 mM NaCl, 2 mM ATP, 250 µM each of dATP, dTTP and dGTP, 10 µM dCTP, 2.5 µCi [α-32P]dCTP (6000 Ci/mmol), 25 fmol of singly primed M13 DNA (primed with 5′-CAAGCTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTCGTAATCATGG-3′), and other proteins as indicated in the figure legend. Reaction mixtures were incubated for 30 min at 65°C, stopped with 10 mM EDTA, and separated in a 1.2% (w/v) alkaline agarose gel followed by autoradiography.
ATPase assays
Generally, the ATPase activity of afRFC was determined using a continuous spectrophotometric assay. This assay measures the loss of NADH absorbance at 340 nm (εM = 6250 cm–1M–1) as ADP is converted to ATP through the coupled action of pyruvate kinase and lactate dehydrogenase in the presence of phosphoenolpyruvate (21). 150 pmol afRFC were allowed to equilibrate at 55°C for 5 min in 1 ml of buffer F containing 2 mM phosphoenolpyruvate, 1 mM d-fructose 1,6-bisphosphate, 125 µM NADH, 50 µg tmLDH and 85 µg tmPK. When indicated, 450 pmol afPCNA or 30 µg DNA were added. The reactions were started by adding ATP (5 µM to 2 mM) and monitored at 340 nm for 15 min. Because of the limited thermal stability of phosphoenolpyruvate, d-fructose 1,6-bisphosphate and NADH the ammonium molybdate–malachite green assay was used to determine the temperature optimum of the afRFC ATPase activity (22). Reactions were carried out in buffer F containing 2 mM ATP. When ATPase rates <0.02 s–1 had to be determined, the turnover of radioactively labelled ATP was analysed. The 20 µl reaction mixtures contained buffer F, 5 µM to 2 mM ATP, 40 pmol afRFC and, where indicated, 50 pmol afPCNA. After various time points of incubation at 55°C, 1 µl aliquots were spotted onto polyethyleneimine–cellulose thin layer plates which were developed in 0.15 M formic acid–0.15 M LiCl (pH 3.0) as the liquid phase and analysed by phosphorimaging.
Determination of afRFC–afPCNA interactions
Pull-down assays and gel filtration were used to determine interactions between afRFC proteins and afPCNA. In the pull-down assays 150 pmol of afRFC or 20 µg of afRFCla were incubated with 400 pmol of afPCNA in a buffer containing 20 mM potassium phosphate (pH 7.5), 500 mM NaCl, 20 mM magnesium chloride, and nucleotides in concentrations ranging from 1 to 10 mM. After 20 min at room temperature, the reaction mixtures were supplemented with 40 µl of Ni-Cam™ HC Resin (Sigma) pre-equilibrated in the same buffer. After 15 min of further incubation at room temperature, the beads were pelleted (30 s, 5000 g) and washed four times with 200 µl of a buffer containing 20 mM potassium phosphate (pH 7.5), 2 M NaCl, 20 mM magnesium chloride and, where indicated, nucleotides. Captured proteins were eluted by boiling in SDS sample buffer and aliquots of the reactions were analysed by SDS–PAGE on 15% gels. Coomassie brilliant blue-stained protein bands were quantified using Image Scanner Software (Amersham Biosciences).
Nucleic acid binding assays
Nucleic acid binding reactions were done in a final volume of 10 µl containing 20 mM Tris–HCl (pH 7.5), 20 mM magnesium chloride, 2 mM DTT, 100 µM EDTA, 5% (v/v) glycerol, 50 fmol of nucleic acid substrate, and afRFC proteins in the range of 0.25–5 pmol. Where indicated 10 pmol of afPCNA and 2 mM ATP, ATPγS or AMP-PNP were added. Binding mixtures were left at room temperature for 15 min, adjusted to 5% glycerol and 0.1% (w/v) bromphenol blue, and loaded onto non-denaturing 8% polyacrylamide–0.8× TBE gels. The gels were electrophoresed at 70 V for 90 min, dried under vacuum and analysed by phosphorimaging.
RESULTS
Expression and purification of afRFC individual subunits, the two-subunit afRFC complex, afPCNA and afPolB1
The aim of this study was to analyse the biochemical properties of the A.fulgidus clamp loader. We chose to express the large subunit of the dimeric afRFC as an N-terminally His-tagged fusion protein in E.coli to allow us to separate complexes containing both large and small subunits from those consisting only of afRFC small subunits. The genes encoding the afRFC large (AF1195; 17) and afRFC small subunits (AF2060; 17) were inserted into E.coli expression vectors. The co-expression of large (57 675 Da) and small (35 994 Da) subunits, as well as the separate expression of the small subunit, resulted in soluble proteins. In contrast, the expression of the large subunit on its own resulted in mostly insoluble protein. In addition, unlike the complex or the small subunit alone, the isolated large subunit was also sensitive to heat denaturation (15 min, 65°C). However, we were able to purify the soluble fraction of the E.coli expressed large subunit, subsequently designated afRFCla, by Ni-chelating chromatography and gel filtration.
