


Abstract
Sequencing of a large number of microbial genomes has led to the discovery of new enzymes involved in tRNA biosynthesis and tRNA function. Preparation of a great variety of RNA molecules is, therefore, of major interest for biochemical characterization of these proteins. We describe a fast, cost-effective and efficient method for in vitro production of tRNA transcripts. T7 RNA polymerase requires a double-stranded DNA promoter in order to initiate transcription; however, elongation does not require a double-stranded DNA template. A partially double-stranded transcription template formed by annealing of a short oligonucleotide, complementary to the T7 promoter, to a larger oligonucleotide is shown to be a good substrate for in vitro transcription. This method allows rapid production of a variety of tRNA transcripts which can be aminoacylated well. This eliminates the need for cloning of tRNA genes, large-scale plasmid preparation and enzymatic digestion.
INTRODUCTION
T7 RNA polymerase has been widely used to produce tRNA transcripts in vitro. In vitro synthesis of tRNA has allowed biochemical characterization of a large number of enzymes and pathways, such as maturation and modification of tRNAs (1), tRNA aminoacylation (2), tRNA export from the nucleus (3) and translation in general (4). A standard method for in vitro transcription is based on recognition of the specific promoter sequence by T7 RNA polymerase and subsequent transcription of tRNA genes encoded in double-stranded plasmid DNA. Synthesis of the tRNA 3′-terminal CCA sequence is accomplished by run-off transcription with T7 RNA polymerase using a template generated by enzymatic digestion (typically BstNI) (5).
This method has been proven to be successful, but it occasionally causes addition or omission of nucleotides at the 3′ end, which impairs the ability of such tRNAs to be aminoacylated (6,7). Another drawback of the method arises from the fact that RNAs can be successfully transcribed in vitro only if the first base is a purine (8,9). If the first base of the RNA is a cytosine or a uracil, the transcription start site is often altered and erroneous nucleotides are likely to be incorporated (9–11). This problem was overcome by including a DNA template sequence starting with a stretch of G residues and encoding a hammerhead ribozyme upstream of the tRNA gene (12). Once transcribed, ribozyme self-cleavage generates the tRNA transcript, starting with the desired, specified nucleotide.
Recently, two efficient methods for tRNA production were proposed (13): preparation of the double-stranded DNA template by PCR or in vitro ligation by T4 RNA ligase of two pieces of chemically synthesized RNA. The PCR method avoids time-consuming cloning and large-scale preparation of DNA templates, but still requires the PCR step prior to the transcription reaction itself. Ligation of two pieces of RNA is straightforward and even allows incorporation of modified bases into the tRNA; however, the cost of chemically synthesized RNAs may limit its use.
Double-stranded DNA is required for recognition and efficient binding of the T7 RNA polymerase to the promoter region, although it is not imperative for extension (8,14–17). T7 RNA polymerase, in contrast to SP6 RNA polymerase (18), can accommodate single-stranded DNA template. It was shown earlier that small RNAs could be transcribed from single-stranded templates (8,14). Here we show a method for in vitro production of tRNA gene transcripts that eliminates the time-consuming DNA maxipreparations, restriction digestions and PCR. The transcription template is formed by annealing two oligonucleotides: the longer oligonucleotide comprises the tRNA gene and at its 3′ end a 23 nt sequence complementary to the shorter oligonucleotide. The resulting double-stranded region forms the T7 promoter (Fig. 1). The tRNA 3′-terminal CCA end is subsequently generated by run-off transcription.
Figure 1.

Diagram of DNA templates used for in vitro generation of tRNA gene transcripts. The shaded portion indicates the two strands of the T7 RNA polymerase promoter (23 nt). The white spotted and the black spotted portions represent sequences complementary to those shown for the B.burgdorferi tRNALys (76 nt for the full-length molecule, 34 and 42 nt for the 5′-half and 3′-half tRNA molecules) and M.jannaschii tRNAPro (78 nt), respectively. The striped portion represents the transcription template encoding the hammerhead ribozyme (HR) (57 nt). The numbers represent the nucleotide positions in the tRNA sequences. The arrow indicates the ligation position of the two half tRNA molecules.
