


Abstract
Rad50/Mre11/NBS1 (R/M/N) is a multi-functional protein complex involved in DNA repair, cell cycle checkpoint activation, DNA replication and replication block-induced responses. Ionizing radiation (IR) induces the phosphorylation of NBS1 and nuclear foci formation of the complex. Although it has been suggested that the R/M/N complex is associated with DNA damage sites, we present here biochemical evidence for chromatin association of the complex. We show that the chromatin association of R/M/N is independent of IR and ataxia telangiectasia mutated (ATM). We also demonstrate that optimal chromatin association of the Rad50/Mre11/NBS1 proteins requires both the conserved forkhead-associated (FHA) and breast cancer C-terminus (BRCT) domains of NBS1. Moreover, both these domains of NBS1 are required for its phosphorylation on Ser343 but not on Ser278. Importantly, both the FHA and BRCT domains are essential for IR-induced foci (IRIF) formation of R/M/N and S phase checkpoint activation, but only the BRCT domain is needed for cell survival after IR. These data demonstrate that the FHA and BRCT domains of NBS1 are crucial for the functions of the R/M/N complex.
INTRODUCTION
A number of intricate networks have evolved in eukaryotic cells to respond to endogenous and exogenous genotoxic stresses. Cell cycle checkpoint control and DNA repair processes play crucial roles in maintaining genomic stability (1,2). A defect in either of these processes often results in hypersensitivity to DNA-damaging agents, chromosomal instability and predisposition to cancer. For example, the inherited cancer-prone syndromes ataxia telangiectasia (A-T), Nijmegen breakage syndrome (NBS) and A-T like disorder (A-TLD) are characterized by radiation sensitivity, chromosomal instability and defects in both checkpoint control and DNA repair.
One of the genes mutated in NBS, NBS1, encodes a 95 kDa protein (NBS1 or nibrin) that is a component of the Rad50/Mre11/NBS1 (R/M/N) complex (3–5). This complex plays important roles in checkpoint activation and repair of DNA double-strand breaks (DSBs) (6,7). Purified Mre11 displays 3′→5′ exonuclease and endonuclease activities (8–10) and, in the presence of Rad50 and NBS1, DNA duplex unwinding and hairpin cleavage activities (11). It has been shown that the R/M/N complex forms nuclear foci after ionizing radiation (IR) (4), suggesting that this complex is directly associated with DSB sites. Although Mre11 exhibits a DNA-binding activity in vitro (9,11–13), it is not yet clear if the R/M/N complex is directly associated with chromatin in vivo. Recently, Maser et al. (14) reported that the R/M/N complex co-localizes with proliferating cell nuclear antigen (PCNA) during S phase, while Franchitto and Pichierri (15) demonstrated that it also assembles into nuclear foci after replication blockage. These studies suggest that the R/M/N complex is associated with DNA replication sites and that this complex plays a regulatory role during DNA synthesis.
In response to IR, NBS1 becomes phosphorylated at Ser278 and Ser343 in an ataxia telangiectasia mutated (ATM)-dependent manner (16–19), which is important for activating the S phase checkpoint. Recent studies have suggested that the ATM/NBS1-dependent activation of the S phase checkpoint is partly through regulating the phosphorylation of structural maintenance of chromosome (SMC) proteins (20,21).
NBS1 contains a forkhead-associated (FHA) domain and a breast cancer C-terminal (BRCT) domain (22). The FHA domain is a phosphoserine/threonine specific protein–protein interaction motif (23), whereas the BRCT domain is also predicted to be a protein–protein interaction domain. His45 of NBS1 is one of the few invariant residues within the FHA domain (22). In Saccharomyces cerevisiae mutation of this conserved histidine in the FHA2 domain of Rad53 abolishes not only the interaction between Rad53 and Rad9, but also DNA damage responses such as the induction of RNR3 transcription and the G2/M checkpoint arrest (24). These observations suggest that this conserved histidine plays a crucial role in the biological activity of the FHA domain. In this study, we have examined chromatin association of the R/M/N complex and analyzed the functional importance of both the FHA and BRCT domains of NBS1. We show that the conserved histidine within the NBS1 FHA domain is critical for its function, and that both the FHA and BRCT domains are required for optimal chromatin association of the R/M/N complex, IR-induced phosphorylation of NBS1 and S phase checkpoint activation.
MATERIALS AND METHODS
Cell culture and retroviral gene expression
The simian virus 40 (SV40)-transformed human fibroblast cell line NBS1-LBI (25), the HeLa S3 cell line and human bladder carcinoma cell line T24 were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies Inc.) supplemented with 10% fetal calf serum (FCS) (Life Technologies Inc.). A-T cells (AT22IJE-T/pEBS7) and A-T cells complemented with ATM (AT22IJE-T/pEBS7-YZ5) were grown in DMEM supplemented with 10% FCS and 100 µg/ml hygromycin B (Life Technologies Inc.). T24 cells were synchronized by density arrest and collected as described (26). Cells were irradiated with 10 Gy γ-irradiation and harvested 1 h later unless otherwise specified. NBS1-LBI cell lines with retroviral expression of NBS1WT, NBS1S278A, NBS1S343A, NBS1S278A/S343A, NBS1H45A, NBS1ΔFHA or NBS1ΔBRCT were established as previously described (20). Briefly, 29310A1 retroviral packaging cells (Imgenex, CA) were transfected with pLXIN vector or pLXIN vectors containing cDNA encoding wild-type NBS1 or mutants. Retroviral supernatants were collected 48 h post-transfection. NBS1-LBI cells were incubated in retroviral supernatant plus fresh DMEM supplemented with 10% FCS (1:1 v/v) for at least 24 h. Seventy-two hours after infection, infected cells were selected with 500 µg/ml G418 (Life Technologies Inc.). Clones with ectopic expression of NBS1 or mutants were identified and maintained in DMEM supplemented with 10% FCS and 400 µg/ml G418.
