


Abstract
Current methods for measuring the efficiency of splicing in mammalian cells rely on either direct analysis of the RNA, which does not lend itself to rapid assays, or on single reporter functions that are subject to numerous intrinsic variables. If two protein activities are encoded within a single reading frame but on separate exons, with an intervening sequence containing termination codons, then the expression of the second activity is dependent on removal of the intervening sequence by pre-mRNA splicing. Thus, the ratio of the activities encoded by exon 2 to exon 1 reflects the ratio of expression from spliced mRNA to the total expression of spliced and unspliced RNA. This provides a rapid and convenient assay for the effects on splicing efficiency of trans-acting factors or of alterations in the sequences of the intron and surrounding exon sequences.
INTRODUCTION
The splicing of mammalian pre-mRNA is complex in terms of both the signals that affect it and the number of factors involved in the reactions. Apart from the consensus sequences around the splice junctions, sequences at relatively distant sites within the exons and the introns are important in determining not just the efficiency of the reactions but also the sites of splicing. Understanding the roles of these sequences is important because the consensus sequences do not suffice to specify the correct sites; there are many additional sites that appear to satisfy the basic requirements and yet are not used. In addition, approximately one-half of all human genes encoding proteins appear to produce several isoforms of mRNA and protein by alternative splicing, often in a regulated way, but very few of the sequences that control the availability and level of use of the alternative sites have been identified yet.
The identification of factors that affect mammalian splicing is generally done by transfection of fragments of a gene into mammalian cells, followed by analysis of sequences included in the mRNA expressed. However, it is not usual for the efficiency of splicing of an intron to be measured directly. In most cases, the assay is based upon the examination of mature mRNA and addresses the mechanisms by which one site is selected rather than another. Thus, for example, preferences among candidate 5′ splice sites or 3′ splice sites are tested by the use of competing alternative sites (1–5) and the sequences that affect the use of an exon are tested by measuring the levels of mRNA in which the exon is included or excluded (skipped). This method has the advantage that many of the other factors that might affect expression, such as variations in transfection efficiency or transcription, are eliminated, while differences in stability of the various forms of mRNA cannot be ignored (2) but can be assumed to be less than might be expected between spliced and unspliced RNA. There are two major disadvantages to such strategies: there is no information bearing directly on the efficiency of splicing, and the direct analysis of mRNA by RT–PCR, RNase protection or northern blotting is relatively laborious and does not lend itself to high throughput analyses. Reporter assays have been used in several cases, but these have been single reporter functions (6,7), in which activity depends on splicing to remove an intron or incorporate an exon, and they are susceptible to variation between samples in the levels of transfection, transcription, etc.
Inferences drawn on the basis of competition between alternative splicing pathways can be misleading. For example, the numerous studies using RT–PCR to analyse incorporation or skipping of an exon after mutagenesis of local sequences are very readily interpreted in terms of exon definition, a widespread and important phenomenon that is of considerable value in understanding splice site usage, but they may not reveal cases in which one of the flanking introns has been spliced, simply because partially spliced RNA is less likely to be detected proportionately.
The most common alternative strategy for identification of factors that affect splicing efficiency is by the analysis of splicing in vitro, in a nuclear extract. This is a powerful and important method that has been used widely. However, it has several limitations: it requires the use of relatively short transcripts that usually lack the complexity and length of the natural pre-mRNA, and thus may not identify the rate-limiting steps for expression in vivo; it is difficult to test a wide range of protein factors because of the labour required to prepare these proteins (many of which, in the case of splicing factors, are not readily prepared in soluble or native form); and there are very few examples of active splicing extracts that have been made from differentiated cells, i.e. it is not a useful route in most cases for identifying sequences that affect tissue-specific splicing.
In contrast to the situation in mammalian cells, it is normal to analyse splicing efficiency as such in yeast. This is generally done by direct analysis of the mRNA and unspliced RNA, often by primer extension without amplification. A more rapid screen is possible with a reporter assay, again based on successful expression of a protein after splicing. A particularly good example is CUP1, in which activity can be measured as the copper resistance of growth and correlates very well with splicing efficiency (3). Many of the variables that affect transient expression assays in mammals are eliminated because stable and well-characterised clones are used.
To develop a simple and reliable assay for splicing efficiency, rather than competition, in mammals, a reporter activity that depends on splicing should be linked to a second activity that corrects for variations in either transfection efficiency or any other steps in transcription and processing. Ideally, the second activity would report the level of unspliced RNA. For this purpose, we have devised a reporter construct in which splicing produces one signal (luciferase activity) but both unspliced and spliced RNA produce β-galactosidase activity. Thus, the ratio of the two activities can be used as a rapid screen to detect mutations or trans-acting factors that might affect the efficiency of pre-mRNA splicing.