MATERIALS AND METHODS
Chemicals, enzymes and oligonucleotides
Plasmid T7-911 for preparation of the His6-tagged recombinant T7 RNA polymerase was a gift from T. Shrader (Department of Biochemistry, Albert Einstein College of Medicine, New York, NY). Purification of the Borrelia burgdorferi lysyl-tRNA synthetase (LysRS) and Methano coccus jannaschii prolyl-tRNA synthetase (ProRS) were performed as described (19,20).The oligonucleotides were synthesized by the Keck Foundation Biotechnology Resource Laboratory at Yale. NTPs were purchased from Sigma.
Oligonucleotides and template preparation
The following DNA oligonucleotides were chemically synthesized (the underlined sequence indicates the double-stranded promoter region of the transcription template): T7 promoter 1 (T7.1), 5′-GGATCCTAATACGACTCACTATA-3′; T7 promoter 2 (T7.2), 5′-GGATCCTAATACGACTCACTATAGGGCT-3′; T7 promoter3 (T7.3), 5′-GGATCCTAATACGACTCACTATAGGGCTCATAG-3′; B.burgdorferi tRNALys (BBLys1), 5′-TGGTGAGCTCAGTAGGACTCGAACCTACGACAAACGCCTTAAAAGGGCGCTGCTCTACCACCTGAGCTATGAGCCCTATAGTGAGTCGTATTAGGATCC-3′; M.jannaschii tRNAPro (MJPro), 5′-TGG TGGGCCTGCCCAGATTTGAACTGGGGTCTCAGGATCCCAAATCCCAAAGGATAGACCAGGCTACCCCACAGGCCCTATAGTGAGTCGTATTAGGATCC-3′.
Transcription templates for production of B.burgdorferi tRNALys were prepared by annealing of the tRNA encoding oligonucleotide (BBLys1) to different T7 promoter oligonucleotides (T7.1, T7.2 or T7.3). Similarly, templates for production of M.jannaschii tRNAPro were constructed by annealing of oligonucleotide MJPro to T7.1. Annealing conditions for all the templates were as follows: a mixture of the long oligonucleotide (BBLys1 for instance, 25 µM) and of the short oligonucleotide (T7.1, 25 µM) was incubated at 95°C for 5 min and allowed to gradually cool to 25°C.
RNA ligation
The following DNA oligonucleotides were chemically synthesized (the underlined sequence indicates the double-stranded promoter region of the transcription template and the bold portion indicates the hammerhead ribozyme sequence): B.burgdorferi tRNALys nucleotides 1–34 (BBLys2), 5′-AAGGGCGCTGCTCTACCACCTGAGCTATGAGCCCGACGGTACCGGGTACCGTTTCGTCCTCACGGACTCATCAGCGGCTCATAGCTCTCCCTATAGTGAGTCGTATTAGGATCC-3′; B.burgdorferi tRNALys nucleotides 35–76 (BBLys3), 5′-TGGTGAGCTCAGTAGGACTCGAACCTACGACAAACGCCTTAAGACGGTACCGGGTACCGTTTCGTCCTCACGGACTCATCAGCGGCTCATAGCTCTCCCTATAGTGAGTCGTATTAGGATCC-3′. Oligonucleotides BBLys2 and BBLys3 were annealed to the T7 promoter oligonucleotide T7.1 and used as a transcription template as described above. Ribozyme cleavage was promoted by incubation at 60°C for 45 min. The two ‘half’ tRNA molecules were purified on a 12% polyacrylamide gel, extracted, ethanol precipitated and ligated using T4 RNA ligase as previously described (13). The ligation product of the expected size was then purified on a 12% polyacrylamide gel, extracted and ethanol precipitated.
Transcription
In order to determine the optimal ratio of T7 RNA polymerase to template DNA necessary for efficient transcription, variable amounts (from 0 to 2.5 µM) of the annealed oligonucleotides were tested in transcription reactions. Reactions were carried out as described before (5) in 40 mM Tris–HCl, pH 8.0, 22 mM MgCl2, 1 mM spermidine, 5 mM DTT, 0.5% Triton-X100, 4 mM each NTP, 5 mM GMP and 30 nM T7 RNA polymerase for 3 h at 37°C. Aliquots (5 µl) of the transcription reaction were run on a 12% polyacrylamide gel containing 8 M urea. The gel was stained with toluidine blue to quantify the yield of the reactions. For tRNA preparation, the transcription reaction products were phenol/chloroform extracted, ethanol precipitated, resuspended in sterile water and purified on a 12% polyacrylamide gel. The purified tRNAs were extracted from the gel as described (12).
