

Abstract
Reverse genetic approaches to understanding gene function would be greatly facilitated by increasing the efficiency of methods for isolating mutants without the reliance on a predicted phenotype. Established PCR-based methods of isolating deletion mutants are widely used for this purpose in Caenorhabditis elegans. However, these methods are inefficient at isolating small deletions. We report here a novel modification of PCR-based methods, employing thermostable restriction enzymes to block the synthesis of wild-type PCR product, so that only the deletion PCR product is amplified. This modification greatly increases the efficiency of isolating small targeted deletions in C.elegans. Using this method six new deletion strains were isolated from a small screen of approximately 400 000 haploid genomes, most with deletions <1.0 kb. Greater PCR detection sensitivity by this modification permitted ∼10-fold greater pooling of DNA samples, reducing the effort and reagents required for screens. In addition, effective suppression of non-specific amplification allowed multiplexing with several independent primer pairs. The increased efficiency of this technique makes it more practical for small laboratories to undertake gene knock-out screens.
INTRODUCTION
The availability of complete genomic sequences from a growing number of species provides the impetus for developing facile and rapid methods of reverse genetics as one means for understanding in vivo gene function. This is particularly true for the nematode Caenorhabditis elegans, whose approximately 19 000 predicted genes may provide valuable insights into highly conserved biological pathways, human disease mechanisms and the control of parasitic nematode species with medical and economic importance (1–3). Large-scale projects are now underway to systematically ascribe mutant phenotypes to either the reduction or loss-of-function for each predicted C.elegans gene, by the techniques of RNAi (4–6) and targeted gene deletion by PCR-based screens (7,8). Though RNAi-mediated suppression of gene function is rapid and highly specific, its usefulness for genetic analysis can be limited by apparent tissue- and stage-specific efficacy, potential mosaicism, and limited hereditability of the suppressive effect (9–11). In contrast, genetic strains created with targeted deletions of individual genes offer permanent and stable null phenotypes that may be more amenable for the discovery of modifier or suppressor genes, and for the phenotypic analysis of transgenes.
Several targeted deletion strategies have been developed for C.elegans, all based upon PCR detection assays. Similar strategies were first described with Drosophila (12,13). A library of randomly mutagenized worms is generated, either by chemical mutagenesis, primarily with ethyl methansulfonate (EMS) (14), or trimethyl psoralen and ultraviolet irradiation (TMP-UV) (15), or by the mobilization of transposable elements, employing either native Tc1 (16,17) or heterologous mariner transposons (18). Mutant worms are arrayed into an ordered library, typically consisting of approximately 1000 wells, each seeded with 20–500 individual mutant worms. This library is amplified through approximately two generations of growth. A portion of worms from each well is then lysed for genomic DNA, while the remaining live worms are maintained, either as viable dauers at 15°C or by freezing (7,19,20). Genomic DNA samples from each well are systematically pooled into fewer samples of higher complexity to represent the entire library. These samples are screened by PCR with primers designed to targeted genes, for the presence of a molecular deletion. Positive reactions direct fractionation and re-screening of the library, until individual wells containing the deletion are identified. Live animals archived from positive wells are recovered in cultures of low complexity, and similarly fractionated and screened to yield clonal cultures of mutant animals.
In these screens, the detection of positive samples by PCR critically depends upon an effective means to distinguish and amplify few copies of shorter amplicons resulting from a deletion, from a background of vastly more wild-type copies. A widely used approach is to limit the extension time of PCR cycles, thus favoring the amplification of shorter products (7,19,20). One limitation of this method is that only relatively large deletions (>1.0 kb) tend to be detected, because smaller deletions result in large PCR products which do not sufficiently differ from the wild-type product; thus they are not preferentially amplified over wild-type (8). Hence, a potentially significant pool of useful small deletions (<1.0 kb) generated by chemical mutagens such as EMS (21) may escape detection. An additional limitation is the relatively low sensitivity inherent with this strategy, since production of any WT product in competition with the deletion product tends to rapidly exhaust reaction substrates, effectively limiting the number of cycles that can be employed. These factors restrict the routine pooling of DNA samples to a complexity of ∼1:3000 (7,8,19,20), and sets the minimum number of samples required for screening a library.
We addressed these limitations by a simple modification, incorporating a digestion step with the thermostable restriction endonuclease PspGI. With this modification, we reliably detected copies of deletion amplicons in mixed DNA samples at a complexity of ∼1:40 000 (deletion:WT ratio). This allows us to greatly reduce the total number of PCRs required to screen our libraries. An additional benefit was a dramatic reduction of spurious amplification products resulting from non-specific priming. The lower backgrounds we achieved allowed us to multiplex up to three independent primer pairs in individual PCRs, increasing our throughput ability. This simple modification is compatible with existing methods for isolating C.elegans deletion mutants employed by the C.elegans Knockout Consortium (7,8,19,20,22), and enables cost-effective small scale efforts, targeting small sets of genes.