Flow cytometric analysis
For each time point after release from density arrest, ∼2 × 106 cells were trypsinized, washed with phosphate-buffered saline (PBS) and fixed with ice-cold 70% ethanol. After at least 2 h fixation at 4°C, cells were collected, washed once with PBS, and incubated in 1 ml staining solution (25 µg/ml propidium iodide and 200 µg/ml DNase-free RNase in PBS) for 30 min at 37°C. Cells were then analyzed with a FACScan (Becton-Dickinson, CA). 10 000 events were counted for each sample.
Plasmids
Full-length cDNAs encoding wild-type NBS1 were generated and cloned into a pCMV vector as described (17). The NBS1S278A, NBS1S343A, NBS1S278A/S343A and NBS1H45A mutants were generated by site-specific mutagenesis using the QuickChange site-directed mutagenesis kit (Stratagene) and confirmed by DNA sequencing. NBS1WT, NBS1S278A, NBS1S343A, NBS1S278A/S343A and NBS1H45A were subcloned from vector pCMV into the XhoI site of the pLXIN retroviral vector (Clontech). Deletions of the FHA and BRCT domains were achieved by ligating cDNA fragments flanking the FHA domain (nucleotides 1–60 and 300–2265) or fragments flanking the BRCT domain (nucleotides 1–300 and 570–2265) and subcloning into the XhoI site of the pLXIN retroviral vector.
Antibodies
The mouse monoclonal antibodies against human NBS1, Mre11, Rad50 and ATM were generated as previously described (10,17,27). Rabbit polyclonal anti-NBS1S278-P and anti-NBS1S343-P phosphoserine-specific antibodies were raised against the following KLH-conjugated peptides: TGITNpSQTLPDCQ (Ser278) and CTPGPSLpSQGVSV(Ser343). The phosphorylated peptide:unphosphorylated peptide reactivity ratio of affinity purified phosphoserine278- and phosphoserine343-specific antibodies were >99:1 and 98.5:1.5, respectively, as determined by ELISA (Bethyl Laboratories Inc.). Rabbit polyclonal anti-hOrc2 antibodies were a gift from Dr B. Stillman (Cold Spring Harbor Laboratory). Mouse anti-MEK2 IgG (catalog no. 610235) was obtained from BD Biosciences Inc. FITC-conjugated anti-mouse and Texas Red-conjugated anti-mouse antibodies were from Jackson Research Laboratories.
Chromatin fractionation
Whole cell extracts (WCE) were obtained by lysis of cells directly in SDS sample buffer and sonication. To isolate chromatin, cells were lysed and fractionated as described by Mendez and Stillman (28). Approximately 2 × 106 cells were washed with PBS and resuspended in 200 µl buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, 5 µg/ml aprotinin, 5 µg/ml leupeptin, 0.5 µg/ml pepstatin A and 0.1 mM phenylmethylsulfonyl fluoride). Triton X-100 (0.1%) was added, mixed gently by inversion and the mixture was incubated on ice for 5 min. Nuclei were collected in pellet 1 (P1) by centrifugation at low speed (1300 g, 5 min, 4°C). The supernatant (S1) was clarified by high speed centrifugation (20 000 g, 15 min, 4°C) to obtain the soluble fraction (S2). Nuclei were then washed once in buffer A, resuspended in 200 µl buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, 5 µg/ml aprotinin, 5 µg/ml leupeptin, 0.5 µg/ml pepstatin A and 0.1 mM phenylmethylsulfonyl fluoride) and incubated on ice for 30 min. Insoluble chromatin was collected by centrifugation (1700 g, 5 min, 4°C), washed once in buffer B, and centrifuged at 1700 g for 5 min. The final chromatin-enriched pellet (P3) was resuspended in SDS sample buffer and subjected to sonication. To release chromatin-bound proteins, nuclei (P1) were digested with 0.2 U of micrococcal nuclease (Sigma) in buffer A plus 1 mM CaCl2. After 1 min incubation at 37°C, the nuclease reaction was stopped by addition of 1 mM EGTA. Nuclei were then lysed and fractionated as above.
