


Abstract
Human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) initiates DNA synthesis from the 3′ end of human tRNALys3. We have used cis-acting hammerhead ribozymes to produce homogeneous-length transcribed tRNALys3 and have developed conditions for purifying highly structured RNAs on a modified tube-gel apparatus. Titration experiments show that this RNA can assemble into an initiation complex that contains equimolar amounts of HIV-1 RT, transcribed tRNALys3, and chemically synthesized template RNA. We have purified this complex using gel-filtration chromatography and have found that it is homogeneous with respect to molecular weight, demonstrating that the initiation complex forms a single discrete species at micromolar concentrations. When this initiation complex is supplied with deoxynucleotides, essentially all of the tRNA is used as a primer by HIV-1 RT and is fully extended to the 5′ end of the template. Thus, in vitro transcribed tRNA can be used efficiently as a primer by HIV-1 RT. We have also obtained crystals of the HIV-1 initiation complex that require the precisely defined ends of this in vitro transcribed tRNALys3 to grow.
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) is responsible for converting the single-stranded viral RNA genome into a double-stranded DNA that is subsequently integrated into the host cell genome (reviewed in 1). The complex series of steps required to complete this process begins with the initiation of minus-strand DNA synthesis from the 3′ end of cellular tRNALys3 that is packaged into the virus. The last 18 nt of the tRNA are unwound and annealed to a complementary primer-binding site (PBS) located near the 5′ end of the viral RNA genome. While HIV-1 RT can add a few nucleotides to tRNALys3 within the virus particle, the bulk of DNA synthesis occurs once the viral nucleocapsid is released within a newly infected cell (2–5).
Initiation of DNA synthesis from tRNALys3 is distinct from other steps of viral DNA synthesis by being the only step in which RNA functions both as a primer and a template. Kinetic measurements indicate that a distinct transition occurs between the initiation and elongation phases of DNA synthesis, subsequent to the addition of 6 nt to the 3′ end of the tRNA (6). The initiation phase is characterized by distributive DNA synthesis with a slow polymerization rate, while in the elongation phase DNA synthesis is processive with a polymerization rate that is approximately 50 times faster than during initiation (6–8). The slower rate of nucleotide incorporation during initiation is not dependent on the full tRNA, as a slower rate is also observed when an 18-nt RNA that is complementary to the PBS is used as a primer (6,9,10). Unlike elongation, initiation of minus-strand DNA synthesis from tRNALys3 annealed to HIV-1 viral RNA is more efficient when performed by HIV-1 RT than by other viral RTs (7,11). This specificity is dependent both on the tRNA, as it is not observed when an RNA oligonucleotide is used as a primer (11), and on the structure of the surrounding viral template RNA (7,11). Another significant functional difference between initiation and elongation is that the initiation step appears to be particularly sensitive to the nucleoside analog azidothymidine (AZT) (12,13). Detailed structural information about the process of initiation will be important for understanding AZT-resistant mutations and for the design of new drugs targeted at HIV-1 RT.
Crystal structures representing several stages of the HIV-1 reverse transcription process have been determined, starting with the structure of HIV-1 RT in complex with a non-nucleoside inhibitor, nevirapine (14). Those with bound nucleic acid include: elongation complexes of HIV-1 RT bound to primer-template DNA duplex with (15) and without incoming dNTP (16,17) HIV-1 RT bound to an RNA–DNA heteroduplex (18); and HIV-1 RT bound to a selected inhibitor pseudoknot RNA (19). These structures have been used to interpret site-specific crosslinking (20) and enzymatic footprinting studies of complexes that initiate DNA synthesis from tRNALys3.
As part of our ongoing efforts to determine the crystal structure of an HIV-1 RT initiation complex, we have used hammerhead (HH) ribozymes to produce transcribed tRNALys3 with precisely defined ends. We demonstrate that this tRNA can be assembled into a complex together with template RNA and HIV-1 RT, that this complex can be purified by gel-filtration chromatography, and that all of the tRNA can be used as a primer for the initiation of DNA synthesis. Finally, we also describe conditions for crystallizing the HIV-1 RT initiation complex.
MATERIALS AND METHODS
Plasmid construction
DNA oligonucleotides (Table 1; HHMI-Keck Biotechnology Center, Yale University) encoding a T7 RNA polymerase promoter, human tRNALys3, and two HH ribozymes (21), were denatured for 2 min at 95°C, then incubated for 1 h at room temperature in a 50 µl reaction containing 4 µM each DNA oligonucleotide, 40 U/µl T4 DNA ligase (New England Biolabs) and 1× DNA ligase buffer (NEB; 50 mM Tris–HCl, pH 7.5, 10 mM MgCl2, 10 mM DTT, 1 mM ATP, 25 µg/ml BSA). The DNA was digested with EcoRI and BamHI (New England Biolabs) and ligated into the high-copy plasmid pUC19 that had been digested with the same enzymes. The sequence of the resulting plasmid, ptRK-HH, was verified (Keck Biotechnology Center). Plasmid DNA was purified from Escherichia coli strain DH5α (Gibco) using a modified alkaline lysis procedure (22) with a typical yield of 20 mg of DNA from a 2 l culture.
Table 1. Oligonucleotides.
| Name | Length (nt) | Sequence |
|---|---|---|
| DNA | ||
| ET7T | 31 | 5′-CTGAATTCTAATACGACTCACTATAGGGAGA-3′ |
| ET7Ba | 26 | 5′-CTATAGTGAGTCGTATTAGAATTCAG-3′ |
| HH5Ta | 51 | 5′-GCTATCCGGGCCTGATGAGTCCGTGAGGACGAAACGGTACCCGGTACCGTC-3′ |
| HH5Ba | 51 | 5′-TACCGGGTACCGTTTCGTCCTCACGGACTCATCAGGCCCGGATAGCTCTCC-3′ |
| TRK5Ta | 34 | 5′-GCCCGGATAGCTCAGTCGGTAGAGCATCAGACTT-3′ |
| TRK5Ba | 34 | 5′-TGATGCTCTACCGACTGAGCTATCCGGGCGACGG-3′ |
| TRK3Ta | 52 | 5′-TTAATCTGAGGGTCCAGGGTTCAAGTCCCTGTTCGGGCGCTAGACGGAGTCT-3′ |
| TRK3Ba | 52 | 5′-CCGTCTAGCGCCCGAACAGGGACTTGAACCCTGGACCCTCAGATTAAAAGTC-3′ |
| HH3Ta | 51 | 5′-AGACTCCGTCCTGATGAGTCCGTGAGGACGAAAGCGCCCAAATGGATCCAG-3′ |
| HH3B | 56 | 5′-CTGGATCCATTTGGGCGCTTTCGTCCTCACGGACTCATCAGGACGGAGTCTAGACT-3′ |
| RNA | ||
| PBS-K2 | 36 | 5′-GGGAGAGCAGUGGCGCCCGAACAGGGACCUGAAAGC-3′ |
| PBS-WT1 | 24 | 5′-CAGUGGCGCCCGAACAGGGACCUG-3′ |
| PBS-CUA | 24 | 5′-CAGUAGCGCCCGAACAGGGACCUG-3′ |
aThese oligonucleotides were synthesized with a 5′ phosphate. PBS sequences in the RNA are shown in bold.