The small subunit (subsequently designated afRFCsm) and the afRFC large/small complex (subsequently designated afRFC) were purified to near homegeneity as described in Materials and Methods. However, afRFCsm eluted from the Superdex 200 gel filtration column as an apparently heterogenous complex (data not shown). The elution volume corresponded, when compared with globular molecular weight standards, to a molecular mass of between 200 000 and 105 000 Da. In contrast, afRFC eluted from native gel filtration columns as a single, symmetrical peak with an elution volume of the peak fraction which corresponded, when compared with globular molecular weight standards, to a molecular mass of 270 000 Da (data not shown). This native gel filtration data would suggest a hexameric (expected molecular mass of two large and four small subunits is 259 326 Da) and not a heteropentameric (expected molecular mass of one large and four small subunits is 201 651 Da) composition of afRFC. However, since the native conformation of afRFC is most likely not globular, we sought to analyse the stoichiometry of afRFC further. We carried out a gel filtration under denaturing conditions as described in Materials and Methods. Denaturing of afRFC by 6 M guanidinium hydrochloride resulted in a good separation of the afRFC large and small subunits (see elution profile in Fig. 1A and compare lane L with lanes 3–8 in Fig. 1B). Peak quantification of the elution profile obtained at 280 nm and dividing by the respective extinction coefficients as described in Materials and Methods resulted in a 1 large:4.2 small afRFC subunit ratio, which favoured a heteropentameric afRFC complex composed of 1 large and 4 small subunits.
Figure 1.

Gel filtration of afRFC under denaturing conditions. (A) Elution profile. Shown is the trace at 280 nm, the fraction numbers are indicated at the x-axis. (B) Coomassie brilliant blue-stained 12.5% SDS–PAGE gel. Lane M, molecular mass marker; lane L, 5 µl of the 100 µl loaded sample; lanes 3–8, 20 µl each of column fractions 18–23.
In order to obtain a different and more precise evaluation of the stoichiometry we employed analytical ultracentrifugation. Sedimentation equilibrium gives molecular weight information in a rigorous, thermodynamic method without the need for standards, as used in gel filtration. Figure 2 shows the results of such an experiment: molecular weight versus absorbance plots were calculated from the data at three concentrations at 7000 r.p.m. It can be seen that the plots are flat and are at a value of ∼200 000 Da: this is consistent with single species corresponding to a stoichiometry of 1 large to 4 small subunit complex. Globally fitting the sedimentation equilibrium data from three loading concentrations, taken at three speeds, yields a molecular weight of 202 496 Da with limits of 201 337 and 203 659 quoted at the 67% confidence limit. This result is again consistent with a subunit stoichiometry of 1 large:4 small (201 651 Da).
Figure 2.
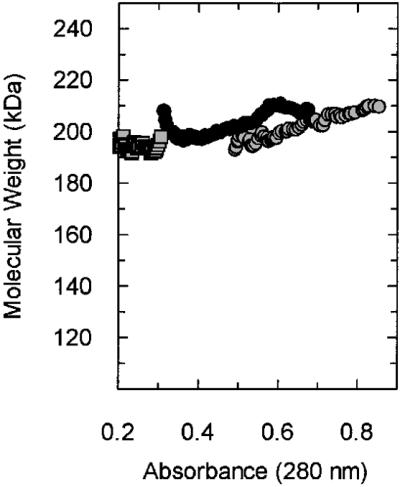
Plots of molecular weight versus concentration for data taken at 7000 r.p.m. The data show a flat profile, distributed around a molecular weight of ∼200 000 Da. This is consistent with a subunit stoichiometry of 1 large:4 small. This result was confirmed by globally fitting the data taken at three concentrations and three speeds (see Results). The loading concentrations were A280 = 0.2 (squares), A280 = 0.4 (black circles) and A280 = 0.6 (grey circles). Molecular weight versus absorbance plots were generated using the software provided with the centrifuge.
afRFC strongly stimulates the processivity of afPolB1 in the presence of afPCNA
To determine whether the purified afRFC was functional, we also expressed and purified the PCNA homologue (AF0335; 17) and a type B polymerase (PolB1, AF0497; 17) from A.fulgidus. Both proteins were overexpressed in E.coli and purified as described in Materials and Methods. Using primed closed circular M13mp18 DNA and mixtures of afRFC, afPCNA and afPolB1, we tested the functionality of the purified proteins in primer extension assays. As shown in Figure 3, in the presence of afPCNA, afRFC strongly stimulated both the amount of overall synthesis and the processivity of afPolB1 compared with reactions containing only afPolB1 (compare lane 1 to lanes 5–7). When used in separate reactions, neither 4 pmol of afPCNA (Fig. 3, lane 3) nor 4 pmol of afRFC (Fig. 3, lane 4) significantly stimulated DNA synthesis by afPolB1. However, at a 3-fold higher afPCNA concentration, some stimulation of DNA synthesis could be observed (Fig. 3, lane 2). The reactions shown in Figure 3 contained 200 mM NaCl since at NaCl concentrations <150 mM, afPolB1 alone showed considerable processivity even in the absence of afPCNA. This phenomenon has already been described for other archaeal family B polymerases (23).
