


Abstract
An intein-mediated approach was developed for expression and affinity purification of a protein that is lethal to Escherichia coli. The protein, I-TevI, is an intron-encoded endonuclease. The approach involved the insertional inactivation of I-TevI with a controllable mini-intein placed in front of a cysteine required for splicing (an I-TevI::intein fusion). The purification was facilitated by a chitin-binding domain inserted into the mini-intein. Affinity purification of the I-TevI::intein fusion precursor on a chitin column was followed by pH-controllable splicing to restore the structure and function of I-TevI. To study the impact of the insertion context on I-TevI inactivation, the chimeric intein was inserted independently in front of seven cysteines of I-TevI. One of the seven intein integrants yielded I-TevI of high activity. This technique is, in principle, generalizable to the expression and purification of other cytotoxic proteins and is amenable to scale-up.
INTRODUCTION
Inteins are the protein analogs of introns that exist as in-frame fusions with flanking protein sequences called exteins (1). Inteins, along with the first residue of the C-terminal extein, are capable of achieving the remarkable chemical feats of breaking two peptide bonds between the exteins and the intein and forming a new peptide bond between the exteins (2). With a few exceptions, these reactions require cysteine, serine or threonine at the N-terminal ends of both the intein and C-extein and an asparagine at the C-terminal end of the intein. The intein-mediated protein splicing mechanism includes four distinct but highly concerted steps that culminate in a new peptide bond between the exteins (3–5). These steps are: (i) formation of a linear (thio)ester intermediate at the N-terminal splice site; (ii) formation of a branched intermediate through trans(thio)esterification by the -OH or -SH side chain at the N-terminus of the C-extein; (iii) detachment of the intein from ligated exteins through cyclization of the terminal asparagine; and (iv) formation of a normal peptide bond between the exteins through a spontaneous acyl shift.
The elucidation of the intein-mediated protein splicing mechanism has facilitated the development of intein applications in some key areas of protein engineering (6,7). These applications include self-cleavable affinity tags to facilitate protein purification (8–11), intein-mediated protein ligation (7,12–14), differential isotope labeling of proteins (15,16) and cyclization of the protein backbone (17). To achieve expression and purification of a full-length cytotoxic protein, we tested the feasibility of a novel intein-mediated approach. The idea involves the insertional inactivation of the toxic protein by an intein. In vivo expression is followed by affinity purification and controllable on-column splicing to restore the native configuration of the target protein (Fig. 1).
Figure 1.
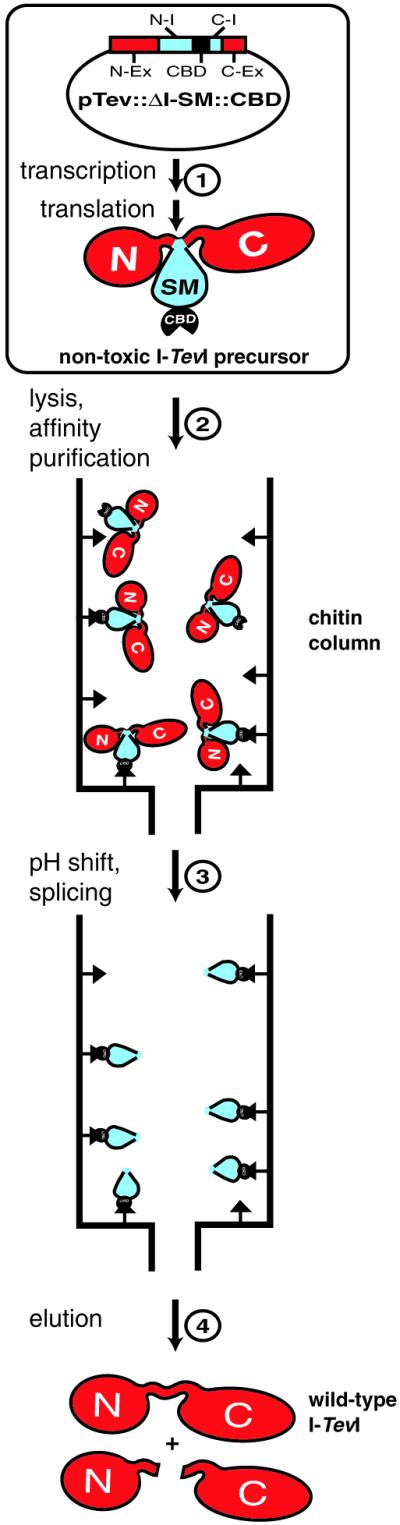
Strategy for intein-mediated purification of cytotoxic endonuclease I-TevI. Step 1, the precursor protein was expressed in vivo after induction of pTevI::ΔI-SM::CBD derivatives with IPTG. N-Ex, C-Ex, N-I and C-I denote N- and C-exteins and N- and C-portions of the intein, respectively. Step 2, after cell lysis and clarification in chitin column buffer (pH 8.5) the cell lysate was loaded onto a chitin column. Step 3, after washing the column, the pH was shifted from 8.5 to 7.5 to trigger splicing. Step 4, the products were eluted after 26 h at 4°C. They included the full-length wild-type I-TevI and the N- and C-terminal cleavage products (see Fig. 5).