Transcription and purification of tRNALys3 and PBS-template RNA
Transcription reactions (25 ml) contained 50 mM Tris–HCl pH 8.0, 6 mM each ATP, CTP, GTP and UTP (Pharmacia), 2 mM spermidine, 100 µg/ml BSA, 40 mM DTT, 32 mM MgCl2, 50 µg/ml BamHI-linearized ptRK-HH and 60 µg/ml T7 RNAP [a generous gift from G. Cheetham, purified as described by Jeruzalmi and Steitz (23)]. Reactions were preheated prior to addition of T7 RNAP, then incubated for 4–8 h at 37–40°C, and terminated by phenol–chloroform extraction and ethanol precipitation. RNA was resuspended in 25 ml of 40 mM Tris–HCl pH 8.0, 100 mM NaCl, 1 mM EDTA, incubated for 10 min at 75°C, cooled to 37°C, then MgCl2 was added to a concentration of 25 mM (21,24) and incubated at 37°C for 30 min to complete the HH ribozyme cleavage. Unincorporated ribonucleotides remaining from the transcription were removed by size-exclusion chromatography using NAP-25 columns (Pharmacia) equilibrated and run in dH2O, by dialysis against dH2O, or by buffer exchange to dH2O using a Centriplus-30 spin concentrator (Millipore).
To open the 2′–3′ cyclic phosphate that results from the ribozyme cleavage, the RNA was adjusted to pH 2 by the addition of 1 M HCl (25) (final concentrations of 10–12.5 mM HCl and ∼2 mg/ml tRNA in a total volume of 25 ml) and incubated for 90 min at room temperature. The acid treatment was stopped by the addition of 1 M Tris-base to pH 8 and the RNA was dephosphorylated for 2 h at 37°C in a 50 ml final volume containing 20 U/ml calf intestinal alkaline phosphatase (New England Biolabs), 10 mM MgCl2, 100 mM NaCl and ∼1 mg/ml tRNA, 100 mM Tris, pH 8. Aliquots from the large-scale reaction were analyzed on a 10% acrylamide (19:1)/0.5× TBE/7 M urea/40% formamide sequencing gel to verify that the dephosphorylation reaction had gone to completion. The RNA was concentrated and the buffer exchanged to dH2O using a Centriplus-30 spin concentrator (Millipore) and was stored at –20°C prior to gel purification. Alternatively, tRNALys3 was gel purified prior to decyclization and dephosphorylation.
The transcribed tRNALys3 was purified by electrophoresis on a 8 cm tall (60 ml volume) 8.5% acrylamide (19:1)/1× TBE/7 M urea/45% formamide gel run in a modified tube-gel format using a Model 491 Prep Cell (Bio-Rad). The gel solution was prepared using a freshly made stock of acryl amide (Bio-Rad) and was thoroughly degassed. Polymer ization was catalyzed with 1 mg/ml ammonium persulfate and 1.5 µl/ml N,N,N′,N′-tetramethylethylenediamine and was allowed to continue overnight. The running and elution buffer (1× TBE) was pre-warmed to 55°C and the gel was run at 12 W constant power for 30–60 min prior to loading the sample. Contrary to the manufacturer’s directions, the running buffer was not recirculated and the lower buffer just barely covered the bottom of the gel. These modifications were required to maintain an even temperature of 50–55°C throughout the gel. Six to 12 mg of transcribed RNA (tRNALys3 plus HH RNAs) in 400 µl of dH2O was mixed with 800 µl of 100% formamide (Ultrapure grade; USB) containing trace amounts of bromophenol blue and xylene cyanol dyes and boiled for 3 min immediately prior to loading onto the Prep-Cell gel. The gel was run at 12 W constant power for 4 h. During the entire run elution buffer was pumped at 1.5 ml/min and monitored by UV absorption at 254 nm. After the bromophenol blue eluted from the gel (∼2 h), 3 ml fractions were collected. Aliquots (7.5 µl) of selected fractions were analyzed on a 10% acrylamide (19:1)/1× TBE/8.3 M urea analytical gel. Fractions containing pure tRNA were pooled, concentrated and the buffer exchanged to dH2O using Centriplus-30 spin concentrators (Millipore). Purified RNA was stored at –20°C.
Template RNAs (Table 1) containing the HIV-1 PBS were either transcribed in vitro from a DNA–oligonucleotide template (26) (K2 RNA) or were chemically synthesized (K2, PBS-WT1 and PBS-CUA RNAs; Dharmacon, Lafayette, CO). Template RNAs were purified by denaturing PAGE on slab gels prior to use. Concentrations of all RNAs were calculated using an extinction coefficient at 260 nm of 40 µg/ml. The identities of the purified tRNALys3 and K2 transcripts were verified by direct RNA sequencing with RNases (data not shown).
HIV-1 reverse transcriptase
HIV-1 RT was produced either from plasmid p6HRT-PROT (27), which co-expresses an N-terminally His6-tagged p66 subunit of HIV-1 RT and HIV-1 protease and results in cleavage of approximately half of the p66 subunit to yield the active p66/p51 RT heterodimer, or from plasmid p66/Δ428(His) (28; P. L. Boyer and S. H. Hughes, unpublished results), which co-expresses p66 and C-terminally His6-tagged p51 (residues 1–428). The enzyme was purified by Ni-NTA (Qiagen), SP-Sepharose (Pharmacia) and Superdex 75 (Pharmacia) column chromatography (19,29). Purified protein was stored at 4°C either in 10 mM bis-Tris-propane (BTP) pH 7.0, 50 mM NaCl, 10% glycerol, 5 mM β-mercaptoethanol, 0.02% β-hexyl-glucoside or in 20 mM Tris–Cl pH 8.0. The concentration of HIV-1 RT was calculated using a UV extinction coefficient of 3.54 mg–1 cm–1 that was experimentally determined (30).