Figure 3.
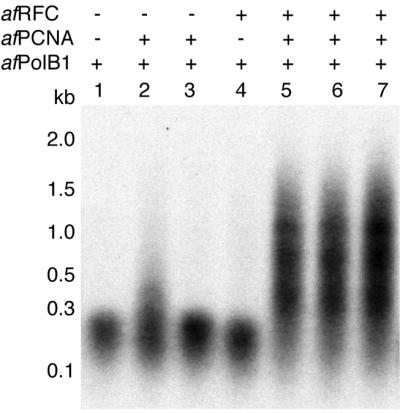
Effects of afRFC and afPCNA on DNA synthesis catalysed by afPolB1. Reaction mixtures (20 µl) were as described in Materials and Methods and contained 200 mM NaCl. All reactions contained 25 fmol of singly primed, closed circular M13mp18 DNA and 12 pmol of afPolB1. Where indicated, 4 pmol (lanes 3 and 5) or 12 pmol (lanes 2, 6 and 7) of afPCNA and 4 pmol (lanes 4, 5 and 6) or 12 pmol (lane 7) of afRFC were added. Reactions were incubated for 30 min at 65°C, precipitated and analysed by alkaline gel electrophoresis. An autoradiogram of a dried gel is shown.
Characterisation of the afRFC ATPase activity
All clamp loader proteins analysed to date possess intrinsic ATPase activity that can be stimulated both by various nucleic acids and by their clamp protein partner (7,13–16). As shown in Table 1, we investigated the ATPase properties of afRFC in the presence or absence of various nucleic acid substrates, and of afPCNA. Using the ammonium molybdate–malachite green assay, we determined 70°C and a magnesium concentration of 20 mM as optimal for the afRFC ATPase activity (data not shown). Even in the presence of saturating concentrations of ATP (2 mM) the rate of ATP hydrolysis was very slow for afRFC alone (kcat = 0.01 s–1 at 55°C, Km for ATP of 10 µM) and could only be determined by using [γ-32P]ATP, the hydrolysis products of which were analysed by thin layer chromatography followed by quantification of released Pi on a phosphorimager. Using a continous coupled ATPase assay we determined that, in the presence of various DNA substrates, kcat was increased at least 50-fold, with a slightly increased Km for ATP of 25 µM. However, we did not find major differences in the ability of various DNA substrates to stimulate the ATPase rate of afRFC. Thus, single-stranded DNA oligonucleotides, double-stranded DNA, 5′- or 3′-tailed DNA substrates, single-stranded or primed M13mp18 DNA, as well as an RNA/DNA heteroduplex gave essentially the same results. However, we could not observe significant stimulation using a single-stranded RNA 21mer. Generally, if afPCNA was added to DNA-containing reactions, a further 2–3-fold increase of the ATPase rate of afRFC was observed. Interestingly, while we observed this afPCNA-dependent increase in the ATPase rate in the presence of both tailed and single-stranded DNA, we did not observe it in the absence of DNA or in the presence of the closed circular M13mp18 DNA. This result suggests that the presence of a non-covalently linked DNA end is essential to obtain the PCNA-dependent afRFC ATPase stimulation. Inhibition of the clamp loader ATPase activity by its clamp in the absence of DNA has also been described for the E.coli γ complex (7,24) and for the human RFC (25), but contrasts with data obtained for yeast RFC (8).
Table 1. Stimulation of afRFC ATPase activity by different DNA effectors and by afPCNA.
| DNA | kcat (per s) | kcat in the presence of afPCNA (per s) |
|---|---|---|
| None | 0.01 | 0.003 |
| ssM13 | 0.50 | 0.37 |
| Primed ssM13 | 1.03 | 2.96 |
| Random ss 30mer | 0.74 | 2.06 |
| Random ds 30mer | 0.48 | 2.59 |
| Random ss 60mer | 1.04 | 2.08 |
| 5′-tailed 60mer | 1.03 | 2.60 |
| 3′-tailed 60mer | 0.99 | 1.96 |
| RNA (21mer) | 0.01 | 0.02 |
| RNA/DNA heteroduplex (21mer/60mer) | 0.94 | 1.94 |
afRFC-catalysed ATP (2 mM) hydrolysis was measured at 55°C, in the absence and presence of afPCNA (450 pmol) and DNA (30 µg). The kcat was determined by dividing the rate of ADP generation by afRFC concentration.
We could not detect a significant ATPase rate of afRFCla and the rate of ATP hydrolysis of afRFCsm was very slow with a kcat = 0.02 s–1 at 55°C in the presence of DNA that increased 2–3-fold in the presence of afPCNA (data not shown).