MATERIALS AND METHODS
Generation of mutant libraries and worm lysates
Libraries of mutant worms were created largely following established protocols (7,19,20,23). Synchronized populations of approximately 30 000 mid- to late-L4 staged N2 C.elegans were mutagenized with 25 mM EMS (Sigma-Aldrich, St Louis, MO) in M9 (24) for 4 h, rocking at room temperature. Animals were allowed to recover and become gravid overnight on OP50 bacteria lawns grown on 80 mm NGM agar plates. Mutagenized P0s were then lysed with 0.25 M KOH, 1.5% NaOCl to release eggs. F1 eggs were rinsed with M9 and allowed to hatch as arrested L1 larvae on NGM agar plates, in the absence of food. These L1s were collected in M9 suspension, counted and diluted to a concentration of approximately 10 animals/µl for plating the library. Approximately 10 000–30 000 mutagenized P0s provided more than sufficient L1 larvae for a library of 100 000 haploid genomes.
EMS mutagenesis rate for each library was determined by scoring an aliquot of L1 larvae for the heterozygous unc-22 ‘twitcher’ phenotype (15) with 3% nicotine (Sigma-Aldrich) in M9. We typically measured a forward mutation rate of ∼1.5 × 10–3 for unc-22, a large gene of ∼36 kb. Extrapolated to a typical gene of ∼5 kb, this represents a forward mutation rate of ∼2.1 × 10–4, comparable with rates previously reported (14,21). However, estimates suggest that only ∼10% of EMS-induced mutations result from deletions (14,20,21), making our forward deletion mutation rate ∼2.1 × 10–5.
Mutant worms were grown on 960 fifty millimeter RNGM agarose plates (7) with OP50 bacterial lawns. Each plate was seeded with approximately 50 mutagenized L1 larvae, for each library representing approximately 100 000 haploid genomes. Animals were grown through two generations at 24°C (∼5–7 days), until bacterial lawns began to clear.
A fraction of worms (∼25%) from each plate were transferred by rinsing with water (supplemented with 12.5 µg/ml nystatin, 100 µg/ml streptomycin, 0.5% Tween-20) (7) and ordered into ten 96-well blocks (1.0 ml well Dyna block; Midwest Scientific, St Louis, MO) for lysis. Crude worm lysates were made by adding proteinase K solution (200 µg/ml proteinase K in 50 mM KCl, 2.5 mM MgCl2, 0.45% NP-40, 0.45% Tween-20, 0.01% gelatin, 10 mM Tris–HCl, pH 8.3) (7). Worm samples were frozen at –80°C for 30 min, digested for 4–8 h at 65°C, then heated at 95°C for 20 min to inactivate proteinase K. Worm lysates were first pooled 10-fold, from ten 96-well samples to one master 96-well tray. DNA samples in this master tray were further pooled to strips of 12 and 8 wells, representing the rows and columns of the 96-well tray (Fig. 1A).
Figure 1.

Schematic of the deletion screen. (A) Method for pooling mutant worm library lysates (7,19,20). A library consisted of 960 individual plates, each seeded with approximately 50 L1 animals and allowed to grow for approximately two generations. One quarter of worms from each plate was transferred for lysis in 96-well blocks, while the remainder were maintained as dauers at 15°C. DNA lysates made from ten 96-well blocks were pooled to one master 96-well tray, increasing the genomic DNA complexity of each sample 10-fold to ∼1:1000. Lysates were further pooled into single 12-well and 8-well strips, each representing the entire library at sample complexities of 1:8000 and 1:12 000, respectively. From these two sample strips, the row and column coordinates for any positive signal were identified. (B) The PCR-screening method made use of nested primers flanking a thermostable endonuclease recognition site in the targeted gene. Wild-type product size was chosen to be ∼3 kb to allow for amplification of deletion products up to ∼2.6 kb. Digestion with thermostable restriction endonuclease allowed only deletion amplicons to be amplified. Deletions may cause a frameshift mutation, leading to premature truncation of the targeted gene, or other deleterious molecular defects. Two rounds of PCR were performed with primers designed with a high annealing temperature (∼65°C). Second round PCRs (outer primers) were performed with a dilution of the first round (inner primers) reaction.
The remaining live worms on each plate were maintained at 15°C for up to 5 weeks, during the screening process.
PCR deletion screen with PspGI digestion
Fifteen different nested primer pairs were designed to screen seven C.elegans genes encoding potassium channel subunits (primer sequences available upon request). These genes were slo-2 (F08B12.3), kqt-1 (C25B8.1), kqt-2 (M60.5), kqt-3 (Y54G9A.3), shl-1 (Y73B6BL.19), shw-3 (R186.5) and shk-1 (ZK1321.2). Primers were chosen with a relatively high annealing temperature (∼65°C), and designed to span one or several PspGI restriction sites within each targeted gene, allowing for ∼3.0 kb of WT genomic sequence to be amplified. Worm DNA lysates were digested with PspGI (0.1 U/25 µl; with 10 mM MgCl2) at 75°C for 1 h, prior to PCR. Following PspGI digestion, samples were screened by PCR following conventional protocols (7), by directly adding PCR substrates at 70°C, utilizing a ‘hot-start’ protocol (25) to minimize non-specific priming. Two rounds of amplification were used, an initial round of 35 cycles with the inner primer set, followed by a second round of 35 cycles with the corresponding outer primer set. For the second round of PCR, a dilution of the first round PCR product (1:200–1:500) was used as template and predigested with PspGI. Positive assays were identified by PCR products of a smaller size than expected for WT fragments, visualized by gel electrophoresis. The frequency of false positives required all initial positive signals to be verified in triplicate by identical PCRs. Only triplicated positive signals were further pursued.