Assembly of RT/tRNALys3/PBS RNA initiation complex
tRNALys3 (25 µM) and PBS-template RNA K2 (12.5–50 µM, as indicated in Fig. 3A) were annealed in a solution containing 50 mM NaCl, 5 mM MgCl2, 5 mM BTP pH 7.0 and 10% glycerol (only included so that reactions could be loaded directly onto a native gel) in a 25 µl reaction volume. The RNAs were denatured for 2 min at 90°C, incubated at 55°C for 5 min, then slow cooled to 25°C at a rate of –0.5°C/min in an MJ Research Minicycler PCR machine and stored at 4°C. Aliquots of the annealing reactions were loaded directly onto a non-denaturing 10% acrylamide (19:1)/1× THE (10 mM Tris, 10 mM HEPES, 0.1 mM EDTA, pH 7)/10 mM MgCl2 gel (31) run at room temperature with recirculation of the top and bottom reservoir buffers. RNAs used for initiation complex formation were annealed at equimolar concentrations (25 µM each). The gel was stained with 0.5 µg/ml ethidium bromide to visualize the RNA.
Figure 3.
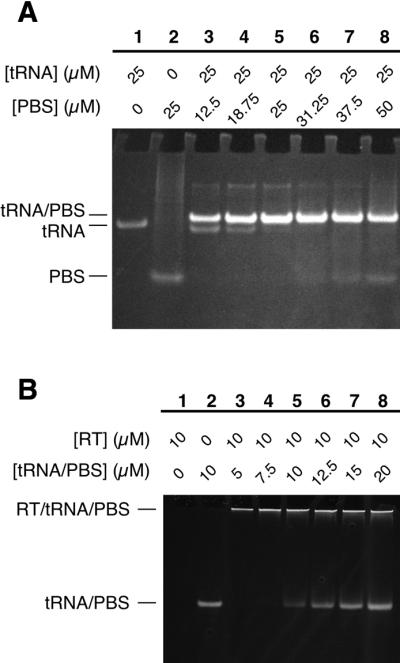
Assembly of an HIV-1 initiation complex containing equimolar concentrations of RT, primer tRNA and template RNA. (A) Native gel showing annealing of purified tRNALys3 (25 µM) to varying concentrations of PBS-K2 template RNA (12.5–50 µM, lanes 3–8) in the presence of 5 mM MgCl2. Migration positions of the complex of annealed RNAs (tRNA/PBS), tRNALys3 alone (tRNA), and PBS-K2 RNA alone (PBS) are indicated. RNAs were visualized by ethidium bromide staining. (B) Native gel showing binding of HIV-1 RT (10 µM) to varying concentrations (5–20 µM, lanes 3–8) of annealed primer-template RNA. Migration positions of the protein–RNA complex (RT/tRNA/PBS) and unbound RNA (tRNA/PBS, lane 2) are indicated. RNAs were visualized by ethidium bromide staining. Presence of protein in the RT/tRNA/PBS complex was confirmed by subsequent staining of the gel with Coomassie blue (data not shown). HIV-1 RT alone (lane 1) migrates in the opposite direction from RNA and the RNA–protein complex and is not retained on the gel (data not shown).
HIV-1 RT (10 µM) was incubated with tRNALys3/PBS-K2 RNA (5–20 µM, as indicated in Fig. 3B) for 20 min at room temperature in a 10 µl reaction volume containing 40 mM NaCl, 4 mM MgCl2, 4 mM BTP pH 7 and 8% glycerol. Aliquots of the binding reactions were loaded directly onto a non-denaturing 5% acrylamide (19:1)/1× THE/5 mM MgCl2 gel run slowly at room temperature. The gel was stained with 0.5 µg/ml ethidium bromide to visualize the RNA, then with Coomassie blue to visualize the protein (data not shown).
Initiation complex activity assay
Initiation reactions (15 µl) contained 5 µM HIV-1 RT, 5 µM tRNALys3/PBS-K2 (annealed as above, without glycerol), 1 mM each dATP, dCTP, dGTP and dTTP (Pharmacia), 50 mM NaCl, 10 mM MgCl2, 50 mM Tris–HCl pH 7.5, and were incubated at 37°C for 15 min. Reactions were stopped by addition of 3 µl of 5× proteinase K mix [250 µg/ml proteinase K (USB), 500 mM Tris–HCl pH 8.0, 0.5% SDS, 25 mM EDTA] and incubation for 10 min at 37°C. Samples were mixed with 30 µl of 100% formamide containing trace amounts of bromophenol blue and xylene cyanol dyes, then boiled for 3 min and immediately loaded onto a 10% acrylamide (19:1)/1× TBE/7 M urea/40% formamide sequencing gel that was pre-warmed and run at 55°C in 1× TBE. RNAs were visualized by staining for 10 min with 0.5 µg/ml ethidium bromide.
Purification of initiation complex
An HIV-1 RT/tRNALys3/PBS-WT1 initiation complex was assembled as above, except that the RNAs were annealed at a concentration of 40 µM each in the absence of glycerol and the protein and RNA were incubated together at concentrations of 20 µM each. The complex (120 µl) was loaded onto a Superdex 200 HR 10/30 column (Pharmacia) equilibrated and run at 25°C in 10 mM BTP pH 7.0, 50 mM NaCl, 2.5 mM MgCl2, 5% glycerol at a flow rate of 0.5 ml/min. The elution profile was monitored by UV absorption at 280 nm and by multiangle laser light scattering at 690 nm using a Mini-DAWN detector (Wyatt Technologies). For comparison, HIV-1 RT (120 µl at 20 µM) and annealed tRNALys3/PBS-WT1 (40 µl at 40 µM) were purified separately, under the same column conditions used for the initiation complex. All samples were filtered (0.2 µm pore size) immediately before loading onto the column.