Immunoblotting, immunoprecipitation and immunostaining
Protein concentration was determined by Bradford assay (Bio-Rad, CA). For immunoprecipitation (IP), cell lysates were incubated with 5 µg antibodies for 4 h followed by addition of 20 µl protein G–Sepharose™ 4 Fast Flow beads (Amersham Pharmacia Biotech Inc., Sweden) for 2 h at 4°C. Cell lysates or IP samples were mixed with SDS sample buffer and analyzed using a SDS–8% polyacrylamide gel. Quantitative analysis of immunoblots was performed using a Personal Densitometer SI (Molecular Dynamics, CA). For immunofluorescence, cells were fixed with 4% paraformaldehyde in PBS (pH 7.2) and permeabilized with 0.5% Triton X-100 in PBS. After 30 min incubation in blocking buffer (10% FCS in PBS), cells were incubated with primary antibodies overnight at 4°C. After being washed three times with PBS, the cells were incubated with FITC- or Texas Red-conjugated secondary antibodies for 2 h at room temperature. The cells were then washed with PBS, counterstained with 0.05 µg/ml 4′,6′-diamidino-2-phenylindole (DAPI) in PBS and mounted in Immunon™ mountant (Shandon, PA). All antibodies were diluted in PBS supplemented with 5% FCS.
Radio-resistant DNA synthesis (RDS) assay
IR-induced inhibition of DNA synthesis was measured as previously described (25,29,30). In brief, 3 × 104 cells were seeded into 35 mm dishes and grown for at least 48 h, and then prelabeled with 10 nCi/ml [14C]thymidine (NEN Life Science Products Inc., Boston, MA) in DMEM for ∼48 h. The cells were subsequently incubated in non-radioactive DMEM for 24 h before treatment with 0, 5, 10 or 15 Gy γ-irradiation. After incubation for 30 min at 37°C, the cells were labeled with 2.5 µCi/ml [3H]thymidine (NEN Life Science Products Inc.) for 30 min. The cells were then washed and incubated in non-radioactive DMEM for 45 min. Then the cells were washed with PBS and lysed with 0.5 ml 0.2 M NaOH. The radioactivity was measured in a liquid scintillation counter. The ratio [3H]:[14C] was calculated and compared with the ratio for non-irradiated cells to determine the rate of inhibition of DNA synthesis.
Colony survival assay
Cells were exposed to 0, 2, 4 or 6 Gy γ-irradiation at 2.44 Gy/min (J.L. Shepherd & Associates, CA) and immediately plated at 800 cells/100 mm diameter dish in triplicate. After incubation at 37°C for 2 weeks, cells were washed with PBS, fixed in ice-cold methanol for 15 min and then stained with Giemsa stain (Sigma Diagnostics, St Louis, MO) for 30 min. Colonies per plate were counted and the means ± standard errors were determined. At least two independent experiments were performed on each cell line.
RESULTS
Rad50/Mre11/NBS1 proteins are chromatin-associated in vivo
The R/M/N complex forms nuclear foci at DNA damage sites after IR treatment (4,31). It has also been shown that R/M/N foci in replicating cells co-localize with PCNA foci (14). Thus, it seems plausible that the R/M/N complex may become chromatin associated after IR treatment and during S phase. However, there is as yet no biochemical evidence to support chromatin association of the R/M/N complex. Using a biochemical fractionation scheme recently described by Mendez and Stillman (28), we examined the chromatin association of the R/M/N complex. Asynchronously growing HeLa S3 cells were fractionated into a cytoplasmic fraction (S2) and a nuclear fraction (P2). The P2 fraction was then divided into a soluble nuclear fraction (S3) and an insoluble chromatin-enriched nuclear fraction (P3). The P2 fraction was also subjected to digestion with micrococcal nuclease to release the insoluble chromatin-bound protein into a soluble fraction (S3′), while the nuclease-resistant fraction (P3′) remained insoluble (Fig. 1A). The subunits of the R/M/N complex were mainly found in the P3 and S3′ fractions, indicating that the R/M/N complex is chromatin-bound. The chromatin association of R/M/N increases moderately following entry into S phase (Fig. 1B and Table 1), which may reflect its interaction with replication factors such as E2F1 and PCNA (14). It is also noteworthy that the chromatin-associated R/M/N decreases significantly during G2/M phases. Chromatin association of the R/M/N complex during the normal cell cycle is consistent with its role in the surveillance of chromosomal integrity.
Figure 1.


Chromatin association of Rad50/Mre11/NBS1. (A) Asynchronous HeLa S3 cells were subjected to biochemical fractionation as described in Materials and Methods. Five different fractions were obtained: soluble cytoplasmic fraction (S2), soluble nuclear fraction (S3), insoluble chromatin-enriched nuclear fraction (P3), nuclease-solubilized chromatin-bound fraction (S3′) and insoluble nuclease-resistant nuclear fraction (P3′). Whole cell extract (WCE) was included as a control. Orc2, a replication initiating factor known to be chromatin bound throughout the cell cycle regardless of DNA damage, served as a control for the chromatin-enriched fractions P3 and S3′. MEK2, a cytoplasmic kinase, was used as a control for the cytoplasmic fraction S2. (B) Cell cycle-dependent chromatin association of R/M/N complex. Cells collected at different time points after release from density arrest were subjected to fractionation. The chromatin-enriched fraction (P3) is shown.