True positive signals from strips of 12 and 8 wells gave coordinates on the master trays representing libraries of 100 000 haploid genomes. Samples representing these coordinates were further fractionated and screened, until one positive well representing a single plate was identified.
Live worms from single positive plates were recovered on NGM plates with OP50 bacterial lawns. One round of sib-selection was performed with approximately 400 clonal cultures of worms from each plate, grown in 24-well culture plates (TPP, Switzerland). The relatively low complexity of initial plates (approximately 50 F1s), allowed us to consistently identify single worms carrying deletions with only one round of sib-selection (Table 1).
Table 1. Summary of deletion screens.
| Screen no. | Sequence screened | Size scanned (kb) | No. of initial positives | No. of positives rescreened | Single plate ID | No. of clonal positives | Allele isolated | Deletion size (bp) |
|---|---|---|---|---|---|---|---|---|
| 1 | kqt-1, 5′ | 2.4 | 4/96 | 0/4 | ||||
| ∼100 000 genomes | slo-2, 5′ | 1.7 | 3/96 | 1/3 | Yes | 2/∼1056 | Nonea | 1554a |
| slo-2, pore | 2.5 | 2/96 | 0/2 | |||||
| slo-2, 3′ | 2.1 | 7/96 | 1/7 | Yes | 3/192 | slo-2(nf100) | 574 | |
| shl-1, 5′ | 2.8 | 0/96 | ||||||
| shl-1, 3′ | 2.4 | 0/96 |
|
|||||
| 16 | 1/16 (2/16)b | |||||||
| kqt-1, 3′ | 3.2 | 1/12 | 0/1 | |||||
| kqt-2, 3′ | 2.1 | 1/12 | 0/1 | |||||
| kqt-2, whole | 3.1 | 0/12 | ||||||
| kqt-3, whole | 3.1 | 2/12 |
1/2 |
Yes | 1/336 | kqt-3(aw1) | 596 | |
| 4 | 1/4 | |||||||
| 2 | shl-1, 5′ | 2.8 | 5/96 | 0/5 | ||||
| ∼100 000 genomes | shl-1, 3′ | 2.4 | 7/96 |
0/7 |
||||
| 12 | 0/12 | |||||||
| kqt-1, 5′ | 2.4 | 0/12 | ||||||
| kqt-1, whole | 7.2 | 1/12 | 0/1 | |||||
| kqt-2, 5′ | 0.9 | 0/12 | ||||||
| kqt-2, whole | 3.1 | 1/12 | 1/1 | Yes | 2/292 | kqt-2(aw2) | 1232 | |
| kqt-3, whole | 3.1 | 1/12 | 0/1 | |||||
| shw-3 | 3.2 | 1/12 | 0/1 | |||||
| slo-2, pore | 2.5 | 0/12 | ||||||
| slo-2, 5′ and 3′ multi | 3.8 | 2/12 | 1/2 | Yes | 2/192 | slo-2(nf101) | 372 | |
| shl-1, 5′ | 2.8 | 0/12 | ||||||
| shl-1, 3′ | 2.4 | 0/12 |
|
|||||
| 6 | 2/6 | |||||||
| 3 | kqt-1, 5′ | 2.4 | 1/12 | 1/1 | Yes | 1/197 | kqt-1(aw3) | 620 |
| ∼200 000 | kqt-1, 3′ | 3.2 | 1/12 | 0/1 | ||||
| genomes | kqt-1, whole | 7.2 | 1/12 | 0/1 | ||||
| kqt-2, 5′ | 0.9 | 2/12 | 0/2 | |||||
| kqt-2, whole | 3.1 | 2/12 | 0/2 | |||||
| kqt-3, whole | 3.1 | 2/12 | 1/2 | Yes | 1/58 | kqt-3(aw4) | 1674 | |
| shw-3 | 3.2 | 1/12 | 0/1 | |||||
| slo-2, pore | 2.5 | 0/12 | ||||||
| shl-1, 5′ | 2.8 | 3/12 | 0/3 | |||||
| shl-1, 3′ | 2.4 | 1/12 | 0/1 | |||||
| shl-1, whole | 4.7 | 0/12 | ||||||
| shl-1, 5′ (TliI) | 1.5 | 0/12 | ||||||
| shk-1, 5′ | 1.9 | 1/12 | 0/1 | |||||
| shk-1, 3′ | 2.2 | 3/12 |
1/3 |
Yes | 0/384c | |||
| 18 | 3/18 |
aStrain failed to show Mendelian segregation.
bIncluding strain that failed to show Mendelian segregation.
cFailed to isolate clonal strain.