Crystallization
An HIV-1 RT/tRNALys3/PBS-CUA initiation complex was assembled as described above except that the RNAs were annealed at a concentration of 200 µM in the absence of glycerol and the final concentration of the complex was 50 µM in 50 mM NaCl, 10 mM MgCl2, 10 mM Tris–HCl pH 8.0. Prior to crystallization, the complex was incubated for 10 min at room temperature either with ddCTP and dTTP or with dCTP, ddTTP and dGTP (2 mM each deoxynucleotide) to trap the complex after the addition of 1 or 2 nt, respectively. Crystals were grown by hanging drop vapor diffusion at 4–16°C by mixing 2 µl of initiation complex with 2 µl of well solution [5–7% PEG-8000 (Fluka), 200 mM NaCl, 50 mM MES pH 5.4–6.6]. Contents of crystals were analyzed by SDS–PAGE (10–20% Tris–HCl Ready Gels; Bio-Rad) followed by silver staining of both protein and RNA (Bio-Rad).
RESULTS
Preparation of in vitro transcribed human tRNALys3
To produce in vitro synthesized human tRNALys3 with precisely defined ends, we constructed a plasmid (ptRK-HH) that yields a primary T7 RNA polymerase transcript containing tRNALys3 sequences preceded and followed by HH ribozyme sequences (Fig. 1). The HH ribozyme design was based on a similar construct used to produce part of the U2 small nuclear RNA (21). As a requirement for efficient cleavage by the 3′ HH, the final tRNA contains a single base substitution (C75U) that changes the sequence at the 3′ end of the tRNA from 5′-CCA-3′ to 5′-CUA-3′. This substitution does not appear to interfere with the use of the tRNA as primer by HIV-1 RT (see below), presumably because the substituted U can still form a wobble pair with its partner G in the HIV-1 PBS. During the course of transcription from linearized ptRK-HH, both the 5′ and 3′ HH ribozymes cleaved in approximately half of the primary transcripts to release free tRNALys3. After phenol–chloroform extraction and ethanol precipitation, the transcribed RNAs were heat denatured then incubated for 30 min at 37°C in the presence of 25 mM Mg2+ to drive the cleavage reaction towards completion (24) (Fig. 2B, ‘load’).
Figure 1.

Secondary structure diagram of tRNALys3 transcript from ptRK-HH. The tRNA is flanked on both ends by HH ribozymes. Dashed lines represent bonds that are cleaved by the ribozymes. Indicated with single and double solid lines are regions of complementarity between the HHs and the tRNA that would form an alternate secondary structure during the ribozyme cleavage reactions. Sequences that are complementary to the HIV-1 PBS are shown in bold letters and boxed, and a C75U mutation in the tRNA (required for 3′ HH cleavage) is in lowercase.
Figure 2.

Purification of tRNALys3 after cleavage by HH ribozymes. (A) Elution profile of RNA separated by denaturing PAGE on a Bio-Rad Model 491 Prep Cell. Fractions collected and time after loading the RNA sample onto the gel are indicated, respectively, below and above the UV absorbance trace. (B) Selected fractions from peaks II (fractions 18 and 22) and III (fractions 28–39), and from between the two peaks (fraction 27), were analyzed for purity on an analytical denaturing gel. Peak I comigrates with the bromophenol blue dye. A previously purified sample (tRNA) and the unpurified RNA sample loaded onto the gel (load) were run for comparison. Migration positions of tRNALys3 (76 nt) and HH ribozymes (HHs; 57 and 58 nt) are indicated.
The tRNALys3 transcript was separated from the other RNAs by electrophoresis on a cylindrical denaturing polyacrylamide tube gel (Model 491 Prep Cell; Bio-Rad). Fractions were monitored by UV absorbance and collected as they electroeluted off the bottom of the gel. The results of a typical separation are shown in Figure 2. Three major peaks were usually visible in the elution profile (Fig. 2A). The first peak (I) comigrates with the bromophenol blue dye and may also contain unincorporated nucleotides and short abortive transcripts (not determined). Peak II contains the 5′ and 3′ HH ribozymes (Fig. 2B, fractions 18 and 22) and peak III contains the tRNA (Fig. 2B, fractions 28–39), as determined by migration on an analytical denaturing slab gel. The identity of tRNALys3 was confirmed by direct sequencing of the purified RNA using RNases. The yield of purified of tRNALys3 transcript is ∼0.5 mg from each milliliter of the transcription reaction.
To achieve the clean separation of the RNAs, we found it necessary to include both formamide (40–45%, v/v) and urea (7 M) in the gel as denaturants and to maintain a gel temperature of 50–55°C. Without these conditions, some of the HH RNA comigrated with the tRNA (data not shown). Because the rest of the HH RNA eluted in a distinct peak, we concluded that the RNA was not fully denatured. The Prep Cell apparatus is designed so that the entire tube gel can be immersed in running buffer and the interior of the cylindrical gel can be cooled with recirculating buffer. While these features allow the gel to be cooled evenly, they made it difficult to heat the gel evenly to the higher temperature range (50–55°C) required to fully denature our RNAs. We found instead that the RNAs eluted in sharper peaks when the interior buffer was not recirculated and when only the very bottom of the gel was covered with running buffer. Longer gels (32) were somewhat helpful, but only when an even gel temperature could be maintained.
The 2′–3′ cyclic phosphate, produced by cleavage of the 3′ HH ribozyme, was removed from the 3′ end of the purified tRNA by acid treatment (25) and subsequent dephosphorylation. We were particularly careful about this step because a 3′ OH is absolutely required for tRNALys3 to be used as a primer for DNA synthesis by HIV-1 RT. Due to the large scale of our transcription reactions, we optimized the dephosphorylation conditions to use a minimal amount of purchased phosphatase. This makes the removal of unincorporated nucleotides from the transcription reaction particularly important, and could be easily accomplished by gel purifying the RNA. However, as the phosphatase preparations sometimes contain small amounts of contaminating RNases, the final RNA preparation is usually more homogeneous if the decyclization and dephosphorylation are done prior to gel purification. In this case, the nucleotides must be removed by gel filtration or extensive dialysis. We monitored the extent of dephosphorylation on high resolution sequencing gels; tRNALys3 migrates slightly slower when the phosphate has been removed (Fig. 4).
Figure 4.