Table 1. The percentage of cells in each phase of the cell cycle as determined by the CellQuest analysis program.
| T8 | T19 | T24 | T28 | T32 | T34 | Asynchronous cells | |
|---|---|---|---|---|---|---|---|
| G1 | 96.3 | 72.9 | 13.1 | 5.24 | 14.5 | 16.9 | 64.6 |
| S | 0.67 | 21.4 | 74.1 | 44.7 | 18.6 | 5.84 | 14.4 |
| G2/M | 1.36 | 2.1 | 12.0 | 48.5 | 65.6 | 75.2 | 19.6 |
Chromatin association of R/M/N is independent of IR and ATM
Since the chromatin association of R/M/N may be important for its functions in response to DNA damage, we next tested whether IR treatment could enhance its association with chromatin. Surprisingly, no significant increase in the amount of R/M/N in the P3 fraction was detected after IR (Fig. 2). Since NBS1 is phosphorylated by ATM after IR on several residues, including Ser278 and Ser343 (16–19), we also examined whether ATM deficiency affects the ability of R/M/N to associate with chromatin. Again, the chromatin association of R/M/N is comparable between A-T cells and A-T cells complemented with ATM, regardless of DNA damage (Fig. 2). Taken together, these data suggest that the chromatin association of R/M/N is not affected by IR or deficiency of ATM.
Figure 2.
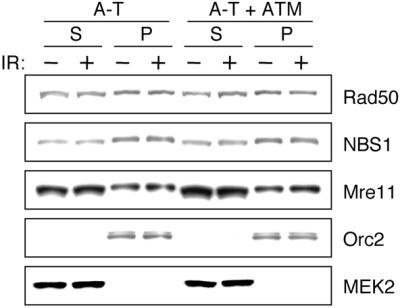
Chromatin association of R/M/N in A-T cells. A-T cells and A-T cells complemented with ATM were fractionated as described. Both soluble fraction S (S2 + S3) and chromatin-enriched fraction P (P3) from IR-treated and non-treated cells are shown.
The FHA and BRCT domains of NBS1 are required for optimal chromatin association of R/M/N
Since NBS1 is crucial for both nuclear localization and formation of IR-induced foci (IRIF) of Rad50 and Mre11 (4), we examined the chromatin association of Rad50 and Mre11 in NBS cells. In NBS1-deficient cells Rad50 and Mre11 were only minimally associated with chromatin, while the majority of them were found in the soluble fractions S2 and S3 (Fig. 3C). The nuclear localization and chromatin association of R/M were fully restored by expression of wild-type NBS1 as well as NBS1S278A/S343A (Fig. 3C, Table 2 and data not shown). These results not only demonstrate that NBS1 is essential for the chromatin-binding activities of the R/M/N but also confirm that the IR-induced phosphorylations of Ser278 and Ser343 by ATM are not required for the chromatin association of R/M/N.
Figure 3.




Chromatin association of R/M/N in NBS cells. (A) Diagram of wild-type NBS1, NBS1H45A, NBS1ΔFHA and NBS1ΔBRCT proteins. Locations of the FHA domain, BRCT domain and phosphorylation sites on Ser278 and Ser343 are shown. The asterisk represents single residue substitution (His→Ala). (B) Alignment of part of the FHA domain of NBS1 among different species. The shaded boxes indicate the regions of identity. The invariant Gly·Arg (GR) pair and His45 (H) are marked with asterisks. (C) Chromatin association of R/M/N in different NBS1-LBI cell clones (with vector only, with expression of NBS1WT, and with expression of NBS1S278A/S343A). (D) Comparison of chromatin association of R/M/N in NBS cells expressing NBS1H45A, NBS1ΔFHA or NBS1ΔBRCT. The soluble fraction (S2 + S3) and chromatin-enriched fraction P (P3) from IR-treated and non-treated cells are shown. MEK2 and Orc2 serve as controls for the soluble fraction and chromatin fraction, respectively.
Table 2. Percentages of chromatin-associated fraction of R/M/N in different NBS cell lines (mean ± SD).
| Vector | Wild-type | S278A/S343A | H45A | ΔFHA | ΔBRCT | ||
|---|---|---|---|---|---|---|---|
| Rad50 | –IR | 9.7 ± 4.7 | 78.0 ± 5.1 | 90.6 ± 3.6 | 61.8 ± 8.7 | 45.2 ± 1.9 | 33.1 ± 7.8 |
| +IR | 10.9 ± 6.9 | 80.2 ± 5.6 | 92.5 ± 3.3 | 45.8 ± 4.2 | 35.6 ± 5.4 | 29.0 ± 9.3 | |
| NBS1 | –IR | NA | 90.2 ± 1.3 | 87.6 ± 8.5 | 70.6 ± 3.2 | 60.2 ± 6.4 | 10.9 ± 6.4 |
| +IR | NA | 91.0 ± 4.8 | 89.2 ± 8.1 | 68.3 ± 5.8 | 56.7 ± 7.2 | 24.2 ± 10.5 | |
| Mre11 | –IR | 8.4 ± 2.3 | 71.0 ± 3.9 | 81.8 ± 4.3 | 47.0 ± 2.7 | 46.4 ± 8.0 | 20.6 ± 2.5 |
| +IR | 4.9 ± 2.1 | 82.0 ± 5.7 | 74.4 ± 3.9 | 26.8 ± 4.9 | 27.5 ± 2.5 | 19.8 ± 3.3 |
In three independent experiments, the protein levels of R/M/N in the soluble fraction and chromatin fraction were quantitatively analyzed; the ratios (%) of P:(S + P) were obtained to indicate the percentage of chromatin-associated R/M/N in the whole cell extract.