Aminoacylation reaction
Aminoacylation reactions were performed as described before (19,20). Reaction mixtures contained 100 mM HEPES– NaOH pH 7.2, 10 mM MgCl2, 50 mM KCl, 5 mM DTT, 10 mM ATP, 30 µM [14C]lysine (300 c.p.m./pmol; NEN), 5 µM tRNALys transcript and 500 nM B.burgdorferi LysRS for 30 min at 37°C. For tRNAPro transcript aminoacylation, reactions were conducted in 50 mM Tris–HCl pH 7.5, 50 mM KCl, 15 mM MgCl2, 5 mM DTT, 10 mM ATP, 19 µM [14C]proline (500 c.p.m./pmol; Amersham), 5 µM tRNAPro transcript and 300 nM M.jannaschii ProRS at 60°C.
Preparation of transcripts from double-stranded plasmid DNA templates
Transcripts of B.burgdorferi tRNALys and M.jannaschii tRNAPro were prepared as previously described (19,20). Plasmid DNA was obtained by large-scale preparation using the Qiagen® Plasmid Maxi Kit and digested overnight with BstNI restriction enzyme (NEB). After phenol/chloroform extraction, the digested DNA was ethanol precipitated and used for in vitro transcription at a concentration of 0.1 mg/ml.
RESULTS AND DISCUSSION
Structural data (21,22) and biochemical evidence show that a double-stranded T7 promoter is a prerequisite for T7 RNA polymerase recognition and formation of the initiation complex. However, this is not the case for the elongation complex, as the role of the non-coding strand is limited to facilitating RNA displacement after transcription (23). T7 RNA polymerase has been shown to transcribe single-stranded DNA templates of up to 40 nt linked to the double-stranded promoter region (8,14). Therefore, we investigated the transcription properties of similar partially double-stranded DNA templates with longer single-stranded template regions sufficient to encode a tRNA gene.
Minimum double-stranded region
Given that the transcription yield depends on the stabilization that the non-coding strand contributes to the transcription complex, we investigated how an extended double-stranded region beyond the promoter would affect the reaction yield. This was tested by comparing two constructs, in which the double-stranded region extended 5 bp (BBLys1/T7.2) or 10 bp (BBLys1/T7.3) beyond the promoter (23 bp in length), with that in which only the promoter region is double stranded (BBLys1/T7.1). In line with earlier data on short RNAs (8), adding only up to 10 bases to the double-stranded DNA portion had little effect on the transcription yield of full-length tRNAs (Fig. 2). As expected, the oligonucleotide (BBLys1) without a double-stranded promoter was not a template for transcription (Fig. 2).
Figure 2.

In vitro transcription of B.burgdorferi tRNALys with double-stranded regions of variable length. Aliquots (5 µl) of the transcription reactions were loaded on 12% polyacrylamide–8 M urea gels. Increasing amounts of template (0–2.5 µM) were used in lanes 1–7. C1, DNA transcription template (10 µl BBLys1); C2, B.burgdorferi tRNALys transcript (1.1 µg) obtained by transcription of digested plasmid DNA. (A) No double-stranded portion (BBLys1). (B) Only the T7 promoter is double-stranded (BBLys1/T7.1). (C) The double-stranded portion extends to +5 on the tRNA gene (BBLys1/T7.2). (D) The double-stranded portion extends to +10 on the tRNA gene (BBLys1/T7.3).
Maximum length of the single-stranded region
We showed that transcription of a 76 nt single-stranded DNA template allowed production of B.burgdorferi tRNALys. Therefore, we investigated if a longer single-stranded DNA template could also be a suitable template for in vitro transcription. Because the addition of 57 nt (the hammerhead ribozyme sequence) is required in the cases of the inappropriate 5′-terminus of the mature tRNA, we tested transcription with a single-stranded DNA portion of up to 133 nt (the ribozyme sequence and the B.burgdorferi tRNALys gene). This did not work satisfactorily, as the yield of the expected product was low and many shorter RNA fragments were formed. Thus, we considered RNA ligation of tRNA halves (13), each generated by transcription from a single-stranded DNA of about 90 nt (the ribozyme construct and half a tRNA gene linked to the double-stranded T7 promoter; see Fig. 1). After self-cleavage of the ribozymes, the two desired RNA fragments were gel purified (Fig. 3). Ligation of the two tRNA halves with T4 RNA ligase generated a full-length tRNA-sized product (Fig. 3) that was subsequently purified and aminoacylated.
Figure 3.