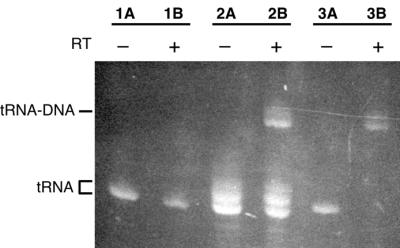
Denaturing gel showing use of transcribed tRNALys3 as a primer for DNA synthesis by HIV-1 RT. A concentration of 5 µM annealed tRNALys3/PBS-K2 RNA was incubated for 15 min at 37°C without (–; lanes 1A, 2A, 3A) or with (+; lanes 1B, 2B, 3B) 5 µM HIV-1 RT in the presence of 1 mM each dNTP. Prior to use in the initiation reaction, the 2′–3′ cyclic phosphate at the 3′ end of tRNALys3, which results from cleavage by the 3′ HH ribozyme, either was not removed (lanes 1A and 1B), was partially removed (lanes 2A and 2B) or was completely removed (lanes 3A and 3B), as described in Materials and Methods. Migration positions of unextended (tRNA) or fully extended (tRNA–DNA) primer, as visualized by ethidium bromide staining, are indicated. The dephosphorylated tRNA is the slower migrating of the two tRNA species.
Assembly of an HIV-1 reverse transcriptase initiation complex
We formed a stoichiometric RNA complex between tRNALys3 and PBS-K2 template by heat denaturing and slow cooling the RNAs, allowing them to anneal (Fig. 3A). PBS-K2 is a 36-nt RNA containing the 18-nt PBS sequences from HIV-1, with an additional 10 nt on the 5′ side and 8 nt on the 3′ side (see Table 1). After annealing, a new band that stains brightly with ethidium bromide is visible on a native gel (Fig. 3A, lanes 3–8), migrating slightly more slowly than tRNA alone (lane 1). When PBS-K2 RNA was present at concentrations above (lanes 6–8) and below (lanes 2 and 3) the 25 µM concentration of tRNALys3, uncomplexed tRNA and PBS-K2 were, respectively, visible. Based on this titration experiment, together with the small difference in mobility compared to tRNA alone, we conclude that the RNA complex contains one molecule each of tRNALys3 and PBS-K2, as we expected. The complex appears to be conformationally homogeneous, as it migrates as a tight, single band on the non-denaturing gel.
We assembled an initiation complex by incubating HIV-1 RT with the annealed tRNALys3/PBS-K2 RNA complex (Fig. 3B). Increasing concentrations of RNA (5–20 µM) were mixed with 10 µM RT, which is at least 200-fold above the apparent dissociation constant (∼10–50 nM; J. D. Pata, unpublished results) for binding tRNA/PBS. Thus, in this titration experiment unbound RNA will only be observed when the RNA binding sites on all the RT molecules are saturated. At RNA concentrations above the 10 µM RT concentration (lanes 6–8), free RNA was visible while at lower concentrations (lanes 3 and 4) none was detected. These results indicate that the initiation complex contains equimolar amounts of each component: HIV-1 RT, tRNALys3 and PBS-K2 RNA.
Initiation complex activity
Essentially all of the tRNALys3 transcript in the HIV-1 RT initiation complex can be extended by RT, as long as the 2′–3′ cyclic phosphate produced during 3′ HH ribozyme cleavage is completely removed (Fig. 4). Equimolar concentrations of tRNALys3 and PBS-K2 template RNA were annealed and then incubated at a concentration of 5 µM with 5 µM HIV-1 RT (lanes 1B, 2B and 3B) for 15 min in the presence of dNTPs to allow the synthesis of DNA. Control reactions (lanes 1A, 2A and 3A) did not contain RT. The tRNALys3 used in these reactions was either not dephosphorylated after HH ribozyme cleavage (lanes 1A and 1B), was partially dephosphorylated (lanes 2A and 2B), or was completely dephosphorylated (lanes 3A and 3B). Only the fully dephosphorylated tRNA was quantitatively used as a primer by HIV-1 RT (lane 3B).
Purification of the initiation complex
The initiation complex, containing HIV-1 RT, tRNALys3 and PBS template RNA, was purified by gel-filtration chromatography, as shown in Figure 5. We assembled an initiation complex as described above, except that the RNA template (PBS-WT1, Table 1) was slightly shorter, with only 3 nt on either side of the 18-nt PBS. Initiation reactions using this template are comparable to those using PBS-K2 template (data not shown). The complex, at a concentration of 20 µM, was loaded onto a Superdex 200 HR 10/30 column for purification. From gel-shift experiments (Fig. 3B) we had been concerned that the initiation complex was forming a heterogeneous aggregate, since the complex barely migrates into the gel. This is not the case, however, as >85% of the complex elutes as a single peak centered at an elution volume of 10.2 ml, well within the separation range of this column (void volume, 8 ml; column volume, 23 ml). Multi-angle laser light scattering measurements indicate that the central portion of this peak is monodisperse (i.e. homogeneous with respect to molecular weight). The absolute molecular weight of the complex was not determined because of technical difficulties in obtaining an accurate value for the refractive index of the initiation complex as a function of concentration. The initiation complex peak is well separated from uncomplexed RT heterodimer (elution volume 12.0 ml) and uncomplexed tRNA/PBS RNA (elution volume 12.5 ml), which were each purified separately for comparison. The purified initiation complex has an A260/A280 ratio of 1.25, which is identical to the value determined for the stoichiometric complex loaded onto the column. This confirms the conclusions from the titration experiments shown in Figure 3, that HIV-1 RT and RNA are present in equimolar concentrations in the initiation complex.
Figure 5.

Purification of the HIV-1 initiation complex by Superdex 200 gel-filtration chromatography. Elution profiles, detected by light scattering at 690 nm, are shown for the initiation complex (RT/tRNA/PBS), RT alone (RT), and tRNALys3 annealed to PBS-WT1 template RNA (tRNA/PBS). The initiation complex (peak at 10.2 ml) cleanly separates from RT alone (peak at 12 ml) and from annealed primer/template RNA (peak at 12.5 ml). Elution positions of BSA monomer (67 kDa) and dimer (134 kDa) standards are shown.