To examine the significance of the FHA and BRCT domains of NBS1 in chromatin association, we constructed mutants with a single deletion of either the FHA or BRCT domain, as well as a substitution of the invariant His45 residue with Ala (Fig. 3A and B). Consistent with previous reports that deletion of the N-terminal region of NBS1 does not affect nuclear localization of R/M/N (32,33), nuclear localization of Mre11 and Rad50 can be fully restored by expression of NBS1H45A, NBS1ΔFHA or NBS1ΔBRCT (data not shown). However, even with restored nuclear localization of R/M, the levels of chromatin-associated R/M/N are significantly lower in NBS cells expressing NBS1H45A, NBS1ΔFHA or NBS1ΔBRCT than those in cells expressing NBS1WT (Fig. 3D and Table 2). For example, only 10.9–24.2% of NBS1ΔBRCT is chromatin bound compared to >90% of wild-type NBS1. Therefore, optimal chromatin association of R/M/N requires intact FHA and BRCT domains.
The FHA and BRCT domains are required for IR-induced phosphorylation of Ser343 of NBS1
Using antibodies specifically against phosphorylated Ser278 or Ser343, we examined the IR-induced phosphorylation of NBS1 and its mutants. The phosphorylation of Ser343 and Ser278 was readily detectable in cells expressing NBS1S278A and NBS1S343A, respectively, indicating that these modifications can occur independently (Fig. 4A). However, while the phosphorylation of Ser278 is normal in NBS1H45A, NBS1ΔFHA and NBS1ΔBRCT, the phosphorylation of Ser343 is greatly diminished (Fig. 4B and C). Therefore, the FHA and BRCT domains play an important role in the phosphorylation of NBS1 at Ser343 in vivo.
Figure 4.



Phosphorylation of Ser278 and Ser343 in wild-type NBS1 and mutants. (A) Immunoprecipitation of NBS1WT, NBS1S278A and NBS1S343A with anti-phosphoserine specific antibodies, α-S278-P and α-S343-P. (B) Immunoprecipitation of NBS1WT, NBS1S278A/S343A, NBS1H45A, NBS1ΔFHA and NBS1ΔBRCT with anti-phosphoserine specific antibodies, α-S278-P and α-S343-P. (C) Comparison of expression levels of wild-type NBS1 and different NBS1 mutants. Immunoblotting of whole cell extracts from different NBS cell clones. All immunoblots were probed with α-NBS1 monoclonal antibody MHN1.
The FHA and BRCT domains are essential for formation of IRIF of R/M/N and S phase checkpoint activation
To determine the importance of the FHA and BRCT domains in other biological functions of NBS1, we examined the IRIF of R/M/N in different NBS cell lines. Both Mre11 and NBS1 (data not shown) antibodies were used to test IRIF to eliminate the possibility that the different expression levels of NBS1WT, NBS1S278A/S343A, NBS1H45A, NBS1ΔFHA and NBS1ΔBRCT may affect foci formation. As shown in Figure 5A, IRIF formation with Mre11 was defective in cells expressing NBS1H45A, NBS1ΔFHA and NBS1ΔBRCT.
Figure 5.



IRIF formation, S phase checkpoint activation and radiation sensitivity of NBS cells expressing NBS1WT, NBS1H45A, NBS1ΔFHA and NBS1ΔBRCT. (A) IRIF formation of Mre11. Four hours after IR (15 Gy), cells were fixed and probed with anti-Mre11 monoclonal antibody, 13H1, followed by FITC-conjugated anti-mouse antibody. Nuclei were counterstained with DAPI. (B) DNA synthesis was measured 30 min after 0, 5, 10 or 15 Gy γ-irradiation. See Materials and Methods for details. (C) Cells were exposed to 0, 2, 4 or 6 Gy γ-irradiation and incubated for 2 weeks before colonies per plate were counted. Post-irradiation survival was assessed by comparison with the non-irradiated control. Experiments were performed in triplicate at least twice. Error bars represent standard deviation.
NBS cells exhibit RDS due to a defective S phase checkpoint (34). While NBS cells expressing NBS1WT were able to inhibit DNA synthesis upon IR, cells expressing the mutant forms of NBS1 failed to do so (Fig. 5B). Taken together, both IRIF and S phase checkpoint activation rely on the FHA and BRCT domains.
The BRCT but not FHA domain is important for cell survival after IR
By measuring cell survival 2 weeks after exposure of cells to a sub-lethal dose of γ-irradiation, we investigated whether the various forms of NBS1 could complement the IR sensitivity of NBS cells. We found that neither His45 nor the FHA domain is essential to complement cell sensitivity to IR. However, the NBS1ΔBRCT allele is less effective in complementing the IR sensitivity of NBS cells, in comparison with other NBS alleles (Fig. 5C).
DISCUSSION
We report here that the R/M/N complex is tightly associated with chromatin in vivo. Moreover, chromatin association is increased following entry into S phase, but is not dependent on IR or ATM kinase, suggesting that this complex plays a role in the maintenance of chromosomal stability even during the normal cell cycle. This suggestion is further supported by the observation that NBS1 forms nuclear foci and interacts with E2F1 and replication origins during S phase (14). The decreased level of chromatin-associated R/M/N during G2/M phases is likely due to its dissociation from replication regions after exit from S phase. However, there may be other mitosis-related mechanisms involved, since the chromatin association of R/M/N in G2/M phases is less than in G1 phase.