MATERIALS AND METHODS
Plasmid construction
The bifunctional reporter plasmid was based upon pBLUGA (8), which contains reading frames for β-galactosidase and luciferase, separated by a termination codon (to assay selenocysteine incorporation) in between SalI and BamHI sites. An exon–intron–exon unit was cloned into these sites. In the case of plasmid pTN23, the exon and intron comprised nucleotides 6084–6237 and 7026–7095 of Ad2, with a novel 5′ splice site at 6171/72 (9), which were joined to nucleotides –5 to +76 of exon 6 of the skeletal muscle-specific mRNA isoform of human αs-tropomyosin gene TPM3. Plasmid pTN24 contained an additional 25 nt from the tropomyosin intron, i.e. nucleotides –30 to +76 of TPM3 exon 6. Exonic (Ex mut) and intronic (In mut) mutations were obtained by site-directed mutagenesis where the (GGA)3 repeat and the nucleotides –9 to –26 of exon 6 were randomly replaced. For in vitro splicing assays, the same Ad2/TPM3 sequences were transcribed, but the adenovirus sequence began at nucleotide 6023 and included a natural 5′ splice site at 6079/80 (TN23ww and TN24ww). Complete reading frames for human Tra2β (Ayres et al., manuscript in preparation), Tra2α, hnRNPG-T and RBMY (Nasim et al., data not shown) were cloned in pEGFP-C1 and pECFP-N1 (Clontech) in frame with the fluorescent protein. Human hnRNPG was cloned in pCG10 vector (10).
Gene transfer, cell culture and enzymatic assay
HEK-293 (human embryonic kidney) cells were transiently transfected with relevant plasmids using Gene Jammer (Stratagene). Cells were harvested 48 h after transfection (11), and β-galactosidase and luciferase activities were measured using Dual Light® System (Biosystems).
Reverse transcription–polymerase chain reaction (RT–PCR)
Total RNA was isolated using TRIreagent (Helena Biosciences). Any contaminating DNA was removed by treatment with restriction endonuclease and DNase, extraction with solvent and ethanol precipitation. After reverse transcription, PCR was done with Extensor Ready mix 1 (ABgene) and primers directed to the β-galactosidase and luciferase sequences for 18, 22 and 26 cycles. One of the primers was labelled with [γ-32P]ATP, so for analysis the products were run onto a denaturing gel and quantified with a PhosphorImager.
RNA processing assay in cell free system
Conventional in vitro transcription and splicing reactions were carried out (12) and the splicing products were analysed on a polyacrylamide gel.
RESULTS
The general principle of the method we have adopted is shown in Figure 1. The reporter system used in this work is based on the genes encoding β-galactosidase and luciferase, which are highly sensitive and linear across several orders of magnitude. We have constructed a set of reporter constructs where these two reporter genes were fused with a recombinant fragment containing two exons and a single intron (Fig. 1A). Upon transfection of these reporter plasmids into a mammalian cell line, transcription from the SV40 promoter would produce a pre-mRNA, which contains three translation stop signals in the intron. This pre-mRNA could be processed in either of two ways. If it is not spliced, the mRNA would contain multiple translation stop signals in the intron. Translation termination would result in the production of the β-galactosidase protein alone. In the event of splicing, the internal stop signals would be removed and a β-galactosidase–luciferase fusion protein would result. Since the luciferase activity would be produced only after splicing, whereas β-galactosidase would be expressed by spliced and unspliced mRNA, the ratio of β-galactosidase and luciferase activities should indicate the proportion of cytoplasmic mRNA that is spliced. Complete splicing would give the highest ratio of luciferase to β-galactosidase. It might be expected that spliced and unspliced RNA would be polyadenylated, exported and degraded with different efficiencies, but the same limitation applies to the analysis of splicing by any other method.
Figure 1.

The test system (A) for in vivo splicing based on the reporter genes encoding β-galactosidase and luciferase, which are fused in frame via a recombinant fragment of adenovirus and human αs-tropomyosin genes. The recombinant fragment contains three in-frame translation stop signals (XXX) in the intronic region. Upon transfection of the respective plasmids into the HEK-293 cell line, the DNA would be transcribed under the control of SV40 promoter. Two different types of mRNAs would be produced. In the event of inefficient splicing the RNA produced would contain pre-mature termination codons. In case of efficient splicing, the translation stop signals would be removed which would lead to the production of a recombinant gal–luc fusion protein. The western blot (B) indicates the presence of gal–luc fusion protein, which was detected by anti luciferase HRP conjugate antibody. The gal–luc fusion protein is absent in the mock-transfected control extracts.