TliI digestion
One deletion screen targeting shl-1 was performed, substituting TliI for PspGI (Table 1, screen 3). Pooled DNA samples were predigested with TliI (0.2 U/25 µl; NEB buffer 3) at 75°C for 1 h, prior to PCR. TliI digests were then supplemented with an additional 16.7 mM KCl for PCR, as described above (7).
Analysis of deletion strains
The molecular lesion associated with each deletion strain was determined by sequencing the PCR product derived from each strain. Each deletion strain was also repeatedly backcrossed to the N2 wild-type strain, and genotyped by PCR to confirm Mendelian segregation of the deletion.
Additional strain
Deletion strain shw-1(r1159) used in multiplexing experiments was provided by Ed Maryon and Phil Anderson (University of Wisconsin-Madison).
RESULTS
Summary of method
We employed the thermostable restriction endonuclease, PspGI (26), in a simple modification of established PCR-based methods for isolating C.elegans deletion strains induced by chemical mutagens (7,19,20,23). This modified strategy is outlined in Figure 1. An entire pooled mutant library of 100 000 haploid genomes was easily screened by PCR assays of either 12 or 8 samples, for each targeted gene (Fig. 1A). Targeted genes were selected for the presence of PspGI sites within either coding or intronic sequences (Fig. 1B). Because the PspGI recognition site is relatively relaxed (CCWGG), this did not pose a severe limitation for the set of genes we targeted. Nested oligonucleotide primers were designed flanking individual or closely spaced PspGI sites in each gene, spanning ∼3.0 kb of genomic DNA screened by PCR. Prior to PCR, DNA samples were digested with PspGI. Conceptually, this digestion selectively restricts all WT copies of targeted genes, allowing only amplicons with missing or destroyed PspGI restriction sites to be subsequently amplified by PCR. Negative samples containing only WT copies of a targeted gene produced no background PCR product, while rare deletion amplicons present in positive samples were preferentially amplified and detected. Figure 2 shows an example of a screen of 100 000 haploid genomes by 12 PCRs with PspGI, detecting the slo-2(nf101) deletion strain. Since PspGI reaction buffer is compatible with Taq polymerase, PCRs were easily assembled by the addition of PCR reagents directly to PspGI digests.
Figure 2.
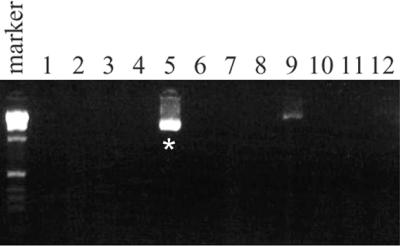
PspGI digestion reduced the total number of PCR screenings and allowed for screening with multiple primer pairs. DNA lysates from a library of mutagenized worms (approximately 100 000 genomes) were pooled and screened with 12 reactions. PCRs were performed with multiplexed slo-2 primers covering the N-terminus and the C-terminus. Two initial positives were detected (lanes 5 and 9). Only one positive (lane 5) verified a 1.7 kb product with triplicated PCRs, and yielded a slo-2 C-terminus deletion strain, slo-2(nf101). PCR products resolved by gel electrophoresis on a 2% agarose gel with a 1 kb DNA size marker (Gibco).
PCR screens with PspGI recover small deletions
Table 1 summarizes the deletion strains we isolated from three independent screens. Six novel deletion strains were isolated in four targeted genes (kqt-1, kqt-2, kqt-3 and slo-2) from a total of approximately 400 000 mutant haploid genomes. Two independent primer sets yielded two deletion alleles each (kqt-3 and slo-2, 3′), while several other primer sets failed to detect deletions. The size distribution of the deletions we obtained is significantly skewed towards small deletions <1.0 kb (372, 574, 596, 620, 1232 and 1674 bp, in ascending order). In contrast, screens by limited PCR extension time (LE) tend to identify strains carrying relatively larger deletions, >1.0 kb (19,20). Detailed characterization of the phenotypes of these deletion strains will be reported elsewhere.
PspGI increases PCR detection sensitivity and permits greater pooling
Inclusion of PspGI digestion prior to PCR greatly increased the sensitivity of our detection assay and lowered spurious non-specific amplification of untargeted genes, compared with the common method of LE (19,20). With model reactions of 250 ng of genomic DNA, we reliably achieved at least a 4-fold increase in detection sensitivity with PspGI digestion over LE, using slo-2(nf100) genomic DNA diluted to 1:40 000 with WT genomic DNA (Fig. 3). We suspect that our present pooling strategy, which results in DNA samples with a complexity of ∼1:10 000 does not fully test the limit of detection by PCR with PspGI digestion.
Figure 3.