As several lines of evidence have suggested that the R/M/N complex is recruited to DNA damage sites, it is puzzling that no increase in chromatin association of the complex was observed after IR. We propose two possible mechanisms by which a change of localization actually occurs without a detectable change in the chromatin-bound fraction of R/M/N. First, there may be a translocalization among chromatin-bound proteins, i.e. similar to the S.cerevisiae Ku and Sir proteins, from telomeric regions to DNA damage sites (35–38). Second, the soluble and chromatin-bound fractions of the R/M/N complex might undergo constant exchange so that relocalization of R/M/N is not necessarily reflected by the relative amounts of protein in each fraction.
Since Rad50 and Mre11 (R/M) are localized in both the cytoplasm and nucleus in NBS cells (4), it is conceivable that the R/M complex fails to bind to chromatin in the absence of NBS1. However, the defective chromatin association cannot be fully corrected by simply restoring nuclear localization of R/M. In fact, intact FHA and BRCT domains seem necessary for not only directing NBS1 itself onto chromatin but also stabilizing the chromatin association of the whole complex. On the other hand, chromatin association of the R/M/N is comparable in NBS cells expressing NBS1WT and NBS1S278A/S343A, suggesting that the phosphorylations at Ser278 and Ser343 of NBS1 are dispensable for its chromatin association. It has been shown that the BRCT regions of the putative DNA-binding proteins TopBP1 and BRCA1 directly bind to DNA breaks and ends (39). Therefore, it is possible that the BRCT domain of NBS1 plays a similar role in the chromatin-binding activity of R/M/N.
Since the FHA and BRCT domains are required for phosphorylation of NBS1 at Ser343 but not Ser278, it appears that there are different levels of regulation of these two phosphorylation events. Moreover, since NBS1H45A, NBS1ΔFHA and NBS1ΔBRCT are defective in binding to chromatin, we speculated that Ser343 may be phosphorylated only when NBS1 is bound to chromatin. However, on immunoprecipitating phosphorylated NBS1 with phosphoserine-specific antibodies, we could not detect a significant difference between the distribution of phosphorylated Ser278 and phosphorylated Ser343 in the soluble and chromatin-bound fractions (data not shown). We therefore hypothesize that there may be a constant exchange between soluble and chromatin-bound fractions of NBS1. Since we could not prevent wild-type NBS1 from binding onto chromatin in vivo, how the FHA and BRCT domains dictate phosphorylation of Ser343 and where phosphorylation occurs remain unknown.
The FHA and BRCT domains are also essential for other DNA damage responses, including IRIF formation and S phase checkpoint activation. Since these mutants are also unable to effectively bind to chromatin in vivo, it seems very likely that the chromatin association of R/M/N is necessary for IRIF formation. However, the precise cause of the defective S phase checkpoint in the NBS1H45A, NBS1ΔFHA and NBS1ΔBRCT mutants remains elusive. Since NBS1S278A/S343A can restore chromatin association of the R/M/N complex but not the S phase checkpoint, chromatin association of the R/M/N is not sufficient for activation of the S phase checkpoint. Therefore, the defective S phase checkpoint observed in the NBS1H45A, NBS1ΔFHA and NBS1ΔBRCT mutants could be simply due to a deficiency in phosphorylation of Ser343 (16–19) or deficiencies in both phosphorylation of Ser343 and chromatin association.
Finally, our results have revealed that the BRCT but not the FHA domain plays a crucial role in cell survival after IR. The lack of correlation between a checkpoint defect and radiosensitivity in the NBS1H45A and NBS1ΔFHA mutants is supported by a previous report that radiosensitivity of NBS cells is not due to a defective S phase checkpoint but rather DNA repair defects (40). Given that multiple checkpoint pathways, i.e. the G1/S checkpoint, G2/M checkpoint and a parallel Chk2-dependent S phase checkpoint (41), can be activated upon DNA damage, abrogation of one checkpoint may not be sufficient to cause hypersensitivity to IR. It has been reported that the N-terminal region (amino acids 1–152) of NBS1, which includes the FHA domain and part of the BRCT domain (amino acids 108–152), is not essential for radiation resistance (32). However, we found that deletion of the entire BRCT domain (amino acids 108–196) results in increased cell sensitivity to IR. The discrepancy is likely due to the fact that different deletion mutants were used in these two studies. We believe that deletion of the BRCT domain may cause additional defects in compensatory checkpoint pathways and/or repair of DNA lesions, thereby sensitizing cells to IR. Since NBS1657del5, the most common mutation found in NBS patients, results in deletion of the BRCT domain, it will be informative to further characterize its function in detail.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs Alan Tomkinson and Patrick Sung for critical reading and comments on the manuscript, Mr Sean M. Post and Drs Juan Mendez, David Levine and Malgorzata Zdzienicka for technical advice, Dr Bruce Stillman for providing anti-hOrc2 antibody, Mei-Hua Song for producing antibodies and Stefan Sigurdsson for constructing the pCMV-NBS1ΔBRCT plasmid. S.Z. is the recipient of a Susan G. Komen Breast Cancer Foundation predoctoral fellowship. E.Y.-H.P.L. is supported by National Cancer Institute grant 1P01CA81020.