To test whether the assay worked, HEK-293 cells were transfected with a plasmid (pTN23) that contained an adenovirus 2 sequence as the 5′ exon and intron, and an alternatively spliced exon from tropomyosin as the 3′ exon. A second plasmid was identical but for the inclusion of 30 nt from the intron preceding the exon in tropomyosin (pTN24). The cells were harvested 48 h after transfection. Analysis by western blotting of cells transfected by pTN23 showed that a protein of a size appropriate for the fusion protein had been made (Fig. 1B). Measurement of the β-galactosidase and luciferase activities showed that the luc/gal ratio in cells transfected with pTN23 plasmid was significantly higher than that from cells transfected with pTN24 (Fig. 2A; all values are means from 6 to 12 independent transfections). Mutations in the exon and intron sequences resulted in significant changes in the luc/gal ratio, suggesting that there had been effects on the splicing efficiency.
Figure 2.

(Previous page) Analyses of the reporter system to determine splicing efficiencies in vivo. The reporter constructs along with plasmids encoding trans-acting factors were transiently transfected into HEK-293 cells and the activities of the β-galactosidase and luciferase proteins were measured and expressed as ratios, normalised to a value of 100 with TN 24 (A). The standard deviations of the normalised ratios are indicated by error bars. The total RNA from (A) was isolated and analysed by RT–PCR (B) where two DNA bands derived from spliced and unspliced RNA. Lanes 1–9 are the same samples as in (A) and (C), in the same order. Lane 10 is same as lane 1 except that there was no reverse transcriptase used. Lane 11 derived from mock-transfected control extracts. The percentages of spliced RNA were quantified (C) and the correlation (D) between the level of splicing measured by RT–PCR and luc/gal ratio was determined.
The sensitivity of the assay to trans-acting modulators of splicing efficiency was assayed by co-transfection with plasmids encoding Tra2α, Tra2β, hnRNP G, hnRNP G-T and RBMY. These produced marked changes in the luc/gal ratio. We have also transfected the plasmids pTN23 and pTN24 in C2C12 (mouse myoblast cell line, ATCC number CRL-1772) and NT2 precursor (human teratocarcinoma cell line, Stratagene) cell lines. The results were similar to those obtained in HEK-293 cells (data not shown).
To confirm that the luc/gal ratio was correlated with the proportion of spliced mRNA, the experiments were repeated and the RNA analysed by RT–PCR (Fig. 2B). The bands corresponding to spliced and unspliced RNA were quantified to measure the apparent proportion of spliced RNA (Fig. 2C). Strikingly, the results from analysis by RT–PCR agreed well with the results obtained using the reporter activities (Fig. 2D). There were parallel changes in the apparent level of splicing by both assays when the substrate was mutated or potential modulating factors were co-transfected. Thus, the absence of some or all of the 30 nt portion of tropomyosin intron in pTN23 and In mut enhanced the proportion of spliced mRNA, whereas an exon mutation reduced it considerably. Similarly, the two Tra2 proteins seemed to stimulate splicing, and the hnRNP G family proteins to reduce it, hnRNP G being most effective. We conclude that the conventional assay of mRNA splicing by RT–PCR validates the reporter assay and that the effects of mutation and trans-acting factors on the proportion of spliced mRNA can be ranked in effectiveness by this assay.
To confirm that the changes in the proportion of spliced RNA inferred from the RT–PCR and reporter assays were produced by changes in splicing efficiency, transcripts containing the adenovirus and tropomyosin portions of pTN23 and pTN24 were tested in standard in vitro splicing reactions. The transcripts contained an additional 60 nt of adenovirus sequence at the 5′ end, which included an alternative 5′ splice site. As with the results from both in vivo assays, it can be seen in Figure 3 that the tropomyosin intron sequence in TN24 inhibited splicing very substantially, particularly the second step. Compared with TN23, there was almost no mRNA, and the intermediates accumulated slightly.
Figure 3.