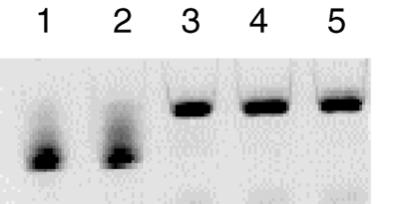
Production of B.burgdorferi tRNALys by in vitro ligation of the two RNA halves. Lane 1, 5′-half of tRNA (nucleotides 1–34); lane 2, 3′-half of tRNA (nucleotides 35–76); lane 3, ligation product (T4 RNA ligase) of the two half molecules of lanes 1 and 2; lanes 4 and 5, in vitro transcript of B.burgdorferi tRNALys obtained by transcription from a single-stranded or from a double-stranded DNA template, respectively.
Transcription yield and biological activity of transcribed RNA
Using two different tRNA genes in multiple reactions we compared (Table 1) the amount of tRNA obtained with our method based on single-stranded DNA transcription to that derived from transcription of double-stranded DNA (5,13). The transcription yield in our method was 40–50% of that obtained with the double-stranded template procedure. Similar yield differences were reached in transcription of other tRNA genes (data not shown). This is not unexpected, as double-stranded DNA is known to be a better template for transcription of short RNAs (15). Nevertheless, our procedure afforded an appreciable amount of tRNA transcript. Judged by their ability to be aminoacylated by the cognate aminoacyl-tRNA synthetase, the tRNA transcription products generated by the two different methods had equal biological activity (Table 1).
Table 1. Comparison of transcript tRNA production methods.
| tRNA | Transcription yield (µg)a | Aminoacylation (pmol/A260) |
|---|---|---|
| B.burgdorferi tRNALys | ||
| dsDNA template | 420 | 400 |
| ssDNA template | 200 | 400 |
| M.jannaschii tRNAPro | ||
| dsDNA template | 350 | 300 |
| ssDNA template | 140 | 300 |
aPer 0.5 ml reaction.
CONCLUSIONS
Single-stranded DNA templates longer than 100 nt are not suitable for in vitro transcription. This limitation can be overcome either by preparation of a double-stranded DNA template (by cloning or by PCR) or ligation of two tRNA half molecules obtained by transcription from a smaller single-stranded DNA template. Despite the fact that the standard in vitro transcription method using double-stranded DNA template yielded higher amounts of tRNA transcript, we find simplicity, quality of the transcript and cost-effectiveness to be the major advantages of the method described here. This single-stranded DNA template method should facilitate the production of the large number of tRNA mutant species that are routinely used for probing protein–RNA interactions.
Acknowledgments
ACKNOWLEDGEMENTS
We thank M. Ibba, S. Bunjun-Srihari and D. Tumbula-Hansen for constructive discussions. This work was supported by a grant from the National Institute for General Medical Sciences.
REFERENCES
- 1.Constantinesco F., Motorin,Y. and Grosjean,H. (1999) Characterisation and enzymatic properties of tRNA(guanine 26, N (2), N (2))-dimethyltransferase (Trm1p) from Pyrococcus furiosus. J. Mol. Biol., 291, 375–392. [DOI] [PubMed] [Google Scholar]
- 2.Sampson J.R., DiRenzo,A.B., Behlen,L.S. and Uhlenbeck,O.C. (1989) Nucleotides in yeast tRNAPhe required for the specific recognition by its cognate synthetase. Science, 243, 1363–1366. [DOI] [PubMed] [Google Scholar]
- 3.Lund E. and Dahlberg,J.E. (1998) Proofreading and aminoacylation of tRNAs before export from the nucleus. Science, 282, 2082–2085. [DOI] [PubMed] [Google Scholar]
- 4.LaRiviere F.J., Wolfson,A.D. and Uhlenbeck,O.C. (2001) Uniform binding of aminoacyl-tRNAs to elongation factor Tu by thermodynamic compensation. Science, 294, 165–168. [DOI] [PubMed] [Google Scholar]
- 5.Sampson J.R. and Uhlenbeck,O.C. (1988) Biochemical and physical characterization of an unmodified yeast phenylalanine transfer RNA transcribed in vitro. Proc. Natl Acad. Sci. USA, 85, 1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nissan T.A., Oliphant,B. and Perona,J.J. (1999) An engineered class I transfer RNA with a class II tertiary fold. RNA, 5, 434–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kholod N., Vassilenko,K., Shlyapnikov,M., Ksenzenko,V. and Kisselev,L. (1998) Preparation of active tRNA gene transcripts devoid of 3′-extended products and dimers. Nucleic Acids Res., 26, 2500–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Milligan J.F. and Uhlenbeck,O.C. (1989) Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol., 180, 51–62. [DOI] [PubMed] [Google Scholar]