REFERENCES
- 1.Dasika G.K., Lin,S.C., Zhao,S., Sung,P., Tomkinson,A. and Lee,E.Y. (1999) DNA damage-induced cell cycle checkpoints and DNA strand break repair in development and tumorigenesis. Oncogene, 18, 7883–7899. [DOI] [PubMed] [Google Scholar]
- 2.Zhou B.B. and Elledge,S.J. (2000) The DNA damage response: putting checkpoints in perspective. Nature, 408, 433–439. [DOI] [PubMed] [Google Scholar]
- 3.Varon R., Vissinga,C., Platzer,M., Cerosaletti,K.M., Chrzanowska,K.H, Saar,K., Beckmann,G., Seemanova,E., Cooper,P.R., Nowak,N.J et al. (1998) Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell, 93, 467–476. [DOI] [PubMed] [Google Scholar]
- 4.Carney J.P., Maser,R.S., Olivares,H., Davis,E.M., Le Beau,M., Yates,J.R.,III, Hays,L., Morgan,W.F. and Petrini,J.H. (1998) The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell, 93, 477–486. [DOI] [PubMed] [Google Scholar]
- 5.O’Driscoll M., Cerosaletti,K.M., Girard,P.M., Dai,Y., Stumm,M., Kysela,B., Hirsch,B., Gennery,A., Palmer,S.E., Seidel,J. et al. (2001) DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol. Cell, 8, 1175–1185. [DOI] [PubMed] [Google Scholar]
- 6.Haber J.E. (1998) The many interfaces of Mre11. Cell, 95, 583–586. [DOI] [PubMed] [Google Scholar]
- 7.D’Amours D. and Jackson,S.P. (2002) The mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nature Rev. Mol. Cell. Biol., 3, 317–327. [DOI] [PubMed] [Google Scholar]
- 8.Paull T.T. and Gellert,M. (1998) The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol. Cell, 1, 969–979. [DOI] [PubMed] [Google Scholar]
- 9.Usui T., Ohta,T., Oshiumi,H., Tomizawa,J., Ogawa,H. and Ogawa,T. (1998) Complex formation and functional versatility of Mre11 of budding yeast in recombination. Cell, 95, 705–716. [DOI] [PubMed] [Google Scholar]
- 10.Trujillo K.M., Yuan,S.S., Lee,E.Y. and Sung,P. (1998) Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11 and p95. J. Biol. Chem., 273, 21447–21450. [DOI] [PubMed] [Google Scholar]
- 11.Paull T.T. and Gellert,M. (1999) Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev., 13, 1276–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furuse M., Nagase,Y., Tsubouchi,H., Murakami-Murofushi,K., Shibata,T. and Ohta,K. (1998) Distinct roles of two separable in vitro activities of yeast Mre11 in mitotic and meiotic recombination. EMBO J., 17, 6412–6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trujillo K.M. and Sung,P. (2001) DNA structure-specific nuclease activities in the Saccharomyces cerevisiae Rad50*Mre11 complex. J. Biol. Chem., 276, 35458–35464. [DOI] [PubMed] [Google Scholar]
- 14.Maser R.S., Mirzoeva,O.K., Wells,J., Olivares,H., Williams,B.R., Zinkel,R.A., Farnham,P.J. and Petrini,J.H. (2001) Mre11 complex and DNA replication: linkage to E2F and sites of DNA synthesis. Mol. Cell. Biol., 21, 6006–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franchitto A. and Pichierri,P. (2002) Bloom’s syndrome protein is required for correct relocalization of RAD50/MRE11/NBS1 complex after replication fork arrest. J. Cell Biol., 157, 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim D.S., Kim,S.T., Xu,B., Maser,R.S., Lin,J., Petrini,J.H. and Kastan,M.B. (2000) ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature, 404, 613–617. [DOI] [PubMed] [Google Scholar]
- 17.Zhao S., Weng,Y.C., Yuan,S.S., Lin,Y.T., Hsu,H.C., Lin,S.C., Gerbino,E., Song,M.H., Zdzienicka,M.Z., Gatti,R.A. et al. (2000) Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature, 405, 473–477. [DOI] [PubMed] [Google Scholar]
- 18.Wu X., Ranganathan,V., Weisman,D.S., Heine,W.F., Ciccone,D.N., O’Neill,T.B., Crick,K.E., Pierce,K.A., Lane,W.S., Rathbun,G. et al. (2000) ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature, 405, 477–482. [DOI] [PubMed] [Google Scholar]
- 19.Gatei M., Young,D., Cerosaletti,K.M., Desai-Mehta,A., Spring,K., Kozlov,S., Lavin,M.F., Gatti,R.A., Concannon,P. and Khanna,K. (2000) ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nature Genet., 25, 115–119. [DOI] [PubMed] [Google Scholar]
- 20.Yazdi P.T., Wang,Y., Zhao,S., Patel,N., Lee,E.Y. and Qin,J. (2002) SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev., 16, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim S.T., Xu,B. and Kastan,M.B. (2002) Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev., 16, 560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Featherstone C. and Jackson,S.P. (1998) DNA repair: the Nijmegen breakage syndrome protein. Curr. Biol., 8, R622–R625. [DOI] [PubMed] [Google Scholar]