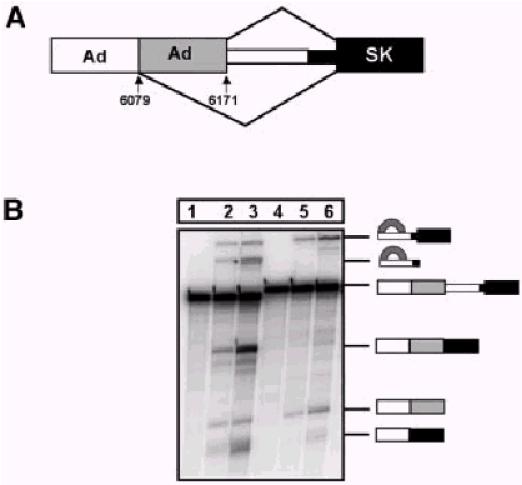
Splicing in vitro of RNA containing the intron and surrounding sequences from TN23 (lanes 1–3) and TN24 (lanes 4–6). (A) Diagram of substrates. (B) Transcripts were incubated in splicing reactions for 0 (lanes 1 and 4), 30 (lanes 2 and 5) or 60 (lanes 3 and 6) min and analysed by electrophoresis on denaturing gels. The precursor is marked, together with the 5′ exon intermediate and mRNA product derived by use of the 5′ splice site at 6171/6172. The lower mobility lariats and the mRNA at the foot of the panel are derived from use of the site at position 6079/6080.
DISCUSSION
The disadvantages of conventional methods for assaying splicing efficiency in mammalian systems prompted us to develop a method based on the use of an indirect reporter system that bypassed a number of the variables that affect single reporter assays. This system comprises two signals, derived from the translation of both spliced and unspliced RNA in mammalian cells. Upon splicing the internal translation stop signals are removed, which leads to the production of a β-galactosidase–luciferase (gal–luc) fusion protein (Fig. 1B). Without splicing, expression of β-galatosidase occurs but translation termination in the intron prevents the production of luciferase.
We have tested the dual-reporter assay in several ways to show that it reflects the efficiency of splicing. The highest ratio of luciferase/galactosidase activity was observed when the intron was removed (data not shown), and luciferase activity was completely abolished when the 5′ splice site was mutated (data not shown). Using conditions or mutations that are known to affect the efficiency of splicing, we found that there is a good correlation between the level of splicing determined by RT–PCR and the luciferase/galactosidase ratio (Fig. 2D).
There are several respects in which it was surprising that such a straightforward assay worked well. The first is that unspliced RNA tends to be retained in the nucleus (13). This is a difficulty that affects any attempt to measure the efficiency of splicing based on measurements of pre-mRNA levels, because the stability of retained RNA is likely to be different from that of exported RNA. One solution, although it does not lend itself to high throughput-type assays, is to measure the level of pre-mRNA for one intron compared with that of another intron in the same gene (14). However, previous investigations (13) and our own results indicate that some unspliced pre-mRNA is exported and translated, and our results suggest that the fraction of unspliced RNA exported might not change with changes in the efficiency of splicing. It is likely that some trans-acting factors or cis-elements being assayed might affect export, and this should be considered when interpreting any apparent increases in splicing efficiency.
Another potential complication envisaged was that the presence of translation termination codons in the pre-mRNA might compromise it. However, nonsense mediated decay (NMD) requires splicing (15); in the reporter assay we describe, the spliced mRNA has no premature termination codons, whereas the mRNA that does contain premature termination codons is, by definition, unspliced. There was no evidence of NMD from experiments (8,11) that investigated selenocysteine incorporation.
Apart from an increase in the proportion of spliced RNA or a decrease in pre-mRNA export, there are other routes by which the luciferase/galactosidase ratio might be increased which need to be borne in mind when appraising the results. For example, the ratio observed for pTN23 was increased dramatically (47-fold, data not shown), beyond that seen with the intron-less constructs, upon co-transfection with SRrp40 (TASR-2) (16,17). Investigation showed that the level of galactosidase activity was depressed in this case because a cryptic upstream 5′ splice site had been activated by SRrp40. Such instances are unlikely to significantly affect the usefulness of the method, as long as a second method is used to validate the most interesting candidates that emerge from a wide initial screen. In addition, it is possible to remove cryptic sites from any substrate of interest by mutagenesis.
This method is very convenient for a rapid screen with a panel of chemicals or co-transfected cDNA. However, a screen for positively acting factors should be done with introns that do not splice with high intrinsic efficiency. For example, a screen of SR proteins with pTN23 showed only modest improvements in the efficiency of splicing (not shown) because the level was high initially (Fig. 2B). It is possible that all assays based upon the analysis of spliced and unspliced or alternative mRNA in vivo might underestimate the effects of positive trans-acting factors. Even when 100% of the mRNA has been spliced, such factors could further increase the rate of splicing and thus reduce the losses of pre-mRNA to degradation in the nucleus. This would be manifest only as an increased level of mRNA.