I-TevI, the T4 td intron-encoded endonuclease (18), was chosen as the subject of our study because of its extreme toxicity and our familiarity with its structure and function. I-TevI binds to its substrate in a sequence-tolerant manner (19) and its toxicity is probably the result of cleavage of the Escherichia coli chromosome at ectopic sites. I-TevI comprises an N-terminal catalytic domain and a C-terminal DNA-binding domain connected by a flexible linker (20–22). The N-terminal domain is particularly toxic, presumably due to its nuclease activity that is even less sequence specific in the absence of its DNA-binding domain. The structures of the C-terminal domain in the presence of its substrate DNA (23) and the N-terminal domain with a catalytic mutation (24) have been solved independently. This knowledge allowed an evaluation of the approach with regard to the extein context of the intein insertion site and facilitated, for the first time, the successful purification of the toxic wild-type I-TevI intron endonuclease.
MATERIALS AND METHODS
Construction of I-TevI-intein fusion constructs
The chitin-binding domain (CBD) was cloned into the ΔI-SM mini-intein (10) using a BssHII site that had been placed at the position where the endonuclease domain of the Mycobacterium tuberculosis (Mtu) RecA intein was removed, to generate ΔI-SM::CBD (10,25). The primers used were as follows: W1200, 5′-CCGTTGGCGCGCACGACAAATCCTGGTGTATCC-3′; W1201, 5′-CCGTTGGCGCGCTTGAAGCTGCCACAAGGCAGGAAC-3′.
For seven intein insertion sites, gene SOEing (26) was used to fuse the ΔI-SM::CBD gene to the I-TevI gene. The DNA for the N-terminal domain of I-TevI, the ΔI-SM::CBD intein and the C-terminal domain of I-TevI were amplified separately by PCR and spliced together sequentially. The PCR conditions for the SOEing reactions were as follows: 1 min at 95°C, 1 min at 50°C and 1.5 min at 72°C for 30 cycles. The SOEing products were cloned into pET28a (Novagen) at the XbaI and HindIII sites to generate pTev::ΔI-SM::CBD. The ligation products were transformed into the non-expression strain JM101 and screened by restriction digestion and sequencing. Isolates were then transformed into BL21(DE3)pLysS for overexpression. SOEing primers carrying alanine mutations for the first and last residues of the ΔI-SM mini-intein were used to make non-spliceable fusion constructs. DNA encoding the catalytic R27A mutant of I-TevI (21) was used as template to make non-toxic fusion genes, which were then cloned into pMAL-c2X (New England Biolabs) using EcoRI and HindIII sites. A typical set of SOEing primers for the construction of pTevC164::ΔI-SM::CBD were as follows: W1504, 5′-CATACAAACTAGTGCTTATACTTGCCTCGCAGAGGGCAC-3′; W1505, 5′-GTGCCCTCTGCGAGGCAAGTATAAGCACTAGTTTGTATG-3′; W1506, 5′-GGTTGTCGTGCACAACTGTTCGAAATGCAGAAATCG-3′; W1507, 5′-CGA TTTCTGCATTTCGAACAGTTGTGCACGACAACC-3′. The following primers were used to clone the I-TevI::ΔI-SM::CBD cartridge into pET28a: W349, 5′-GGCGGCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATACATATGAAAAGCGGAATTTATCAG-3′; W476, 5′-CGGGGT ACCCGAAGCTTTTAGGCATTTATGTAGAACCAATTC-3′.
Overexpression, purification and mass spectrometry of wild-type I-TevI
pTevC164::ΔI-SM::CBD was transformed into BL21(DE3) pLysS. The cells were grown in L broth (2% tryptone, 1% yeast extract and 1% w/v NaCl) with 50 µg/ml kanamycin at 20°C and induced with 1 mM IPTG at an OD600 of 0.4 for 2 h. The cells were collected by centrifugation and the pellet was resuspended and sonicated in pH 8.5 chitin column buffer (20 mM Tris–HCl, 500 mM NaCl, 1 mM DTT, 0.1 mM EDTA and 0.1% Triton X-100). The clarified cell lysate was loaded onto a chitin column equilibrated with pH 8.5 column buffer and the column was washed with 20 bed volumes of pH 8.5 buffer. The column pH was then rapidly changed to pH 7.5 to trigger splicing. After 26 h, the splicing and cleavage products were eluted. In order to separate full-length I-TevI from N- and C-terminal cleavage products, the eluted fractions were pooled and concentrated to 200 µl using a Centricon 10 concentrator (Amicon) and then applied to a Superose 12 gel filtration column (Pharmacia Biotech). The entire purification process was conducted at 4°C. The molecular mass of ligated I-TevI was analyzed by infusion on a Finnigan-MAT TSQ-700 triple quadrupole mass spectrometer equipped with a Finnigan ESI source.
Quantitation of intein reaction products
All seven pMBP-TevR27A::ΔI-SM::CBD derivatives were transformed into strain JM109. Cells were grown in L broth with 200 µg/ml ampicillin at 20°C to an OD600 of 0.4 and expression was induced by the addition of 1 mM IPTG. After 2 h at 20°C cells were harvested as above. The cell pellet was resuspended in pH 8.5 amylose column buffer (500 mM NaCl, 20 mM 2-amino-2-methyl-1,3-propanediol, 20 mM PIPES, 2 mM EDTA and 5% glycerol). Purification of the precursor was carried out using amylose resin (NEB) according to the manufacturer’s instructions. The peak fractions from all seven constructs were immediately analyzed by SDS–PAGE to approximate the in vivo distribution of reaction products.