Digestion with PspGI increased PCR detection sensitivity and specificity, compared with limiting extension time. Various dilutions (1:1000– 1:40 000) of genomic DNA from slo-2(nf100) were added to wild-type N2 (250 ng) and used as model templates to test the ability of PspGI to boost the sensitivity of PCR deletion assays. Each dilution was assayed with and without PspGI. With a 2 min extension time without PspGI, the 1.5 kb deletion product was detected at a maximal dilution of 1:1000 (right). By limiting the extension time to 1 min, sensitivity for detecting the deletion product was increased to a dilution of 1:10 000 (left). Using identical model templates, sensitivity was further increased with PspGI digestion. With PspGI, robust detection of deletion product was obtained at a dilution of 1:40 000, regardless of the extension time, and background the WT product was completely suppressed. In addition to increasing the sensitivity of the PCR assay, PspGI improved specificity. Without PspGI, multiple bands are generated, whereas only specific deletion bands are observed with PspGI. PCR products resolved on a 1.2% agarose gel with a 1 kb DNA size marker (Gibco).
Multiplexing primer sets with PspGI
We tested the ability to multiplex several independent primer pairs in PCR deletion assays, following PspGI digestion. Primer pairs targeting three genes for which deletion strains are available [slo-2(nf100), kqt-3(aw1) and shw-1(r1159)] were combined together and tested against model template DNAs consisting of WT genomic DNA (50 ng) and combinations of genomic DNA from each of these deletion strains (5 pg of each deletion strain). The results are shown in Figure 4. PspGI digestion effectively suppressed spurious background bands due to non-specific priming, presumably by restricting all potential amplicons containing PspGI sites (lanes 15 and 16). All three deletions were robustly detected individually in PCR screenings, multiplexed with all three primer pairs (compare lanes 1–6 with lanes 9–14). Further more, the presence of all three deletion DNAs in a single reaction could be unambiguously detected using identical multiplexed PCR conditions (lanes 7 and 8). These results suggest that further improvements in throughput may be achieved, using multiple primer pairs for screening each library in conjunction with PspGI digestion. The slo-2(nf101) deletion strain was obtained by screening with multiplexed PCRs combining two primer pairs (Table 1, screen 2 and Fig. 2).
Figure 4.

Multiplexing multiple primer sets in PCR assays with PspGI digestion. PCR deletion assays were performed with three primer pairs (lanes 1–8, and 15 and 16) or single primer pairs (lanes 9–14), with and without PspGI digestion. Model templates consisted of 5 pg of genomic DNA from deletion strains slo-2(nf100) (lanes 1, 2, 9 and 10), kqt-3(aw1) (lanes 3, 4, 11 and 12) and shw-1(r1159) (lanes 5, 6, 13 and 14) added singly, or combined together (lanes 7, 8, 15 and 16), in the background of 50 ng of WT genomic DNA. PspGI digestion resolved single deletion bands corresponding to each deletion product [slo-2(nf100), 1.5 kb; shw-1(r1159), 0.55 kb; kqt-3(aw1), 0.5 kb] when all three primer pairs were multiplexed (lanes 1–8), with a specificity comparable with single primer pair controls (lanes 9–14). The presence of all three deletion templates could be unambiguously detected in single multiplexed reactions (lanes 7 and 8). PCR products resolved on a 1.2% agarose gel with a 1 kb DNA size marker (Gibco).
PCR with other thermostable restriction endonucleases
We tested the ability of other thermostable restriction endonucleases to substitute for PspGI, and thus extend the potential range of restriction recognition sites that may be targeted by our strategy. One thermostable restriction enzyme, TliI, was found to be as effective as PspGI in completely suppressing a PCR using WT genomic DNA (data not shown and Table 1, screen 3). TliI shares with PspGI an optimal reaction temperature of 75°C, extreme thermostability and compatibility with Taq polymerase activity. The future commercial availability of other equally thermostable restriction endonucleases may extend the range of recognition sites that may be targeted by this method.
PspGI reduced false positives through greater pooling
A high proportion of false positives is consistently reported for PCR-based C.elegans deletion screens (7,8,19,20,23). These are defined as samples that initially produce positive PCRs, but fail to subsequently replicate. Reported proportions of false positives range from ∼80 to ∼90% of total positive reactions. Authenticating positive signals can thus represent a significant expenditure of resources and effort.
We observed lower rates of false positives when we pooled each library to 12 reactions, compared with screens with 96 reactions (Table 1). Our first library had a false positive rate of 93% (15/16) when screened in 96 reactions. This same library produced a false positive rate of 75% (3/4) when screened in 12 reactions. Similarly, our second library had a false positive rate of 100% (12/12) when screened in 96 reactions and 67% (4/6) when screened in 12 reactions. In both screens, the total number of positive signals we had to authenticate was significantly reduced when we pooled the library to 12 reactions (4- and 2-fold reductions in the first and second screens, respectively). Direct sequencing of PCR products from several false positive reactions confirmed that they amplified from targeted genes, and thus could not have resulted from non-specific priming events (data not shown). The origin of false positive products remains unknown, but our results suggest that greater dilution in DNA samples of higher pooled complexity may reduce the frequency of false positive signals.