ATP binding, but not hydrolysis, is essential for afRFC–afPCNA interactions
In light of the stimulation of the afRFC ATPase activity by both DNA and afPCNA, we sought to analyse these interactions further. First, we investigated the interaction between afRFC and afPCNA using both pull-down assays and gel filtration. For the pull-down assays, afRFC was coupled to Ni–agarose beads via the His-tag located at the N-terminus of the large subunits. This complex was incubated with a molar excess of afPCNA in the presence or absence of ATP, ADP, or the ATP analogues AMP-PNP or ATPγS. Because of its imido bond, AMP-PNP is a completely non-hydrolysable analogue of ATP, whereas ATPγS can be hydrolysed by some enzymes. However, we could not detect any ATPγS hydrolysis by afRFC even after prolonged incubation (data not shown) and therefore regard ATPγS as non-hydrolysable in our studies. To determine the level of non-specific binding, afPCNA was incubated with the beads in the absence of afRFC (Fig. 4C, lanes 5–8). The observed amount of non-specifically bound afPCNA (Fig. 4C, lanes 7 and 8) was then subtracted from all reactions. The amount of afPCNA bound in the presence of afRFC and 1 mM ATP was taken to be 100% and all other values were normalised to this value. As shown in Figure 4A, only about one-quarter (27%) of the amount of afPCNA that binds to afRFC in the presence of 1 mM ATP also binds in the absence of nucleotide (Fig. 4A, compare lanes 3 and 4 with 7 and 8). The non-hydrolysable analogues ATPγS and AMP-PNP supported the interaction between afRFC and afPCNA, albeit to differing extents. Hence, whereas 1 mM ATPγS was able to substitute for ATP (88%; Fig. 4B, lanes 7 and 8), 1 mM AMP-PNP stimulated the interaction between afRFC and afPCNA only marginally (36%; Fig. 4B, lanes 3 and 4). However, at 10 mM AMP-PNP, the highest concentration analysed, this value increased to 72% (data not shown), indicating that 1 mM AMP-PNP was not saturating under our assay conditions. In contrast, ADP did not stimulate the interaction between afRFC and afPCNA at concentrations ranging from 1 mM (27%; Fig. 4C, lanes 3 and 4) to 10 mM (data not shown). We sought to verify these results by a different method. Since gel filtration is a non-equilibrium technique, only tight interactions are detected. We incubated afRFC and afPCNA in buffers containing no nucleotide, 1 mM ATP or 1 mM ADP and ran the mixtures on 24-ml gel filtration columns at room temperature (data not shown). Significant afRFC–afPCNA co-elution could only be detected when ATP was present in the running buffer, but not in the presence of ADP or in the absence of nucleotide. Since afRFC has only very low ATPase activity at room temperature (kcat = 0.02 s–1 in the presence of ssM13), this gel filtration data favours the conclusion drawn from the pull-down assays, that ATP binding rather than hydrolysis drives formation of afRFC–afPCNA complexes. When ATP was present only in the initial sample, but not in the running buffer, the interaction with afPCNA was lost on the column, indicating that the Kd of afRFC for ATP was too high to keep ATP bound during the gel filtration run.
Figure 4.

ATP binding, but not hydrolysis, is needed to stimulate afRFC–afPCNA interactions. 150 pmol of afRFC were incubated with 400 pmol of afPCNA in the presence of magnesium and various nucleotides. afRFC–afPCNA complexes were captured by incubation with Ni–agarose beads as described in Materials and Methods. Captured proteins were eluted by boiling in SDS sample buffer and aliquots of the reactions were analysed by SDS–PAGE on 15% gels followed by staining with Coomassie brilliant blue dye. To improve quantification, 30% as well as 70% of the eluates were run on the gels. Where indicated above the respective lanes, nucleotides (ATP, AMP-PNP, ATPγS or ADP) at final concentrations of 1 mM were added to both the reaction mixtures and the washing buffers. After correction for non-specifically bound afPCNA [(C), lanes 7 and 8], the amount of afPCNA bound to afRFC in the presence of ATP was taken to be 100% and all other values were normalised to this value. The results of these calculations are shown below the respective lanes. Lanes M, molecular mass markers; lanes 1 and 5, 10% of reaction mixtures; lanes 2 and 6, 10% of supernatants after incubation with Ni–agarose beads; lanes 3 and 7, 30% of eluates; lanes 4 and 8, 70% of eluates.
Using the gel filtration assay, we also detected ATP-dependent afRFCsm–afPCNA interactions. However, when afPCNA binding of the same amounts of afRFC small subunit present on its own or in the large/small subunit complex were compared, only about one-fifth of afPCNA that bound to the afRFC complex also bound to afRFCsm (data not shown).
As Figure 5 shows, using the pull-down technique, we could also detect significant interaction between afRFCla and afPCNA. However, unlike the afRFC–afPCNA and afRFCsm–afPCNA interactions, this complex formation was not stimulated by ATP (Fig. 5, compare lanes 3 and 4 with 7 and 8).
Figure 5.

ATP does not stimulate afRFCla–afPCNA interactions. Aliquots of 20 µg of afRFCla were incubated with 400 pmol of afPCNA in the absence (lanes 1–4) or presence of ATP (lanes 5–8). AfRFCla–afPCNA complexes were captured and analysed as described in Materials and Methods and in the legend to Figure 4. Lane M, molecular mass markers; lanes 1 and 5, 10% of reaction mixtures; lanes 2 and 6, 10% of supernatants after incubation with Ni–agarose beads; lanes 3 and 7, 30% of eluates; lanes 4 and 8, 70% of eluates.