In addition, samples were immediately subjected to in vitro reaction at pH 7.4 and 37°C to investigate in vitro splicing and cleavage. Aliquots were taken at different times and resolved by 8% SDS–PAGE. The gels were scanned with a Microtek Scanmaker 3750i and quantitated with ImageQuant (Molecular Dynamics). The measured intensities of bands were normalized to their molecular weights.
Determination of optimal pH for intein-mediated protein splicing at Cys164 of I-TevI
The precursor of MBP-TevR27ACys164::ΔI-SM::CBD was purified as described in the previous section. The purified protein was stored at 4°C overnight before being subjected to in vitro reaction at 10 different pH values. The reaction buffer was the same as the column buffer and the pHs were adjusted at 4°C, the temperature at which the in vitro reaction was conducted. Aliquots were taken at various times and resolved by SDS–PAGE. The gels were scanned and quantified as described in the previous section.
Specific activity determination of purified I-TevI and its H40Y derivative
One unit of I-TevI activity is defined as the amount of enzyme required to cleave 250 ng of linearized target DNA (5 kb) to 50% completion in 1 min at 37°C (27). Protein concentrations of purified wild-type I-TevI and its H40Y derivative were determined using a modified Lowry assay (DC Protein Assay; Bio-Rad). The concentration of wild-type I-TevI eluted from the chitin column was estimated by multiplying the concentration of total protein and the mass percentage of the full-length species as determined by scanning and quantitating SDS–PAGE gels. Cleavage assays were carried out with serial dilutions of the enzymes.
RESULTS
Insertional inactivation of I-TevI by a mini-intein mutant
Because I-TevI is lethal when expressed in E.coli, its overexpression for purification has been an insurmountable challenge since its discovery in 1989 (18). An intein-mediated affinity purification system developed in our laboratory (11), which is based on pH-sensitive C-terminal intein cleavage, also failed due to the toxicity of the side-by-side I-TevI–intein fusion. Therefore, a mini-intein mutant (ΔI-SM) of the full-length Mtu RecA intein (10) was used to insertionally inactivate I-TevI in vivo (an I-TevI::intein fusion) and trigger splicing after purification in an affinity column in vitro. The ΔI-SM mini-intein splices as efficiently as the 440 amino acid wild-type intein and yet is only one-third the size (168 amino acids), thereby facilitating the production of fusion precursors. Furthermore, the splicing of this intein can be activated by a downshift in pH (see below).
About 90% of the inteins identified so far have a bipartite domain structure, which comprises a protein-splicing domain and an optional central endonuclease domain (25,28). The endonuclease domain of the Mtu RecA intein was dispensable for intein-mediated protein splicing (25,29), facilitating the isolation of the above-mentioned mini-intein. To allow the purification of the I-TevI::ΔI-SM fusion proteins, the CBD from Bacillus circulans was inserted into ΔI-SM where the endonuclease domain had been removed, giving rise to I-TevI::ΔI-SM::CBD. The properties of this fusion indicated that insertion of the CBD had negligible effects on intein-mediated protein splicing or cleavage (data not shown).
The chemistry of splicing usually requires that an intein be positioned in front of a cysteine or serine, to allow nucleophilic attack on the N-extein–intein junction by the -SH or -OH side chain (3). Because the Mtu RecA intein occurs naturally before a cysteine, seven cysteines were selected as insertion sites throughout I-TevI, for comparison in the purification scheme. Cys164 of I-TevI was chosen as the prototypic site of insertion. Fortuitously, this construct turned out to be the most successful for expression and purification purposes. The ΔI-SM::CBD cassette was positioned in front of Cys164 of I-TevI. This entire cartridge was cloned into pET28a (Novagen) behind a T7 promoter to yield pTevC164:: ΔI-SM::CBD (Fig. 1, top). This construct was transformed into BL21(DE3)pLysS, which produces T7 RNA polymerase under control of the plac promoter and T7 lysozyme, an inhibitor of T7 RNA polymerase conferring more stringent control over leaky expression from the pET vectors (30).
The fact that pTevC164::ΔI-SM::CBD transformants were viable only in the presence of pLysS suggested that the fusion construct was toxic. This toxicity might be ascribed to three factors: (i) the toxicity of the fusion protein per se; (ii) the restoration of I-TevI activity through in vivo intein-mediated protein splicing; or (iii) the unleashing of the hyper-toxic catalytic domain of I-TevI through isolated cleavage at the N-terminus of the intein. To distinguish among these possibilities, a non-spliceable fusion construct was made, pTevC164:: ΔIAA-SM::CBD, in which the first and last residues of the intein were mutated to alanines (10). This construct could be readily transformed into BL21(DE3) without hampering the growth of the host strain. The innocuousness of pTevC164:: ΔIAA-SM::CBD compared with the toxicity of pTevC164::ΔI-SM::CBD suggests that the precursor per se is non-toxic, that I-TevI can be inactivated by intein insertion and that the inactivation is reversible through in vivo intein-mediated protein splicing and/or cleavage.