Crystallization of the HIV-1 RT initiation complex
We have grown crystals of the HIV-1 RT initiation complex that contain protein and RNA in stoichiometric amounts (Fig. 6). The crystals were grown by hanging drop vapor diffusion out of a solution containing 50 µM pre-assembled initiation complex (HIV-1 RT/tRNALys3/PBS-CUA), 200 mM NaCl, 10 mM MgCl2, 50 mM MES (pH 5.4–6.6), 5–7% PEG-8000. The crystals are small (dimensions up to 100 × 50 × 25 µm) and grow as thin rods or plates that appear in 3 days to several weeks. The growth characteristics of the crystals vary consistently depending on the combination of nucleotides included (Fig. 6A and B, and data not shown); the growth rate and number of nucleations in the drop decreased as the number of nucleotides included increased. Using synchrotron radiation (Beamline X25, Brookhaven National Laboratory) the crystals diffracted to 8 Å. The crystals are orthorhombic, with C centered lattice spacings of a = 166 Å, b = 167 Å and c = 321 Å. The diffraction was most likely limited by the small size of the crystals, combined with the large unit cell dimensions; the ultimate diffraction limit of this crystal form remains to be determined. Silver-stained SDS–PAGE analysis of washed crystals (Fig. 6C) shows that the crystals contain intact RNA and protein in the same equimolar ratios used to assemble the initiation complex.
Figure 6.

HIV-1 RT initiation complex crystals. Initiation complex containing HIV-1 RT, tRNALys3 and PBS-CUA was incubated for 10 min (A) with ddCTP and dTTP or (B) with dCTP, ddTTP and dGTP prior to crystallization to trap the complex after the addition of 1 or 2 nt, respectively. Crystals were grown by hanging drop vapor diffusion by mixing equal volumes of initiation complex and well solution (5.5% PEG-8000, 200 mM NaCl, 50 mM MES pH 5.7). (C) Silver-stained SDS–PAGE analysis of initiation complex crystals grown in the presence of ddCTP and dTTP. Crystals were washed three times (W1, W2 and W3) in stabilizing solution (well solution with 10% PEG-8000), then dissolved in water (X) and compared to the remaining solution in the drop (S) and the complex used to set up the drop (RT/tRNA/PBS). Markers: HIV-1 RT p66/p51 heterodimer (RT), annealed primer/template RNAs (tRNA/PBS), and primer alone (tRNA). An additional band (*) is probably an alternate conformation, possibly a dimer, of the primer/template RNA that results from incomplete denaturation of the RNA. Other gels of similar crystals do not show this species (data not shown).
DISCUSSION
Structural studies of the initiation of minus-strand DNA synthesis by HIV-1 RT have been limited, to a large extent, by difficulties in obtaining sufficient quantities of pure tRNALys3 that is fully active as a primer. In vitro transcription of tRNALys3 typically results in a heterogeneous mixture of RNA lengths (20; J. D. Pata, unpublished results) due to the tendency of T7 RNA polymerase to add non-templated nucleotides to the 3′ end of transcripts. Yields of tRNALys3 isolated from tissues such as calf liver tend to be low, requiring extreme efforts to purify enough for crystallographic studies (33), and overexpression of human tRNALys3 in E.coli produces a heterogeneous mixture of submodified species that, when purified, hybridize to HIV-1 viral RNA with 70–80% efficiency (34).
Here we show that we can produce tRNALys3 transcripts of homogeneous length by using HH ribozymes to cleave the primary transcript. This RNA can assemble into an initiation complex containing equimolar amounts of HIV-1 RT, tRNALys3 and PBS-template RNA, and a homogeneous complex can be purified by gel-filtration chromatography. Virtually all of the tRNA in the complex can be used by HIV-1 RT to prime DNA synthesis, indicating that we have produced an active initiation complex and that transcribing tRNALys3 in vitro is a viable alternative to purifying tRNALys3 from cells. Finally, we have obtained crystals of the HIV-1 RT initiation complex and have confirmed that they contain intact tRNA. To our knowledge, this is the first report describing conditions for co-crystallizing HIV-1 RT with primer tRNA.
We have developed conditions for purifying highly structured RNAs using the Bio-Rad Prep Cell, and are able to isolate ∼3 mg of tRNALys3 from one-third of a 25 ml transcription reaction on each run. The Prep Cell has been previously used to purify a 34-nt HH ribozyme RNA away from N + 1 and N – 1 transcription products (32,35). The principle advantages of this purification method over traditional slab gel purification methods are that minimal handling of the sample is required and that the gels can be reused two to three times. We initially found the Prep Cell difficult to use because the tRNA and HH RNAs are surprising difficult to denature. We were able to overcome this problem by including formamide in the running buffer and by altering the set-up of the apparatus gel so that we could maintain a constant high temperature across the gel. The modifications required for separating tRNALys3 from the HH RNAs are generally applicable for purifying other highly structured RNAs.
A precisely defined 3′ end was critical for obtaining fully active transcribed tRNALys3. We observe that ∼100% of transcribed tRNALys3 can be fully extended by HIV-1 RT in a 15 min reaction, using an RNA oligonucleotide template. In a comparable experiment Yusupova et al. (36) had previously found that only about half of their transcribed tRNALys3 could be extended in a 30 min reaction. Two differences in our experimental design may explain why were able to substantially increase the priming efficiency, assuming that there was no significant difference between the transcribed RNAs. First, we used a template with overhanging sequences on the 3′ side of the PBS, to avoid competition from the template being used as an alternate primer. Secondly, we used much higher concentrations of HIV-1 RT, tRNALys3 and template RNA (10 µM compared to 30 nM), so that the protein and RNA were both at concentrations well above the binding constant (10–50 nM; J. D. Pata, unpublished results). Regardless of the explanation, producing transcribed tRNALys3 with a homogeneous 3′ end was an extremely important step forward because we have only been able to grow initiation complex crystals using tRNALys3 that can be fully extended by HIV-1 RT.