Influence of nucleotides on afRFC–DNA–afPCNA interactions and specificity of afRFC–afPCNA for DNA versus RNA substrates
We analysed the influence of ATP and its non-hydrolysable analogues AMP-PNP and ATPγS on the interaction between afRFC and DNA in the absence or presence of PCNA. Clamp loaders are believed to load their clamp onto the free 5′ single strand of primer–template substrates (26). Therefore, in the experiments shown in Figure 6, we used a DNA substrate that consisted of a 30-nt duplex and a (dT)30 tail attached to one of the 5′ ends. If afPCNA was not present in the reactions (Fig. 6A) and at 2 mM final concentration of nucleotide, ATP (triangles) resulted in the greatest amount of shifted DNA, followed by ATPγS (circles) and AMP-PNP (diamonds). Using afRFCsm, we did not detect significant amounts of shifted DNA in these assays (data not shown), suggesting that the main DNA interacting domain of afRFC resides in the large subunit. afPCNA stimulated the interaction of afRFC with DNA significantly, both in the absence and presence of nucleotide. However, in the presence of afPCNA, ATPγS resulted in the highest amount of shifted DNA (Fig. 6B, circles). It has been shown in other systems that clamp loaders are released from ternary clamp loader–clamp–DNA complexes after ATP hydrolysis, leaving the clamp assembled on DNA (7,8,27,28). Since the substrate we used in our gel shift assays was not circular, once loading of afPCNA was completed, afPCNA should be able to slip off the free ends of the DNA substrate. Therefore, the result that ATPγS gave a higher amount of shifted DNA compared with ATP indicates that the loading of afPCNA by afRFC was stalled in the presence of ATPγS.
Figure 6.

Effects of nucleotides on afRFC–DNA interactions in the absence (A) or presence of 10 pmol afPCNA (B). Increasing amounts of afRFC (0.25–5 pmol) were incubated as described in Materials and Methods with 50 fmol of a 5′-tailed DNA substrate consisting of a 30mer (5′-CTGACCGTCGAGCACCGCTGCGGCTGCACC-3′) annealed to a 60mer (5′-T30GGTGCAGCCGCAGCGGTGCTCGACGGTCAG-3′). In addition, the reactions either contained 2 mM ATP (triangles), 2 mM ATPγS (circles), 2 mM AMP-PNP (diamonds), or were carried out in the absence of nucleotide (squares). Reaction products were separated by native gel electrophoresis in 8% polyacrylamide gels. The amount of shifted DNA (fmol) was quantitated by phosphorimager analysis and plotted against the amount of afRFC (pmol) in the reactions.
After synthesising an RNA–DNA hybrid primer of 35–50 residues (29), the eukaryotic polymerase/primase, Pol α, is replaced by Pol δ in a process that involves RFC–PCNA (30,31). Therefore we also investigated the interaction of afRFC–afPCNA with RNA and RNA/DNA heteroduplexes. As shown in Figure 7, in the presence of ATPγS, we observed comparable amounts of shifted single-stranded DNA (circles) and 5′-tailed DNA duplex (diamonds). In comparison, slightly more RNA/DNA heteroduplex (triangles) and significantly less single-stranded RNA (squares) were bound.
Figure 7.

Comparison of afRFC–afPCNA binding to single-stranded DNA, single-stranded RNA, a 5′-tailed DNA duplex or an RNA/DNA heteroduplex. Increasing amounts of afRFC (0.25–5 pmol) were incubated as described in Materials and Methods with 50 fmol of the polynucleotides in the presence of 10 pmol afPCNA and 2 mM ATPγS. The single-stranded DNA substrate (circles) was a 30mer (5′-CTGACCGTCGAGCACCGCTGCGGCTGCACC-3′), the RNA substrate (squares) was a 21mer (5′-GAGCACCGCUGCGGCUGCACC-3′), and the RNA/DNA heteroduplex (triangles) consisted of the RNA 21mer annealed to the DNA 60mer already used for the generation of the 5′-tailed DNA substrate of Figure 6. Thus, the RNA/DNA heteroduplex was composed of 21 bp attached to a 5′ (dT)30 tail and a 3′ tail of nine deoxyribonucleotides. The data for the 5′-tailed DNA duplex (diamonds) was obtained from Figure 6B. Reaction products were separated by native gel electrophoresis in 8% polyacrylamide gels. The amount of shifted polynucleotide (fmol) was quantitated by phosphorimager analysis and plotted against the amount of afRFC (pmol) in the reactions.