Probing intein insertion sites in front of different cysteines of I-TevI
To test different intein insertion sites for inactivation in the target protein, ΔI-SM::CBD was positioned in front of six additional cysteines of I-TevI. All of the insertion sites, which precede cysteines 39, 58, 100, 153, 164, 207 and 214, are displayed in the context of the I-TevI structure in Figure 2A. The N-terminal catalytic domain of I-TevI is compactly folded (21,24), whereas the C-terminal domain in the presence of its DNA substrate has an extended structure comprising three subdomains: a zinc finger, an α-helix and a helix–turn–helix (23). The α-helix and helix–turn–helix modules constitute the DNA-binding domain, whereas the zinc finger module has recently been shown to be a component of the linker (22). Among these cysteines, Cys39 is located in a surface loop of the catalytic domain of I-TevI, Cys58 is situated in the middle of a β-strand, Cys100 is in an unstructured or loosely structured section of the linker between the domains, Cys153 and Cys164 are involved in the formation of the zinc finger, while Cys207 is the last residue of a β-strand and Cys214 precedes an α-helix, both in the helix–turn–helix module.
Figure 2.

(A) Insertion contexts of ΔI-SM::CBD in I-TevI relative to I-TevI structure. Cysteines before which ΔI-SM::CBD was inserted are presented in space-filling configuration in the context of a composite I-TevI ribbon diagram derived from the independently solved crystal structures of the N- and C-terminal domains of the enzyme (residues 1–97 and 130–245, respectively) (23,24). Residues between 93 and 147 are likely unstructured or loosely structured. (B) Properties of fusion derivatives. Viability of pTev::ΔI-SM::CBD fusions are denoted as follows: =, lethality in non-expression strain JM101; –, + and ++, no growth, slow growth and normal growth, respectively, in BL21(DE3)pLysS. The approximate in vivo distribution of precursors and reaction products for all seven MBP-TevR27A:: ΔI-SM::CBD constructs is shown. (See also Fig. 3B.) For fusions at C207 and C214, the percentages for precursor and C-extein were not independently quantifiable and were combined (brackets). LE+N-ex, combined ligated exteins and N-terminal extein (bold).
Wild-type fusion constructs at Cys100 and Cys153 were never obtained. The derivatives always contained mutations in the I-TevI sequence, in contrast to the generation of wild-type fusion constructs at the other five cysteines (Fig. 2B). While all five of these constructs were lethal to BL21(DE3), fusion constructs at Cys164, Cys207 and Cys214 could be rescued by pLysS, but those at Cys39 and Cys58 could not. The fusion construct at Cys207 imparts a slow growth phenotype compared to fusions at Cys164 and Cys214. As for the Cys164 fusion, non-spliceable fusions with the terminal intein residues mutated to alanines at the other six integration sites (pTev::ΔIAA-SM::CBD) were non-toxic, indicating again that the toxicity was caused by in vivo intein-mediated splicing and/or cleavage.
To further study the behavior of the intein at different positions, the coding sequences for all seven Tev::ΔI-SM:: CBD proteins were fused to the 3′ end of the maltose-binding protein (MBP) gene, using a catalytic mutant of I-TevI (R27A) to eliminate toxicity. MBP facilitated purification of the fusion precursors by affinity chromatography with an amylose column, for in vitro analysis. Two affinity modules were necessary because CBD fusions cannot be eluted from chitin without denaturation, whereas MBP fusions elute readily in native form from an amylose column. All seven MBP-TevR27A::ΔI-SM::CBD fusion proteins were purified on amylose resin. The maltose eluate had all the MBP-containing species: the precursors, the splicing products and the N-terminal portion of the N- and C-terminal cleavage products (Figs 2B and 3A and B). The molar percentages of the molecular species immediately after purification, which approximate the in vivo distribution, are listed (Fig. 2B).
Figure 3.

Characterization of ΔI-SM::CBD in front of different cysteines of I-TevI. (A) Ligation and cleavage products of pMBP-TevR27A::ΔI-SM::CBD. N-Ex, C-Ex and N-Ex/C-Ex are the N-extein, C-extein and ligated exteins of the I-TevI derivatives, respectively. N-I and C-I are the N- and C-terminal halves of the ΔI-SM intein, respectively. (B) SDS–PAGE of MBP-I-TevR27A::ΔI-SM::CBD at seven different cysteines of I-TevI after affinity chromatography on amylose resin, before in vitro splicing. M1, protein molecular weight standards, high range (Gibco BRL); M2, broad range protein markers (NEB). Lane 1, Cys39; lane 2, Cys58; lane 3, Cys100; lane 4, Cys153; lane 5, Cys164; lane 6, Cys207; lane 7, Cys214. Symbols for precursor and products are as designated in (A).
The relative cleavage and splicing efficiency in vivo varies among the derivatives, but there is no strict correlation between the amounts of ligated exteins plus N-extein products and toxicity (Figs 2B and 3B). The combination of these products ranges between 51 and 90% for all of the fusion derivatives except Cys164, where they amount to only 15%. Whereas Cys100 and Cys153 are lethal even in a non-expression strain, Cys39 and Cys58 can grow only in a non-expression host, while Cys164, Cys207 and Cys214 can grow even in an expression host (Figs 2B and 3B). Consistent with the extreme toxicity of the Cys100 and Cys153 fusions, splicing was so efficient that the precursors were absent (Fig. 3B, lanes 3 and 4). The Cys164 fusion has two striking properties that likely contribute to its being the optimal fusion. First, it has the lowest amount of ligated extein and N-extein products (11 and 4%, respectively) and, second, the Cys164 fusion yields the highest amount of C-terminal cleavage product (Figs 2B and 3B, lane 5). Thus, the relatively low toxicity may be due to the least efficient in vivo splicing and to the efficient C-terminal cleavage, which generates an intact DNA-binding domain to compete with full-length I-TevI for binding to potential cleavage sites on the E.coli chromosome.