- 23.Durocher D., Henckel,J., Fersht,A.R. and Jackson,S.P. (1999) The FHA domain is a modular phosphopeptide recognition motif. Mol. Cell, 4, 387–394. [DOI] [PubMed] [Google Scholar]
- 24.Sun Z., Hsiao,J., Fay,D.S. and Stern,D.F. (1998) Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science, 281, 272–274. [DOI] [PubMed] [Google Scholar]
- 25.Kraakman-van der Zwet M., Overkamp,W.J., Friedl,A.A., Klein,B., Verhaegh,G.W., Jaspers,N.G., Midro,A.T., Eckardt-Schupp,F., Lohman,P.H. and Zdzienicka,M.Z. (1999) Immortalization and characterization of Nijmegen Breakage syndrome fibroblasts. Mutat. Res., 434, 17–27. [DOI] [PubMed] [Google Scholar]
- 26.Chen P.L., Scully,P., Shew,J.Y., Wang,J.Y. and Lee,W.H. (1989) Phosphorylation of the retinoblastoma gene product is modulated during the cell cycle and cellular differentiation. Cell, 58, 1193–1198. [DOI] [PubMed] [Google Scholar]
- 27.Chen G. and Lee,E. (1996) The product of the ATM gene is a 370-kDa nuclear phosphoprotein. J. Biol. Chem., 271, 33693–33697. [DOI] [PubMed] [Google Scholar]
- 28.Mendez J. and Stillman,B. (2000) Chromatin association of human origin recognition complex, cdc6 and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol. Cell. Biol., 20, 8602–8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Painter R.B. and Young,B.R. (1980) Radiosensitivity in ataxia-telangiectasia: a new explanation. Proc. Natl Acad. Sci. USA, 77, 7315–7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morgan S.E., Lovly,C., Pandita,T.K., Shiloh,Y. and Kastan,M.B. (1997) Fragments of ATM which have dominant-negative or complementing activity. Mol. Cell. Biol., 17, 2020–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nelms B.E., Maser,R.S., MacKay,J.F., Lagally,M.G. and Petrini,J.H. (1998) In situ visualization of DNA double-strand break repair in human fibroblasts. Science, 280, 590–592. [DOI] [PubMed] [Google Scholar]
- 32.Tauchi H., Kobayashi,J., Morishima,K., Matsuura,S., Nakamura,A., Shiraishi,T., Ito,E., Masnada,D., Delia,D. and Komatsu,K. (2001) The forkhead-associated domain of NBS1 is essential for nuclear foci formation after irradiation but not essential for hRAD50·hMRE11·NBS1 complex DNA repair activity. J. Biol. Chem., 276, 12–15. [DOI] [PubMed] [Google Scholar]
- 33.Desai-Mehta A., Cerosaletti,K.M. and Concannon,P. (2001) Distinct functional domains of nibrin mediate Mre11 binding, focus formation and nuclear localization. Mol. Cell. Biol., 21, 2184–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Young B.R. and Painter,R.B. (1989) Radioresistant DNA synthesis and human genetic diseases. Hum. Genet., 82, 113–117. [DOI] [PubMed] [Google Scholar]
- 35.Martin S.G., Laroche,T., Suka,N., Grunstein,M. and Gasser,S.M. (1999) Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell, 97, 621–633. [DOI] [PubMed] [Google Scholar]
- 36.Mills K.D., Sinclair,D.A. and Guarente,L. (1999) MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell, 97, 609–620. [DOI] [PubMed] [Google Scholar]
- 37.Wu G., Lee,W.H. and Chen,P.L. (2000) NBS1 and TRF1 colocalize at promyelocytic leukemia bodies during late S/G2 phases in immortalized telomerase-negative cells. Implication of NBS1 in alternative lengthening of telomeres. J. Biol. Chem., 275, 30618–30622. [DOI] [PubMed] [Google Scholar]
- 38.Zhu X.D., Kuster,B., Mann,M., Petrini,J.H. and Lange,T. (2000) Cell-cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nature Genet., 25, 347–352. [DOI] [PubMed] [Google Scholar]
- 39.Yamane K. and Tsuruo,T. (1999) Conserved BRCT regions of TopBP1 and of the tumor suppressor BRCA1 bind strand breaks and termini of DNA. Oncogene, 18, 5194–5203. [DOI] [PubMed] [Google Scholar]
- 40.Girard P.M., Foray,N., Stumm,M., Waugh,A., Riballo,E., Maser,R.S., Phillips,W.P., Petrini,J., Arlett,C.F. and Jeggo,P.A. (2000) Radiosensitivity in Nijmegen Breakage Syndrome cells is attributable to a repair defect and not cell cycle checkpoint defects. Cancer Res., 60, 4881–4888. [PubMed] [Google Scholar]
- 41.Falck J., Petrini,J.H., Williams,B.R., Lukas,J. and Bartek,J. (2002) The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nature Genet., 30, 290–294. [DOI] [PubMed] [Google Scholar]