DISCUSSION
We describe a simple technique incorporating thermostable restriction endonucleases with established PCR-based screens (7,19,20,23), which offers advantages of improved detection sensitivity and throughput screening efficiency for the isolation of targeted C.elegans deletion strains. Present PCR-based detection assays for deletions primarily rely upon preferential amplification of shorter deletion amplicons over WT copies, using LE. Potential drawbacks of this method include a limited ability to detect short deletions (<1.0 kb) which may provide insufficient selective advantage over WT during PCR amplification, and a relatively low detection sensitivity (∼1:2000 ratio of deletion to WT DNA) (7,19,20), limiting the maximal extent that DNA samples can be pooled. Both factors may limit the speed and efficacy of deletion screens. One recent approach to address these limitations utilizes a ‘poison primer’ to simultaneously target small deletions and compete for the production of WT PCR products (8). This method allows detection of small deletions at a dilution of ∼1:5000 with WT DNA in model reactions, though significant residual background WT PCR product may mask detection of small deletion PCR products that electrophorese near the same size as WT PCR product.
Our modification makes use of a relatively common recognition site (5′ CCWGG 3′) used by the thermostable restriction endonuclease PspGI. This restriction enzyme isolated from Pyrococcus spp. is highly thermostable with a half-life of >2 h at 95°C, and is compatible with Taq DNA polymerase reaction buffer (26). A PspGI predigestion step prior to PCR effectively cleaves all WT amplicons, blocking their subsequent amplification. Any deletion that removes or destroys the PspGI sites within the screened region escapes restriction and is permitted to amplify preferentially. Due to the extreme thermostability of PspGI, its additional activity during subsequent PCR cycles further ensures restriction of all residual WT amplicons. We believe this may contribute significantly to the effective suppression of background amplification by WT template. Against this background, deletions of any size removing the PspGI sites are thus preferentially amplified, with no background contribution from WT copies.
Digestion of template DNAs with PspGI prior to PCR allowed us to easily detect copies of deletion amplicons at a dilution of at least 1:40 000 (deletion to WT ratio). This high sensitivity permitted us to pool DNA samples to ∼10-fold greater complexity over typical samples screened by LE, thus greatly reducing the total number of PCRs per targeted gene needed to screen an entire library. A secondary benefit is that relatively small deletions (<1.0 kb) as well as larger deletions were detected equally well. Since EMS mutagenesis may create a useful pool of both small and large deletions (8,21), our modification may allow the recovery of a greater fraction of total available deletions in each library. Thus, a smaller number of mutagenized haploid genomes may need to be screened to ensure a high likelihood of isolating a targeted deletion.
A rough estimate of our success rate can be determined by the fraction of total PCR screenings yielding a deletion allele, normalized to the size of the library screened. For the first screen of approximately 100 000 haploid genomes, 2/10 screening reactions yielded a deletion allele. Similarly, in the second screen (approximately 100 000 haploid genomes), 2/12 screening reactions yielded a deletion allele, and in the third screen (approximately 200 000 haploid genomes), 2/18 screening reactions yielded a deletion allele. This suggests that approximately 1/5 to 1/9 screening reactions is likely to yield a deletion allele from a library of approximately 100 000–200 000 haploid genomes. Extra polated for any single primer set, we can reasonably assume the isolation of one deletion from a screen of approximately 500 000–1 800 000 haploid genomes. This compares favorably with estimates of approximately 1 000 000–4 000 000 haploid genomes required for isolating a targeted deletion by LE (7,8,19,20,23).
Our modification requires the presence of a PspGI restriction site within the sequence of the targeted gene. The PspGI recognition site is relatively relaxed, so on average one site should be expected every 512 bp. For a typical 2–5 kb gene in C.elegans, multiple PspGI sites are likely to be present. In practice, we had no difficulty identifying useful PspGI sites in the six genes we targeted. However, one other thermostable restriction endonuclease, TliI (recognition site 5′ CTCGAG 3′) was found to be equally effective in suppressing WT amplicons. Two additional highly thermostable endonucleases have been cloned which may also be adaptable for this screen, TmaI (from Thermotoga, recognition site 5′ CGCG 3′) and PhoI (from Pyrococcus horikoshii, recognition site 5′ GGCC 3′) (R.J.Morgan and S.-Y.Xu, unpublished results). The use of these and other thermostable restriction endonucleases may increase the potential range of genes that can be targeted by our method.