All of these purified fusion proteins were subjected to in vitro reaction at pH 7.4 and 37°C. Aliquots were taken at different times, resolved by SDS–PAGE and the molar percentages of the precursor and the reaction products determined for all seven derivatives as a function of time (Fig. 4A and data not shown). Under most circumstances, intein-mediated protein splicing among these derivatives reached equilibrium either in vivo or during purification, as for example with Cys214 (Fig. 4A, right). The exceptions were Cys164 and Cys207. In vitro splicing reactions carried out at 37°C showed significant increases in ligated exteins for both of these derivatives over the course of 4 h (Fig. 4A, left and middle). The data indicate that the in vitro splicing of the Cys164 derivative is more rapid than that of the Cys207 derivative, although both carry out ∼20% splicing in vitro. Subsequent experiments with Cys164 showed that in vitro reactions at 4°C, although slower than at 37°C, produced higher yields of ligated exteins (Fig. 4B). This could be related to the instability of the complex MBP-Tev::ΔI-SM::CBD protein at 37°C. Regardless, although both constructs seemed suitable for I-TevI purification, we selected the prototypic pTevC164::ΔI-SM::CBD fusion because of its much faster growth rate.
Figure 4.

In vitro splicing. (A) Intein-mediated protein splicing products of pMBP-TEVR27A::ΔI-SM::CBD fusion constructs at Cys164, Cys207 and Cys214. Ligated exteins are shown as a molar percentage of total material as a function of time at 37°C and pH 7.4. (B) pH-dependent protein processing. SDS–PAGE gel showing different molecular species derived from in vitro splicing of the fusion derivative at Cys164 after incubation at 4°C for 24 h at 10 different pHs. Bands were not sufficiently resolved to allow quantitation, but the dramatic enhancement of splicing is evident from the reduction in precursor and increase in ligated exteins with decreasing pH, from 8.3 to 7.2. Filled square, precursor; filled circle, spliced product (ligated exteins); open square, C-terminal cleavage product; filled triangle, N-terminal cleavage product.
Purification of wild-type I-TevI by pH-controllable intein-mediated protein splicing
Previous work in our laboratory showed that C-terminal cleavage of the Mtu RecA intein was favored at pH <6.5 (10). To determine whether the splicing of this intein could also be affected by pH, the plasmid encoding the fusion derivative MBP-TevR27AC164::ΔI-SM::CBD was transformed into JM109 and protein expression was induced with IPTG. The cell pellet was sonicated at 4°C in pH 8.5 column buffer to minimize intein-mediated protein splicing and cleavage. The fusion precursor was purified on an amylose column and eluted with maltose. The fusion protein was then subjected to in vitro splicing and cleavage at 10 different pHs, ranging from 8.3 to 7.2, at 4°C (Fig. 4B). Both splicing and C-terminal cleavage were slow at pH 8.3 and both increased as the pH was lowered. However, at pH <7.4 C-terminal cleavage dominated over splicing, with the optimal pH for maximum yield and greatest purity of ligated exteins being 7.5.
A purification scheme was devised making use of the controllability of intein-mediated protein splicing by pH shift from pH 8.5 to 7.5 (Fig. 1), with the pTevC164::ΔI-SM::CBD plasmid in BL21(DE3)pLysS. Pure spliced I-TevI and cleavage products were eluted from the chitin resin after 26 h at 4°C (Fig. 5A, lanes 5–16). The yield of full-length I-TevI was ∼0.2 mg/l culture. The presence of 1 mM DTT in the chitin column buffer was essential for maximal yield. Purification of the protein was completed using a gel filtration column (Fig. 5B).
Figure 5.

Intein-mediated expression and purification of I-TevI. (A) Affinity column purification by pH shift. The SDS–PAGE shows the efficacy of the purification scheme illustrated in Figure 1. Lane M, protein size markers; lane 1, uninduced sample; lane 2, induced sample, filled square, the fusion precursor Tev::ΔI::CBD of 52.7 kDa; lane 3, clarified cell lysate; lane 4, chitin column flow-through; lanes 5–16, eluted fractions after on-column splicing at pH 7.5 for 26 h at 4°C. The splice product at 28.3 kDa and cleavage products at 18.9 and 9.4 kDa are indicated. The inset shows a DNA cleavage assay with the eluted I-TevI fractions. M, λ DNA HindIII markers; C, control with no enzyme added; S, ScaI-linearized plasmid substrate containing the td homing site; P, cleavage products with purified I-TevI fraction. (B) Gel filtration. The SDS–PAGE gel shows fractions eluted from a Superose 12 gel filtration column. Lane 1, load; lanes 2–18, eluted fractions. Labels as in (A).