HIV-1 RT forms a discrete complex with tRNALys3 and template RNA, as demonstrated by our purification of the initiation complex by gel-filtration chromatography and subsequent analysis by dynamic light scattering. While perhaps not surprising at first glance, this result was not a forgone conclusion. When we analyze binding of HIV-1 RT to tRNALys3/PBS primer-template RNA using gel band shift assays, we find either that multiple bands form at nanomolar concentrations (J. D. Pata, unpublished results) or that the RNA and protein shift together into the wells (at micromolar concentrations; see Fig. 3B). Similar results have been reported for complexes of HIV-1 RT bound to primer tRNALys3 alone (37,38). The slower migrating complex has been interpreted as being a dimer of the binary complex (37). The complex we have isolated may similarly be a dimer of the HIV-1 RT/tRNA/PBS initiation complex. Our gel filtration (Fig. 5) and light scattering studies have not yet been able to distinguish whether the complex is a monomer or a dimer, although in either case the protein and the RNA are present in equimolar amounts. Each HIV particle contains approximately 80 molecules of HIV-1 RT (39), equivalent to a concentration of ∼100 µM (40). Because addition of the first few deoxynucleotides to the tRNA primer can occur in virus particles (5), micromolar concentrations of the initiation complex are biologically relevant and warrant further investigation.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Stuart LeGrice, Stephen Hughes and Paul Boyer for the generous gifts of HIV-1 RT expression clones; Satwik Kamtekar and Joe Jäger for helpful discussions and critical reading of the manuscript; Bill Eliason and Joe Watson for technical assistance; Bo-Lu Zhou for communication of unpublished results; and Tobias Restle for helpful discussions in the early stages of this project. This work was supported by NIH Postdoctoral Fellowship AI09693 to J.D.P. and by NIH Grant AI43896 to T.A.S.
REFERENCES
- 1.Arts E.J. and Le Grice,S.F. (1998) Interaction of retroviral reverse transcriptase with template-primer duplexes during replication. Prog. Nucleic Acid Res. Mol. Biol., 58, 339–393. [DOI] [PubMed] [Google Scholar]
- 2.Arts E.J. and Wainberg,M.A. (1996) Human immunodeficiency virus type 1 reverse transcriptase and early events in reverse transcription. Adv. Virus Res., 46, 97–163. [DOI] [PubMed] [Google Scholar]
- 3.Zhang H., Dornadula,G., Wu,Y., Havlir,D., Richman,D.D. and Pomerantz,R.J. (1996) Kinetic analysis of intravirion reverse transcription in the blood plasma of human immunodeficiency virus type 1-infected individuals: direct assessment of resistance to reverse transcriptase inhibitors in vivo. J. Virol., 70, 628–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Auxilien S., Keith,G., Le Grice,S.F. and Darlix,J.L. (1999) Role of post-transcriptional modifications of primer tRNALys3 in the fidelity and efficacy of plus strand DNA transfer during HIV-1 reverse transcription. J. Biol. Chem., 274, 4412–4420. [DOI] [PubMed] [Google Scholar]
- 5.Oude Essink B.B., Das,A.T. and Berkhout,B. (1996) HIV-1 reverse transcriptase discriminates against non-self tRNA primers. J. Mol. Biol., 264, 243–254. [DOI] [PubMed] [Google Scholar]
- 6.Lanchy J.M., Keith,G., Le Grice,S.F., Ehresmann,B., Ehresmann,C. and Marquet,R. (1998) Contacts between reverse transcriptase and the primer strand govern the transition from initiation to elongation of HIV-1 reverse transcription. J. Biol. Chem., 273, 24425–24432. [DOI] [PubMed] [Google Scholar]
- 7.Isel C., Lanchy,J.M., Le Grice,S.F., Ehresmann,C., Ehresmann,B. and Marquet,R. (1996) Specific initiation and switch to elongation of human immunodeficiency virus type 1 reverse transcription require the post-transcriptional modifications of primer tRNALys3. EMBO J., 15, 917–924. [PMC free article] [PubMed] [Google Scholar]
- 8.Lanchy J.M., Ehresmann,C., Le Grice,S.F., Ehresmann,B. and Marquet,R. (1996) Binding and kinetic properties of HIV-1 reverse transcriptase markedly differ during initiation and elongation of reverse transcription. EMBO J., 15, 7178–7187. [PMC free article] [PubMed] [Google Scholar]
- 9.Vaccaro J.A., Singh,H.A. and Anderson,K.S. (1999) Initiation of minus-strand DNA synthesis by human immunodeficiency virus type 1 reverse transcriptase. Biochemistry, 38, 15978–15985. [DOI] [PubMed] [Google Scholar]
- 10.Thrall S.H., Krebs,R., Wohrl,B.M., Cellai,L., Goody,R.S. and Restle,T. (1998) Pre-steady-state kinetic characterization of RNA-primed initiation of transcription by HIV-1 reverse transcriptase and analysis of the transition to a processive DNA-primed polymerization mode. Biochemistry, 37, 13349–13358. [DOI] [PubMed] [Google Scholar]
- 11.Arts E.J., Stetor,S.R., Li,X., Rausch,J.W., Howard,K.J., Ehresmann,B., North,T.W., Wohrl,B.M., Goody,R.S., Wainberg,M.A. et al. (1996) Initiation of (–) strand DNA synthesis from tRNALys3 on lentiviral RNAs: implications of specific HIV-1 RNA-tRNALys3 interactions inhibiting primer utilization by retroviral reverse transcriptases. Proc. Natl Acad. Sci. USA, 93, 10063–10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rigourd M., Lanchy,J.M., Le Grice,S.F., Ehresmann,B., Ehresmann,C. and Marquet,R. (2000) Inhibition of the initiation of HIV-1 reverse transcription by 3′-azido-3′-deoxythymidine. Comparison with elongation. J. Biol. Chem., 275, 26944–26951. [DOI] [PubMed] [Google Scholar]
- 13.Vaccaro J.A. and Anderson,K.S. (1998) Implication of the tRNA initiation step for human immunodeficiency virus type 1 reverse transcriptase in the mechanism of 3′-azido-3′-deoxythymidine (AZT) resistance. Biochemistry, 37, 14189–14194. [DOI] [PubMed] [Google Scholar]
- 14.Kohlstaedt L.A., Wang,J., Friedman,J.M., Rice,P.A. and Steitz,T.A. (1992) Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science, 256, 1783–1790. [DOI] [PubMed] [Google Scholar]
- 15.Huang H., Chopra,R., Verdine,G.L. and Harrison,S.C. (1998) Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science, 282, 1669–1675. [DOI] [PubMed] [Google Scholar]