The high proportion of false positives seen with initial screening reactions is a factor limiting the throughput rate of deletion screens. We consistently observed a lower false positive rate when our sample DNAs were pooled to ∼10-fold greater complexity (∼1:8000) than that typically used for screening by LE (1:1000). Identical libraries screened either in 96 wells (at a complexity of 1:1000) or in a set of 12 wells (at a complexity of 1:8000) yielded true positive rates of ∼6 and ∼30%, respectively. Though the origin of false positive signals remains unknown, one potential explanation may be somatic mutations generated either at the normal basal rate or at an elevated rate by EMS-induced mutations of DNA mismatch repair genes (27). Somatic mutations may be more tolerated in polyploid cells, such as multinucleated hypodermal cells or endoreplicated intestinal cells (28). These somatic mutations might be expected in far fewer copy numbers than propagated germline mutations in pooled DNA samples. The inability to replicate false positives with repeated aliquots from individual DNA samples may reflect the stochastic nature of sampling very few or single copies of somatic mutations. More extensive pooling of sample DNAs may dilute the concentration of amplicons derived from somatic mutations, below the threshold for their stochastic detection. Our observations are consistent with this hypothesis. However, other explanations such as Taq-mediated slippage across gaps due to hairpin secondary structure (20) are equally plausible. The high detection sensitivity we obtained with PspGI, allowed us to routinely pool libraries to a high complexity (1:8000). We suggest this reduced our false positive rate, in addition to reducing the total number of reactions needed for screening a library.
Further improvements in the efficiency of this screen may be achieved by several additional modifications. Our ability to multiplex several primer pairs in single PCR assays with PspGI suggests that at least a 3-fold gain in throughput may be expected by multiplexing primer sets. The use of other common chemical mutagens, including TMP-UV (15,21), ethylnitrosourea (29) or diepoxyoctane (20) may normalize the regions of the genome susceptible to mutagenesis and increase the proportional rate of deletions produced. Additional gains may be achieved by the growth and handling of mutant libraries entirely in liquid culture, in 96-well micro-trays (7,20).
Though the methods described here use C.elegans, other organisms that can be grown in small cultures may be equally adaptable. This technique may be particularly suitable for the reverse genetics of single-cell organisms whose genomes are the subject of genome sequencing projects, including Dictyostelium, Leishmania and Chlamydomonas. The effort and cost required to implement a successful PCR-based deletion screen, targeting only a small set of genes, may presently restrict the feasibility of this technique for some laboratories. Our modifications may permit the broader adoption of PCR-based deletion screens as a routine and general technique for reverse genetics.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Jessica Goldstein and Emily Cartwright who assisted in some aspects of the screens. We are indebted to Michael Nonet for generous use of equipment, and initial discussions leading to the development of these techniques. We also wish to thank Mark Edgley and Tim Schedl for helpful discussions, David Thaler and Sandhya Koushika for useful comments on this manuscript, and Don Combs for support. This work was supported by grants from the National Science Foundation (A.W.) and the National Institutes of Health (L.S.), and a pilot grant from the Digestive Diseases Research Core Center and Clinical Nutrition Research Unit at Washington University School of Medicine (A.W.).
REFERENCES
- 1.Waterston R. and Sulston,J. (1995) The genome of Caenorhabditis elegans. Proc. Natl Acad. Sci. USA, 92, 10836–10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The C. elegans Sequencing Consortium (1998) Genome sequence for the nematode C. elegans: a platform for investigating biology. Science, 282, 2012–2018. [DOI] [PubMed] [Google Scholar]
- 3.Blaxter M.L., De Ley,P., Garey,J.R., Liu,L.X., Scheldeman,P., Vierstraete,A., Vanfleteren,J.R., Mackey,L.Y., Dorris,M., Frisse,L.M., Vida,J.T. and Thomas,W.K. (1998) A molecular evolutionary framework for the phylum Nematoda. Nature, 392, 71–75. [DOI] [PubMed] [Google Scholar]
- 4.Fraser A.G., Kamath,R.S., Zipperlen,P., Martinez-Campos,M., Sohrmann,M. and Ahringer,J. (2000) Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature, 408, 325–330. [DOI] [PubMed] [Google Scholar]
- 5.Gonczy P., Echeverri,G., Oegema,K., Coulson,A., Jones,S.J., Copley,R.R., Duperon,J., Oegema,J., Brehm,M., Cassin,E., Hannak,E., Kirkham,M., Pichler,S., Flohrs,K., Goessen,A., Leidel,S., Alleaume,A.M., Martin,C., Ozlu,N., Bork,P. and Hyman,A.A. (2000) Functional genomic analysis of cell division in C. elegans using RNAi of genes on chromosome III. Nature, 408, 331–336. [DOI] [PubMed] [Google Scholar]
- 6.Maeda I., Kohara,Y., Yamamoto,M. and Sugimoto,A. (2001) Large-scale analysis of gene function in Caenorhabditis elegans by high-throughput RNAi. Curr. Biol., 11, 171–176. [DOI] [PubMed] [Google Scholar]
- 7.Barstead R.J. (2000) Reverse genetics. In Hope,I. (ed.), C. elegans, A Practical Approach. Oxford University Press, New York, pp. 97–118.