Several lines of evidence indicate that the I-TevI purified with this novel intein-mediated method was wild-type. First, the molecular weight of the ligated I-TevI was determined by mass spectrometry to be 28 226.5 Da, within 5 Da of its predicted molecular mass of 28 231 Da. Second, the pTevC164::ΔI-SM::CBD plasmid extracted from induced BL21(DE3)pLysS cells was sequenced and verified to be wild-type. Third, cleavage assays were performed on the purified I-TevI (Fig. 5A, inset), both before and after gel filtration chromatography, and the enzyme was shown to be extremely active, with a specific activity of 14.2 ± 0.7 × 103 U/µg. To confirm that this is a reasonable value for the wild-type activity, we compared the activity with that of a reduced activity derivative, H40Y, previously studied in our laboratory (27). We determined that the protein isolated by intein-mediated purification was ∼620-fold more active than the H40Y mutant. These results are consistent with data derived from assays comparing the activity of the same proteins expressed in vitro, where wild-type I-TevI is ∼100-fold more active than the H40Y derivative (D. Smith, unpublished results).
DISCUSSION
This work demonstrates the feasibility of purifying a cytotoxic protein through intein inactivation followed by pH- controllable splicing. The success of the method requires the identification of a cysteine in the target protein where the insertion of an intein can substantially alleviate the toxicity in vivo and generate spliced product in vitro. In the case of I-TevI, intein insertion at Cys164 gave the lowest toxicity and the highest in vitro:in vivo splicing ratios, which made it the most appropriate site for intein inactivation.
Although firm context rules could not be established, the extein environment had a significant impact on the relative efficiency of in vivo intein-mediated protein splicing and extein cleavage without ligation, as demonstrated in Figures 2B, 3B and 4B. The last residue of the N-extein plays a very important role because its carbonyl carbon is the target of three of the four nucleophilic displacements in the splicing reaction (5). Most inteins have their own preferred set of –1 residues for efficient N-terminal cleavage. For the Mxe GyrA and Sce VMA inteins, aspartate at –1 leads to nearly complete N-terminal cleavage in vivo (31,32). Similarly, MBP-TevR27AC214::ΔI-SM::CBD, with the intein located downstream of an aspartate residue, had the highest percentage of in vivo N-terminal cleavage, consistent with other recent work with the Mtu RecA intein (33). However, factors other than the proximal extein sequence can have a dramatic effect on the splicing efficiency. Intein fusions at Cys100 and at Cys164 had exactly the same –1 and +2 extein residues (threonine and serine), yet they had the highest (73%) and lowest (11%) in vivo splicing efficiencies, respectively, among all seven fusion derivatives. When an intein is inserted into an ectopic site, the host protein and the intein must fold individually. Whereas insertion into an unstructured region of the extein may be energetically less costly for the intein to adopt the optimal configuration for splicing, integration into a structured region may favor splicing by bringing the intein ends into juxtaposition. For the above reasons, coupled with the idiosyncrasies of the toxicity of different proteins and their needs for folding, it is not possible to predict optimal insertion sites. Several sites should be sampled and the most favorable selected for purification of a toxic protein. The best insertion site would be the one that not only inactivates the target protein and thereby facilitates cell growth, but also yields the highest in vitro:in vivo splicing levels. In our case, two of the seven insertion sites were acceptable for subsequent purification of the toxic protein, with the Cys164 derivative being our choice over the Cys207 fusion because of its more rapid growth rate.
As mentioned in the Results, pTevC164::ΔI-SM::CBD is still somewhat toxic and this can compromise overexpression. We anticipate that ΔI-SM derivatives with reduced splicing activity would be useful in situations where this becomes a problem. Furthermore, inteins with reduced activity might yield a higher fraction of fusions with the host protein that are productive in relieving toxicity and resulting in purified proteins of high yield.
Curiously, neither Cys164 nor the next most favorable construct, Cys207, are fusions in the catalytic domain, where one would predict an insertion to be maximally effective in rendering I-TevI non-toxic. Presumably the efficient splicing from the well-folded catalytic domain results in persistence of endonuclease-mediated toxicity. In contrast, the intein in the less compact linker or DNA-binding domain splices less efficiently and perhaps sterically hinders an intact N-terminal catalytic domain thereby inhibiting its lethal cleavage activity.
There are several alternative methods for the expression and purification of cytotoxic proteins. Tightly regulated promoters of various types have been unsuccessful in our hands in generating wild-type I-TevI. In vitro translation systems such as wheatgerm extract have worked well for I-TevI (27,34). Although they are usually limited by yield and impurity, recent reports suggest they can be used for large-scale production of some proteins (35,36). Intein-mediated protein ligation has also been used to make semi-synthetic cytotoxic proteins. First, an innocuous truncated version of the toxic protein is expressed as a fusion, target protein–intein–CBD. Then the fusion protein is affinity purified and cleaved by a sulfhydryl-containing reagent to form a thioester at the C-terminus of the target protein. Finally, the thioester-tagged truncated protein is ligated to a synthetic peptide with an N-terminal cysteine to restore the full-length protein (12). This approach has promise but also limitations. The most serious of these is the requirement for an extra renaturation step for the ligated toxic protein to restore activity (12). Protein trans-splicing systems with natural (37) or artificial (38) split inteins also have the potential to be applied to purification of cytotoxic proteins. However, the engineered split Mtu RecA intein requires an extra denaturation/renaturation step for the split parts to assemble into a functional intein (38). The naturally occurring Synechocystis DnaE intein obviates the requirement for denaturation/renaturation, but is also limited by its requirement for native proximal extein residues (37). Our approach has the advantage of expressing the toxic protein in its entirety, with the intein as a compactly folded module, which does not appear to interfere with the folding of the protein of interest here, I-TevI. In both the intein-mediated protein ligation system and this insertional inactivation method, a sizing column or other additional purification step is also required if the full-length splice product must be free of ligation substrates or cleavage products, respectively.