DISCUSSION
The process by which replicative DNA polymerases obtain processivity has been conserved among prokaryotes, eukaryotes and archaea both mechanistically and in terms of the participating proteins (see 1 for a recent review). Thus, generally, replicative DNA polymerases are tethered to DNA by ring-shaped proteins, called sliding clamps. To overcome the problem of loading a toroidal protein onto DNA, sliding clamps depend on multisubunit protein complexes, called clamp loaders. Both the clamp loader of eukaryotes, RFC, and the minimal clamp loader of E.coli, the γ3δδ′ complex, are heteropentamers (3,10). Deletion analysis in yeast showed that each of the five different subunits of RFC is essential (9). In contrast, in all archaea analysed to date only two different proteins constitute the RFC-like clamp loader (14–16).
In this study, we describe the biochemical characterisation of a clamp–loader complex from the euryarchaeon A.fulgidus (afRFC). Our analysis showed that afRFC, like the eukaryotic and the minimal E.coli clamp loader, is a heteropentamer, consisting of one large and four identical small subunits. In primer extension assays, afRFC stimulated the processivity of purified A.fulgidus polymerase B1 in afPCNA-dependent reactions. We were also able to purify the small afRFC subunit and, albeit only to a limited extent, the large afRFC subunit on their own. Both subunits interact individually with afPCNA. However, unlike the afRFC small subunit and the afRFC complex, the afPCNA interaction of the large subunit was not stimulated by nucleotides. Multiple contact sites between the clamp loader and the sliding clamp have also been shown for human RFC (32) and have been suggested in the E.coli system (4). We could not detect significant DNA binding of afRFCsm, indicating that the main DNA-interacting domain of afRFC resides in the large subunit. In human RFC only the large subunit, but none of the small subunits, could be crosslinked to DNA (26). The DNA binding activity of the small human RFC subunits analysed so far (p40, p37 and p36) is very weak and could only be detected at low ionic strength (32). For the A.fulgidus RFC, like in the E.coli and eukaryotic systems (7,26,33), only ATP binding, but not hydrolysis, proved to be essential to stimulate clamp loader– clamp interactions as well as clamp loader–clamp–DNA interactions.
The interaction of afRFC–afPCNA with RNA was significantly lower than the interaction detected with DNA or RNA/DNA substrates. To date, the length of the predicted RNA/DNA primers generated during initiation of DNA replication is not known for archaea. However, archaeal primases show similar in vitro activities as eukaryotic primases (34,35). Thus, like the eukaryotic primase/Pol α (29), archaeal primases might generate a similar RNA/DNA primer of ∼40 nt in length, including ∼10 nt of RNA. Assuming a similar recognition of primer–template junctions as eukaryotic RFC–PCNA (26), afRFC–afPCNA might rather contact the DNA than the RNA moiety of RNA/DNA primers generated during the initiation of DNA replication.
The afRFC ATPase activity depended strongly on the presence of DNA (at least 50-fold stimulation was observed). However, as described for other clamp loaders (7), we could not detect a significant preference for any of the DNA substrates. The ATPase activity of afRFC was a further 2–3-fold stimulated by afPCNA. Interestingly, in the absence of DNA, we detected a 3-fold inhibition of the already very low afRFC ATPase activity (kcat = 0.01/s at 55°C), if afPCNA was added to the reactions (kcat = 0.003/s at 55°C). This result is in agreement with a previous study of human RFC (25), in which it was suggested that the ATPase activity of RFC is stalled in RFC–PCNA complexes and the stalling is only released when PCNA is loaded onto DNA. Furthermore, we also determined a slight inhibition of the afRFC ATPase activity in the presence of single-stranded closed circular M13 DNA. This result provides additional evidence for the model presented by Shiomi et al. (25), since an unprimed closed circular DNA should not support RFC-dependent PCNA loading. A similar inhibition of the E.coli γ complex ATPase activity in the presence of unprimed ssM13 DNA by the β sliding clamp has been described (7).
Low- and high-resolution structures of E.coli, P.furiosus and human clamp loader proteins have provided fundamental insights into the function of these ATP-driven molecular machines (4,5,25,36,37). Overall, they showed a high extent of conservation, which is in good agreement with the observed similar biochemical properties. The biochemical data, however, also pinpoint important differences between the different systems. For example, whereas the E.coli clamp loader is released from the clamp–DNA composite after ATP hydrolysis (7), this is apparently not the case for the human RFC (31). Moreover, the individual roles that the five different eukaryotic RFC subunits play during the clamp loading process, for example which of them hydrolyse nucleotide and which are only essential for nucleotide binding, remains elusive at the moment. The similar, yet simplified, two-subunit archaeal clamp loader should provide a good model system to address these questions.
Acknowledgments
ACKNOWLEDGEMENTS
We thank N. Raven for providing A.fulgidus and T.maritima cells and S. Sandler for providing plasmid pSJS1240. This work was supported by the Medical Research Council, Cancer Research UK (formerly Imperial Cancer Research Fund), and a postdoctoral fellowship from the European Molecular Biology Organization (to A.S.).