- 9.Imburgio D., Rong,M., Ma,K. and McAllister,W.T. (2000) Studies of promoter recognition and start site selection by T7 RNA polymerase using a comprehensive collection of promoter variants. Biochemistry, 39, 10419–10430. [DOI] [PubMed] [Google Scholar]
- 10.Pleiss J.A., Derrick,M.L. and Uhlenbeck,O.C. (1998) T7 RNA polymerase produces 5′ end heterogeneity during in vitro transcription from certain templates. RNA, 4, 1313–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Helm M., Brule,H., Giegé,R. and Florentz,C. (1999) More mistakes by T7 RNA polymerase at the 5′ ends of in vitro-transcribed RNAs. RNA, 5, 618–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fechter P., Rudinger,J., Giegé,R. and Theobald-Dietrich,A. (1998) Ribozyme processed tRNA transcripts with unfriendly internal promoter for T7 RNA polymerase: production and activity. FEBS Lett., 436, 99–103. [DOI] [PubMed] [Google Scholar]
- 13.Sherlin L.D., Bullock,T.L., Nissan,T.A., Perona,J.J., Lariviere,F.J., Uhlenbeck,O.C. and Scaringe,S.A. (2001) Chemical and enzymatic synthesis of tRNAs for high-throughput crystallization. RNA, 7, 1671–1678. [PMC free article] [PubMed] [Google Scholar]
- 14.Milligan J.F., Groebe,D.R., Witherell,G.W. and Uhlenbeck,O.C. (1987) Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res., 21, 8783–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mentesana P.E., Chin-Bow,S.T., Sousa,R. and McAllister,W.T. (2000) Characterization of halted T7 RNA polymerase elongation complexes reveals multiple factors that contribute to stability. J. Mol. Biol., 302, 1049–1062. [DOI] [PubMed] [Google Scholar]
- 16.Krupp G. and Söll,D. (1987) Simplified in vitro synthesis of mutated RNA molecules. An oligonucleotide promoter determines the initiation site of T7 RNA polymerase on ss M13 phage DNA. FEBS Lett., 212, 271–275. [DOI] [PubMed] [Google Scholar]
- 17.Moran S., Ren,R.X., Sheils,C.J., Rumney,S.,IV and Kool,E.T. (1996) Non-hydrogen bonding ‘terminator’ nucleosides increase the 3′-end homogeneity of enzymatic RNA and DNA synthesis. Nucleic Acids Res., 24, 2044–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stump W.T. and Hall,K.B. (1993) SP6 RNA polymerase efficiently synthesizes RNA from short double-stranded DNA templates. Nucleic Acids Res., 21, 5480–5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ibba M., Bono,J.L., Rosa,P.A. and Söll,D. (1997) Archaeal-type lysyl-tRNA synthetase in the Lyme disease spirochete Borrelia burgdorferi. Proc. Natl Acad. Sci. USA, 94, 14383–14388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stathopoulos C., Jacquin-Becker,C., Becker,H.D., Li,T., Ambrogelly,A., Longman,R. and Söll,D. (2001) Methanococcus jannaschii prolyl-cysteinyl-tRNA synthetase possesses overlapping amino acid binding sites. Biochemistry, 40, 46–52. [DOI] [PubMed] [Google Scholar]
- 21.Cheetham G.M. and Steitz,T.A. (1999) Structure of a transcribing T7 RNA polymerase initiation complex. Science, 286, 2305–2309. [DOI] [PubMed] [Google Scholar]
- 22.Cheetham G.M. and Steitz,T.A. (2000) Insights into transcription: structure and function of single-subunit DNA-dependent RNA polymerases. Curr. Opin. Struct. Biol., 10, 117–123. [DOI] [PubMed] [Google Scholar]
- 23.Gopal V., Brieba,L.G., Guajardo,R., McAllister,W.T. and Sousa,R. (1999) Characterization of structural features important for T7 RNAP elongation complex stability reveals competing complex conformations and a role for the non-template strand in RNA displacement. J. Mol. Biol., 290, 411–431. [DOI] [PubMed] [Google Scholar]