Our approach of intein-mediated insertional inactivation and pH-controllable splicing is, in theory, readily generalizable to the purification of other cytotoxic proteins. This was indeed the case with our intein-based pH-controllable cleavage system for non-toxic proteins, which has been successful for single step purification of many different products (11). Furthermore, the effort of up-front strain construction in surveying for the best insertional inactivation site is handily rewarded by the ease of purification of a lethal product, the potential for scale-up and the high quality of the protein preparation.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Maryellen Carl for expert secretarial assistance, Carol Lyn Engel for preparation of the I-TevI H40Y mutant, Patrick Van Roey for atomic coordinates of I-TevI and for providing Figure 2A, John Dansereau for expert assistance in preparing the figures, and Joe Kowalski and Kenji Ichiyanagi for thoughtful comments on the manuscript. The Molecular Genetics Core facility at the Wadsworth Center provided DNA oligonucleotides and DNA sequencing services and the Mass Spectrometry Core performed molecular mass determinations. This work was supported by National Institutes of Health grants GM39422 and GM44844 to M.B.
REFERENCES
- 1.Perler F.B., Davis,E.O., Dean,G.E., Gimble,F.S., Jack,W.E., Neff,N., Noren,C.J., Thorner,J. and Belfort,M. (1994) Protein splicing elements: inteins and exteins—a definition of terms and recommended nomenclature. Nucleic Acids Res., 22, 1125–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Paulus H. (2000) Protein splicing and related forms of protein autoprocessing. Annu. Rev. Biochem., 69, 447–496. [DOI] [PubMed] [Google Scholar]
- 3.Perler F.B., Xu,M.-Q. and Paulus,H. (1997) Protein splicing and autoproteolysis mechanisms. Curr. Opin. Chem. Biol., 1, 292–299. [DOI] [PubMed] [Google Scholar]
- 4.Shao Y., Xu,M.-Q. and Paulus,H. (1997) Protein splicing: estimation of the rate of O-N and S-N acyl rearrangements, the last step in the splicing process. J. Peptide Res., 50, 193–198. [DOI] [PubMed] [Google Scholar]
- 5.Noren C.J., Wang,J. and Perler,F.B. (2000) Dissecting the chemistry of protein splicing and its applications. Angew. Chem. Int. Ed., 39, 450–466. [PubMed] [Google Scholar]
- 6.Xu M.W., Paulus,H. and Chong,S. (2000) Fusions to self-splicing inteins for protein purification. Methods Enzymol., 326, 376–418. [DOI] [PubMed] [Google Scholar]
- 7.Xu M.Q. and Evans,T.C.J. (2001) Intein-mediated ligation and cyclization of expressed proteins. Methods, 24, 257–277. [DOI] [PubMed] [Google Scholar]
- 8.Chong S., Mersha,F.B., Comb,D.G., Scott,M.E., Landry,D., Vence,L.M., Perler,F.B., Benner,J., Kucera,R.B., Hirvonen,C.A. et al. (1997) Single-column purification of free recombinant proteins using a self-cleavage affinity tag derived from a protein splicing element. Gene, 192, 271–281. [DOI] [PubMed] [Google Scholar]
- 9.Chong S., Montello,G.E., Zhang,A., Cantor,E.J., Liao,W., Xu,M.-Q. and Benner,J. (1998) Utilizing the C-terminal cleavage activity of a protein splicing element to purify recombinant proteins in a single chromatographic step. Nucleic Acids Res., 26, 5109–5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wood D., Wu,W., Belfort,G., Derbyshire,V. and Belfort,M. (1999) A genetic system yields self-cleaving inteins for bioseparations. Nat. Biotechnol., 17, 889–892. [DOI] [PubMed] [Google Scholar]
- 11.Wood D.W., Derbyshire,V., Wu,W., Cartrain,M., Belfort,M. and Belfort,G. (2000) Optimized single-step affinity purification with a self-cleaving intein applied to human acidic fibroblast growth factor. Biotechnol. Prog., 16, 1055–1063. [DOI] [PubMed] [Google Scholar]
- 12.Evans T.C.J., Benner,J. and Xu,M.-Q. (1998) Semisynthesis of cytotoxic proteins using a modified protein splicing element. Protein Sci., 7, 2256–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evans T.C.J., Benner,J. and Xu,M.Q. (1999) The in vitro ligation of bacterially expressed proteins using an intein from Methanobacterium thermoautotrophicum. J. Biol. Chem., 274, 3923–3926. [DOI] [PubMed] [Google Scholar]
- 14.Mathys S., Evans,T.C.J., Chute,I.C., Wu,H., Chong,S., Benner,J., Liu,X.-Q. and Xu,M.-Q. (1999) Characterization of a self-splicing mini-intein and its conversion into autocatalytic N- and C-terminal cleavage elements: facile production of protein building blocks for protein ligation. Gene, 231, 1–13. [DOI] [PubMed] [Google Scholar]
- 15.Otomo T., Teruya,K., Uegaki,K., Yamazaki,T. and Kyogoku,Y. (1999) Improved segmental isotope labeling of proteins and application to a larger protein. J. Biomol. NMR, 14, 105–114. [DOI] [PubMed] [Google Scholar]