REFERENCES
- 1.Jeruzalmi D., O’Donnell,M. and Kuriyan,J. (2002) Clamp loaders and sliding clamps. Curr. Opin. Struct. Biol., 12, 217–224. [DOI] [PubMed] [Google Scholar]
- 2.Pritchard A.E., Dallmann,H.G., Glover,B.P. and McHenry,C.S. (2000) A novel assembly mechanism for the DNA polymerase III holoenzyme DnaX complex:association of δδ′ with DnaX4 forms DnaX3δδ′. EMBO J., 19, 6536–6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Onrust R. and O’Donnell,M. (1993) DNA polymerase III accessory proteins. II. Characterization of δ and δ′. J. Biol. Chem., 268, 11766–11772. [PubMed] [Google Scholar]
- 4.Jeruzalmi D., O’Donnell,M. and Kuriyan,J. (2001) Crystal structure of the processivity clamp loader gamma (γ) complex of E. coli DNA polymerase III. Cell, 106, 429–441. [DOI] [PubMed] [Google Scholar]
- 5.Jeruzalmi D., Yurieva,O., Zhao,Y., Young,M., Stewart,J., Hingorani,M., O’Donnell,M. and Kuriyan,J. (2001) Mechanism of processivity clamp opening by the δ subunit wrench of the clamp loader complex of E. coli DNA polymerase III. Cell, 106, 417–428. [PubMed] [Google Scholar]
- 6.Naktinis V., Onrust,R., Fang,L. and O’Donnell,M. (1995) Assembly of a chromosomal replication machine: two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. II. Intermediate complex between the clamp loader and its clamp. J. Biol. Chem., 270, 13358–13365. [PubMed] [Google Scholar]
- 7.Turner J., Hingorani,M.M., Kelman,Z. and O’Donnell,M. (1999) The internal workings of a DNA polymerase clamp-loading machine. EMBO J., 18, 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gomes X.V. and Burgers,P.M. (2001) ATP utilization by yeast replication factor C.I. ATP-mediated interaction with DNA and with proliferating cell nuclear antigen. J. Biol. Chem., 276, 34768–34775. [DOI] [PubMed] [Google Scholar]
- 9.Cullmann G., Fien,K., Kobayashi,R. and Stillman,B. (1995) Characterization of the five replication factor C genes of Saccharomyces cerevisiae. Mol. Cell. Biol., 15, 4661–4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee S.H., Kwong,A.D., Pan,Z.Q. and Hurwitz,J. (1991) Studies on the activator 1 protein complex, an accessory factor for proliferating cell nuclear antigen-dependent DNA polymerase delta. J. Biol. Chem., 266, 594–602. [PubMed] [Google Scholar]
- 11.Cann I.K. and Ishino,Y. (1999) Archaeal DNA replication: identifying the pieces to solve a puzzle. Genetics, 152, 1249–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uhlmann F., Cai,J., Gibbs,E., O’Donnell,M. and Hurwitz,J. (1997) Deletion analysis of the large subunit p140 in human replication factor C reveals regions required for complex formation and replication activities. J. Biol. Chem., 272, 10058–10064. [DOI] [PubMed] [Google Scholar]
- 13.Podust V.N., Tiwari,N., Ott,R. and Fanning,E. (1998) Functional interactions among the subunits of replication factor C potentiate and modulate its ATPase activity. J. Biol. Chem., 273, 12935–12942. [DOI] [PubMed] [Google Scholar]
- 14.Pisani F.M., De Felice,M., Carpentieri,F. and Rossi,M. (2000) Biochemical characterization of a clamp-loader complex homologous to eukaryotic replication factor C from the hyperthermophilic archaeon Sulfolobus solfataricus. J. Mol. Biol., 301, 61–73. [DOI] [PubMed] [Google Scholar]
- 15.Kelman Z. and Hurwitz,J. (2000) A unique organization of the protein subunits of the DNA polymerase clamp loader in the archaeon Methanobacterium thermoautotrophicum ΔH. J. Biol. Chem., 275, 7327–7336. [DOI] [PubMed] [Google Scholar]
- 16.Cann I.K., Ishino,S., Yuasa,M., Daiyasu,H., Toh,H. and Ishino,Y. (2001) Biochemical analysis of replication factor C from the hyperthermophilic archaeon Pyrococcus furiosus. J. Bacteriol., 183, 2614–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klenk H.P., Clayton,R.A., Tomb,J.F., White,O., Nelson,K.E., Ketchum,K.A., Dodson,R.J., Gwinn,M., Hickey,E.K., Peterson,J.D. et al. (1997) The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature, 390, 364–370. [DOI] [PubMed] [Google Scholar]
- 18.DelTito B.J. Jr, Ward,J.M., Hodgson,J., Gershater,C.J., Edwards,H., Wysocki,L.A., Watson,F.A., Sathe,G. and Kane,J.F. (1995) Effects of a minor isoleucyl tRNA on heterologous protein translation in Escherichia coli. J. Bacteriol., 177, 7086–7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gill S.C. and von Hippel,P.H. (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem., 182, 319–326. [DOI] [PubMed] [Google Scholar]
- 20.Laue T.M., Shah,B.D., Ridgeway,T.M. and Pelletier,S.L. (1992) Computer-aided interpretation of analytical sedimentation data for proteins. In Harding,S.E., Rowe,A.J. and Horton,J.C. (eds), Analytical Ultracentrifugation in Biochemistry and Polymer Science. Royal Society of Chemistry, Cambridge, UK, pp. 90–125.