- 8.Edgley M., D’Souza,A., Moulder,G., McKay,S., Shen,B., Gilchrist,E., Moerman,D. and Barstead,R. (2002) Improved detection of small deletions in complex pools of DNA. Nucleic Acids Res., 30, e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fire A., Xu,S., Montgomery,M.K., Kostas,S.A., Driver,S.E. and Mello,C.C. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature, 391, 806–811. [DOI] [PubMed] [Google Scholar]
- 10.Fire A. (1999) RNA-triggered gene silencing. Trends Genet., 15, 358–363. [DOI] [PubMed] [Google Scholar]
- 11.Grishok A. and Mello,C.C. (2002) RNAi (Nematodes: Caenorhabditis elegans). Adv. Genet., 46, 339–360. [DOI] [PubMed] [Google Scholar]
- 12.Ballinger D.G. and Benzer,S. (1989) Targeted gene mutations in Drosophila. Proc. Natl Acad. Sci. USA, 86, 9402–9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaiser K. and Goodwin,S.F. (1990) ‘Site-selected’ transposon mutagenesis of Drosophila. Proc. Natl Acad. Sci. USA, 87, 1686–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson P. (1995) Mutagenesis. In Epstein,H.F. and Shakes,D.C. (eds), Caenorhabditis elegans: Modern Biological Analysis of an Organism. Methods in Cell Biology. Academic Press, pp. 31–58.8531732 [Google Scholar]
- 15.Yandell M.D., Edgar,L.G. and Wood,W.B. (1994) Trimethylpsoralen induces small deletion mutations in Caenorhabditis elegans. Proc. Natl Acad. Sci. USA, 91, 1381–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rushforth A.M., Saari,B. and Anderson,P. (1993) Site-selected insertion of the transposon Tc1 into a Caenorhabditis elegans myosin light chain gene. Mol. Cell. Biol., 13, 902–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zwaal R.R., Broeks,A., van Meurs,J., Groenen,J.T. and Plasterk,R.H. (1993) Target-selected gene inactivation in Caenorhabditis elegans by using a frozen transposon insertion mutant bank. Proc. Natl Acad. Sci. USA, 90, 7431–7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bessereau J.L., Wright,A., Williams,D.C., Schuske,K., Davis,M.W. and Jorgensen,E.M. (2001) Mobilization of a Drosophila transposon in the Caenorhabditis elegans germ line. Nature, 413, 70–74. [DOI] [PubMed] [Google Scholar]
- 19.Jansen G., Hazendonk,E., Thijssen,K.L. and Plasterk,R.H. (1997) Reverse genetics by chemical mutagenesis in Caenorhabditis elegans. Nature Genet., 17, 119–121. [DOI] [PubMed] [Google Scholar]
- 20.Liu L.X., Spoerke,J.M., Mulligan,E.L., Chen,J., Reardon,B., Westlund,B., Sun,L., Abel,K., Armstrong,B., Hardiman,G., King,J., McCague,L., Basson,M., Clover,R. and Johnson,C.D. (1999) High-throughput isolation of Caenorhabditis elegans deletion mutants. Genome Res., 9, 859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gengyo-Ando K. and Mitani,S. (2000) Characterization of mutations induced by ethyl methanesulfonate, UV and trimethylpsoralen in the nematode Caenorhabditis elegans. Biochem. Biophys. Res. Commun., 269, 64–69. [DOI] [PubMed] [Google Scholar]
- 22.Jansen G., Thijssen,K.L., Werner,P., van der Horst,M., Hazendonk,E. and Plasterk,R.H. (1999) The complete family of genes encoding G proteins of Caenorhabditis elegans. Nature Genet., 21, 414–419. [DOI] [PubMed] [Google Scholar]
- 23.Dong M.Q., Chase,D., Patikoglou,G.A. and Koelle,M.R. (2000) Multiple RGS proteins alter neural G protein signaling to allow C. elegans to rapidly change behavior when fed. Genes Dev., 14, 2003–2014. [PMC free article] [PubMed] [Google Scholar]
- 24.Sulston J. and Hodgkin,J. (1988) Methods. In Wood,W.B. (ed.), The Nematode Caenorhabditis elegans. Cold Spring Harbor Press, Cold Spring Harbor, NY, pp. 587–607.
- 25.Nuovo G.J., Gallery,F., MacConnell,P., Becker,J. and Bloch,W. (1991) An improved technique for the in situ detection of DNA after polymerase chain reaction amplification. Am. J. Pathol., 139, 1239–1244. [PMC free article] [PubMed] [Google Scholar]
- 26.Morgan R., Xiao,J. and Xu,S. (1998) Characterization of an extremely thermostable restriction enzyme, PspGI, from a Pyrococcus strain and cloning of the PspGI restriction-modification system in Escherichia coli. Appl. Environ. Microbiol., 64, 3669–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tijsterman M., Pothof,J. and Plasterk,R.H. (2002) Frequent germline mutations and somatic repeat instability in DNA mismatch-repair-deficient Caenorhabditis elegans. Genetics, 161, 651–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White J. (1988) The anatomy. In Wood,W.B. (ed.), The Nematode Caenorhabditis elegans. Cold Spring Harbor Press, Cold Spring Harbor, NY, pp. 81–122.
- 29.De Stasio E.A. and Dorman,S. (2001) Optimization of ENU mutagenesis of Caenorhabditis elegans. Mutat. Res., 495, 81–88. [DOI] [PubMed] [Google Scholar]