- 16.Ding J., Das,K., Hsiou,Y., Sarafianos,S.G., Clark,A.D.,Jr, Jacobo-Molina,A., Tantillo,C., Hughes,S.H. and Arnold,E. (1998) Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 Å resolution. J. Mol. Biol., 284, 1095–1111. [DOI] [PubMed] [Google Scholar]
- 17.Jacobo-Molina A., Ding,J., Nanni,R.G., Clark,A.D.,Jr, Lu,X., Tantillo,C., Williams,R.L., Kamer,G., Ferris,A.L., Clark,P. et al. (1993) Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 Å resolution shows bent DNA. Proc. Natl Acad. Sci. USA, 90, 6320–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarafianos S.G., Das,K., Tantillo,C., Clark,A.D.,Jr, Ding,J., Whitcomb,J.M., Boyer,P.L., Hughes,S.H. and Arnold,E. (2001) Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J., 20, 1449–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jäger J., Restle,T. and Steitz,T.A. (1998) The structure of HIV-1 reverse transcriptase complexed with an RNA pseudoknot inhibitor. EMBO J., 17, 4535–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mishima Y. and Steitz,J.A. (1995) Site-specific crosslinking of 4-thiouridine-modified human tRNALys3 to reverse transcriptase from human immunodeficiency virus type I. EMBO J., 14, 2679–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Price S.R., Ito,N., Oubridge,C., Avis,J.M. and Nagai,K. (1995) Crystallization of RNA-protein complexes. I. Methods for the large-scale preparation of RNA suitable for crystallographic studies. J. Mol. Biol., 249, 398–408. [DOI] [PubMed] [Google Scholar]
- 22.Lee S.Y. and Rasheed,S. (1990) A simple procedure for maximum yield of high-quality plasmid DNA. Biotechniques, 9, 676–679. [PubMed] [Google Scholar]
- 23.Jeruzalmi D. and Steitz,T.A. (1998) Structure of T7 RNA polymerase complexed to the transcriptional inhibitor T7 lysozyme. EMBO J., 17, 4101–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shields T.P., Mollova,E., Ste Marie,L., Hansen,M.R. and Pardi,A. (1999) High-performance liquid chromatography purification of homogenous-length RNA produced by trans cleavage with a hammerhead ribozyme. RNA, 5, 1259–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forster A.C., Davies,C., Hutchins,C.J. and Symons,R.H. (1990) Characterization of self-cleavage of viroid and virusoid RNAs. Methods Enzymol., 181, 583–607. [DOI] [PubMed] [Google Scholar]
- 26.Milligan J.F., Groebe,D.R., Witherell,G.W. and Uhlenbeck,O.C. (1987) Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res., 15, 8783–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Le Grice S.F. and Gruninger-Leitch,F. (1990) Rapid purification of homodimer and heterodimer HIV-1 reverse transcriptase by metal chelate affinity chromatography. Eur. J. Biochem., 187, 307–314. [DOI] [PubMed] [Google Scholar]
- 28.Boyer P.L., Ding,J., Arnold,E. and Hughes,S.H. (1994) Subunit specificity of mutations that confer resistance to nonnucleoside inhibitors in human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother., 38, 1909–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Le Grice S.F., Cameron,C.E. and Benkovic,S.J. (1995) Purification and characterization of human immunodeficiency virus type 1 reverse transcriptase. Methods Enzymol., 262, 130–144. [DOI] [PubMed] [Google Scholar]
- 30.Kohlstaedt L.A. and Steitz,T.A. (1992) Reverse transcriptase of human immunodeficiency virus can use either human tRNALys3 or Escherichia coli tRNAGln2 as a primer in an in vitro primer-utilization assay. Proc. Natl Acad. Sci. USA, 89, 9652–9656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferre-d’Amare A.R. and Doudna,J.A. (1997) Establishing suitability of RNA preparations for crystallization. Determination of polydispersity. Methods Mol. Biol., 74, 371–377. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen T.H., Cunningham,L.A., Hammond,K.M. and Lu,Y. (1999) High-resolution preparative-scale purification of RNA using the Prep Cell. Anal. Biochem., 269, 216–218. [DOI] [PubMed] [Google Scholar]
- 33.Benas P., Bec,G., Keith,G., Marquet,R., Ehresmann,C., Ehresmann,B. and Dumas,P. (2000) The crystal structure of HIV reverse-transcription primer tRNALys3 shows a canonical anticodon loop. RNA, 6, 1347–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tisne C., Rigourd,M., Marquet,R., Ehresmann,C. and Dardel,F. (2000) NMR and biochemical characterization of recombinant human tRNALys3 expressed in Escherichia coli: identification of posttranscriptional nucleotide modifications required for efficient initiation of HIV-1 reverse transcription. RNA, 6, 1403–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cunningham L., Kittikamron,K. and Lu,Y. (1996) Preparative-scale purification of RNA using an efficient method which combines gel electrophoresis and column chromatography. Nucleic Acids Res., 24, 3647–3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yusupova G., Lanchy,J.M., Yusupov,M., Keith,G., Le Grice,S.F., Ehresmann,C., Ehresmann,B. and Marquet,R. (1996) Primer selection by HIV-1 reverse transcriptase on RNA-tRNALys3 and DNA-tRNALys3 hybrids. J. Mol. Biol., 261, 315–321. [DOI] [PubMed] [Google Scholar]
- 37.Arts E.J., Miller,J.T., Ehresmann,B. and Le Grice,S.F. (1998) Mutating a region of HIV-1 reverse transcriptase implicated in tRNALys3 binding and the consequences for (–)-strand DNA synthesis. J. Biol. Chem., 273, 14523–14532. [DOI] [PubMed] [Google Scholar]
- 38.Barat C., Le Grice,S.F. and Darlix,J.L. (1991) Interaction of HIV-1 reverse transcriptase with a synthetic form of its replication primer, tRNALys3. Nucleic Acids Res., 19, 751–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Layne S.P., Merges,M.J., Dembo,M., Spouge,J.L., Conley,S.R., Moore,J.P., Raina,J.L., Renz,H., Gelderblom,H.R. and Nara,P.L. (1992) Factors underlying spontaneous inactivation and susceptibility to neutralization of human immunodeficiency virus. Virology, 189, 695–714. [DOI] [PubMed] [Google Scholar]
- 40.Lebowitz J., Kar,S., Braswell,E., McPherson,S. and Richard,D.L. (1994) Human immunodeficiency virus-1 reverse transcriptase heterodimer stability. Protein Sci., 3, 1374–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]