- 21.Norby J.G. (1988) Coupled assay of Na+,K+-ATPase activity. Methods Enzymol., 156, 116–119. [DOI] [PubMed] [Google Scholar]
- 22.Lanzetta P.A., Alvarez,L.J., Reinach,P.S. and Candia,O.A. (1979) An improved assay for nanomole amounts of inorganic phosphate. Anal. Biochem., 100, 95–97. [DOI] [PubMed] [Google Scholar]
- 23.Kelman Z., Pietrokovski,S. and Hurwitz,J. (1999) Isolation and characterization of a split B-type DNA polymerase from the archaeon Methanobacterium thermoautotrophicum deltaH. J. Biol. Chem., 274, 28751–28761. [DOI] [PubMed] [Google Scholar]
- 24.Hingorani M.M., Bloom,L.B., Goodman,M.F. and O’Donnell,M. (1999) Division of labor-sequential ATP hydrolysis drives assembly of a DNA polymerase sliding clamp around DNA. EMBO J., 18, 5131–5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shiomi Y., Usukura,J., Masamura,Y., Takeyasu,K., Nakayama,Y., Obuse,C., Yoshikawa,H. and Tsurimoto,T. (2000) ATP-dependent structural change of the eukaryotic clamp-loader protein, replication factor C. Proc. Natl Acad. Sci. USA, 97, 14127–14132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsurimoto T. and Stillman,B. (1991) Replication factors required for SV40 DNA replication in vitro. I. DNA structure-specific recognition of a primer-template junction by eukaryotic DNA polymerases and their accessory proteins. J. Biol. Chem., 266, 1950–1960. [PubMed] [Google Scholar]
- 27.Hingorani M.M. and O’Donnell,M. (1998) ATP binding to the Escherichia coli clamp loader powers opening of the ring-shaped clamp of DNA polymerase III holoenzyme. J. Biol. Chem., 273, 24550–24563. [DOI] [PubMed] [Google Scholar]
- 28.Gomes X.V., Schmidt,S.L. and Burgers,P.M. (2001) ATP utilization by yeast replication factor C. II. Multiple stepwise ATP binding events are required to load proliferating cell nuclear antigen onto primed DNA. J. Biol. Chem., 276, 34776–34783. [DOI] [PubMed] [Google Scholar]
- 29.Murakami Y., Eki,T. and Hurwitz,J. (1992) Studies on the initiation of simian virus 40 replication in vitro: RNA primer synthesis and its elongation. Proc. Natl Acad. Sci. USA, 89, 952–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsurimoto T. and Stillman,B. (1991) Replication factors required for SV40 DNA replication in vitro. II. Switching of DNA polymerase α and δ during initiation of leading and lagging strand synthesis. J. Biol. Chem., 266, 1961–1968. [PubMed] [Google Scholar]
- 31.Yuzhakov A., Kelman,Z., Hurwitz,J. and O’Donnell,M. (1999) Multiple competition reactions for RPA order the assembly of the DNA polymerase delta holoenzyme. EMBO J., 18, 6189–6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cai J., Gibbs,E., Uhlmann,F., Phillips,B., Yao,N., O’Donnell,M. and Hurwitz,J. (1997) A complex consisting of human replication factor C p40, p37, and p36 subunits is a DNA-dependent ATPase and an intermediate in the assembly of the holoenzyme. J. Biol. Chem., 272, 18974–18981. [DOI] [PubMed] [Google Scholar]
- 33.Gerik K.J., Gary,S.L. and Burgers,P.M. (1997) Overproduction and affinity purification of Saccharomyces cerevisiae replication factor C. J. Biol. Chem., 272, 1256–1262. [DOI] [PubMed] [Google Scholar]
- 34.Desogus G., Onesti,S., Brick,P., Rossi,M. and Pisani,F.M. (1999) Identification and characterization of a DNA primase from the hyperthermophilic archaeon Methanococcus jannaschii. Nucleic Acids Res., 27, 4444–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu L., Komori,K., Ishino,S., Bocquier,A.A., Cann,I.K., Kohda,D. and Ishino,Y. (2001) The archaeal DNA primase: biochemical characterization of the p41-p46 complex from Pyrococcus furiosus. J. Biol. Chem., 276, 45484–45490. [DOI] [PubMed] [Google Scholar]
- 36.Oyama T., Ishino,Y., Cann,I.K., Ishino,S. and Morikawa,K. (2001) Atomic structure of the clamp loader small subunit from Pyrococcus furiosus. Mol. Cell., 8, 455–463. [DOI] [PubMed] [Google Scholar]
- 37.Mayanagi K., Miyata,T., Oyama,T., Ishino,Y. and Morikawa,K. (2001) Three-dimensional electron microscopy of the clamp loader small subunit from Pyrococcus furiosus. J. Struct. Biol., 134, 35–45. [DOI] [PubMed] [Google Scholar]