This method would be useful to screen chemical libraries to identify drugs that affect specific splicing reactions. In this case it would be advantageous to set up stable incorporation of the bi-functional reporter into a mammalian cell line. This would allow screening for chemicals that would rescue or suppress particular splicing reactions in cases where there would be therapeutic implications, as in SMN2 and dystrophin. It would also allow screening of libraries to identify potentially toxic chemicals that might inhibit or induce aberrant patterns of splicing.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr G. Screaton (Oxford), Mr D. Hayward and Ms B. Dal Zilio for plasmids expressing some of the splicing activators. This work was supported by the Wellcome Trust.
REFERENCES
- 1.Eperon L.P., Estibeiro,J.P. and Eperon,I.C. (1986) The role of nucleotide sequences in splice site selection in eukaryotic pre-messenger RNA. Nature, 324, 280–282. [DOI] [PubMed] [Google Scholar]
- 2.Lear A.L., Eperon,L.P., Wheatley,I.M. and Eperon,I.C. (1990) Hierarchy for 5′ splice site preference determined in vivo. J. Mol. Biol., 211, 103–115. [DOI] [PubMed] [Google Scholar]
- 3.Lesser C.F. and Guthrie,C. (1993) Mutational analysis of pre-mRNA splicing in Saccharomyces cerevisiae using a sensitive new reporter gene, CUP1. Genetics, 133, 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nelson K.K. and Green,M.R. (1988) Splice site selection and ribonucleoprotein complex assembly during in vitro pre-mRNA splicing. Genes Dev., 2, 319–329. [DOI] [PubMed] [Google Scholar]
- 5.Reed R. and Maniatis,T. (1986) A role for exon sequences and splice-site proximity in splice-site selection. Cell, 46, 681–690. [DOI] [PubMed] [Google Scholar]
- 6.Roberts G.C., Gooding,C. and Smith,C.W. (1996) Smooth muscle alternative splicing induced in fibroblasts by heterologous expression of a regulatory gene. EMBO J., 15, 6301–6310. [PMC free article] [PubMed] [Google Scholar]
- 7.Woerfel G. and Bindereif,A. (2001) In vitro selection of exonic splicing enhancer sequences: identification of novel CD44 enhancers. Nucleic Acids Res., 29, 3204–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kollmus H., Flohe,L. and McCarthy,J.E. (1996) Analysis of eukaryotic mRNA structures directing cotranslational incorporation of selenocysteine. Nucleic Acids Res., 24, 1195–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Mullane L. and Eperon,I.C. (1998) The pre-mRNA 5′ cap determines whether U6 small nuclear RNA succeeds U1 small nuclear ribonucleoprotein particle at 5′ splice sites. Mol. Cell. Biol., 18, 7510–7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caceres J.F., Screaton,G.R. and Krainer,A.R. (1998) A specific subset of SR proteins shuttles continuously between the nucleus and the cytoplasm. Genes Dev., 12, 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nasim M.T., Jaenecke,S., Belduz,A., Kollmus,H., Flohe,L. and McCarthy,J.E. (2000) Eukaryotic selenocysteine incorporation follows a nonprocessive mechanism that competes with translational termination. J. Biol. Chem., 275, 14846–14852. [DOI] [PubMed] [Google Scholar]
- 12.Eperon I.C., Ireland,D.C., Smith,R.A., Mayeda,A. and Krainer,A.R. (1993) Pathways for selection of 5′ splice sites by U1 snRNPs and SF2/ASF. EMBO J., 12, 3607–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Legrain P. and Rosbash,M. (1989) Some cis- and trans-acting mutants for splicing target pre-mRNA to the cytoplasm. Cell, 57, 573–583. [DOI] [PubMed] [Google Scholar]
- 14.Patel A.A., McCarthy,M. and Steitz,J.A. (2002) The splicing of U12-type introns can be a rate-limiting step in gene expression. EMBO J., 21, 3804–3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maquat L.E. and Li,X. (2001) Mammalian heat shock p70 and histone H4 transcripts, which derive from naturally intronless genes, are immune to nonsense-mediated decay. RNA, 7, 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang L., Embree,L.J., Tsai,S. and Hickstein,D.D. (1998) Oncoprotein TLS interacts with serine-arginine proteins involved in RNA splicing. J. Biol. Chem., 273, 27761–27764. [DOI] [PubMed] [Google Scholar]
- 17.Cowper A.E., Caceres,J.F., Mayeda,A. and Screaton,G.R. (2001) Serine-arginine (SR) protein-like factors that antagonize authentic SR proteins and regulate alternative splicing. J. Biol. Chem., 276, 48908–48914. [DOI] [PubMed] [Google Scholar]