- 16.Cowburn D. and Muir,T.W. (2001) Segmental isotopic labeling using expressed protein ligation. Methods Enzymol., 339, 41–54. [DOI] [PubMed] [Google Scholar]
- 17.Evans T.C.J., Benner,J. and Xu,M.Q. (1999) The cyclization and polymerization of bacterially expressed proteins using modified self-splicing inteins. J. Biol. Chem., 274, 18359–18363. [DOI] [PubMed] [Google Scholar]
- 18.Quirk S.M., Bell-Pedersen,D. and Belfort,M. (1989) Intron mobility in the T-even phages: high frequency inheritance of group I introns promoted by intron open reading frames. Cell, 56, 455–465. [DOI] [PubMed] [Google Scholar]
- 19.Bryk M., Quirk,S.M., Mueller,J.E., Loizos,N., Lawrence,C. and Belfort,M. (1993) The td intron endonuclease makes extensive sequence tolerant contacts across the minor groove of its DNA target. EMBO J., 12, 2141–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Derbyshire V., Kowalski,J.C., Dansereau,J.T., Hauer,C.R. and Belfort,M. (1997) Two-domain structure of the td intron-encoded endonuclease I-TevI correlates with the two-domain configuration of the homing site. J. Mol. Biol., 265, 494–506. [DOI] [PubMed] [Google Scholar]
- 21.Kowalski J.C., Belfort,M., Stapleton,M.A., Holpert,M., Dansereau,J.T., Pietrokovski,S., Baxter,S.M. and Derbyshire,V. (1999) Configuration of the catalytic domain of intron endonuclease I-TevI: coincidence of computational and molecular findings. Nucleic Acids Res., 27, 2115–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dean A.B., Stanger,M.J., Dansereau,J.T., Van Roey,P., Derbyshire,V. and Belfort,M. (2002) Zinc finger as distance determinant in the flexible linker of intron endonuclease I-TevI. Proc. Natl Acad. Sci. USA, 99, 8554–8561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Roey P., Waddling,C.A., Fox,K.M., Belfort,M. and Derbyshire,V. (2001) Intertwined structure of the DNA-binding domain of intron endonuclease I-TevI with its substrate. EMBO J., 20, 3631–3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Roey P., Meehan,L., Kowalski,J., Belfort,M. and Derbyshire,V. (2002) Catalytic domain structure and hypothesis for function of GIY-YIG intron endonuclease I-TevI. Nature Struct. Biol., 9, 806–811. [DOI] [PubMed] [Google Scholar]
- 25.Derbyshire V., Wood,D.W., Wu,W., Dansereau,J.T., Dalgaard,J.Z. and Belfort,M. (1997) Genetic definition of a protein-splicing domain: functional mini-inteins support structure predictions and a model for intein evolution. Proc. Natl Acad. Sci. USA, 94, 11466–11471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vallejo A.N., Pogulis,R.J. and Pease,L.R. (1995) PCR Primer: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Plainview, NY, pp. 603–612.
- 27.Bell-Pedersen D., Quirk,S.M., Bryk,M. and Belfort,M. (1991) I-TevI, the endonuclease encoded by the mobile td intron, recognizes binding and cleavage domains on its DNA target. Proc. Natl Acad. Sci. USA, 88, 7719–7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perler F.B. (2000) InBase, the intein database. Nucleic Acids Res., 28, 344–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shingledecker K., Jiang,S.-Q. and Paulus,H. (1998) Molecular dissection of the Mycobacterium tuberculosis RecA intein: design of a minimal intein and of a trans-splicing system involving two intein fragments. Gene, 207, 187–195. [DOI] [PubMed] [Google Scholar]
- 30.Studier F.W., Rosenberg,A.H., Dunn,J.J. and Dubendorff,J.W. (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol., 185, 60–89. [DOI] [PubMed] [Google Scholar]
- 31.Southworth M.W., Amaya,K., Evans,T.C., Xu,M.Q. and Perler,F.B. (1999) Purification of proteins fused to either the amino or carboxy terminus of Mycobacterium xenopi gyrase A intein. Biotechniques, 27, 110–120. [DOI] [PubMed] [Google Scholar]
- 32.Chong S., Williams,K.S., Wotkowicz,C. and Xu,M.-Q. (1998) Modulation of protein splicing of the Saccharomyces cerevisiae vacuolar membrane ATPase intein. J. Biol. Chem., 273, 10567–10577. [DOI] [PubMed] [Google Scholar]
- 33.Lew B.M. and Paulus,H. (2002) An in vivo screening system against protein splicing useful for the isolation of non-splicing mutants or inhibitors of the RecA intein of Mycobacterium tuberbulosis. Gene, 282, 169–177. [DOI] [PubMed] [Google Scholar]
- 34.Bryk M., Belisle,M., Mueller,J.E. and Belfort,M. (1995) Selection of a remote cleavage site by I-TevI, the td intron-encoded endonuclease. J. Mol. Biol., 247, 197–210. [DOI] [PubMed] [Google Scholar]
- 35.Kigawa T., Yabuki,T., Yoshida,Y., Tsutsui,M., Ito,Y., Shibata,T. and Yokoyama,S. (1999) Cell-free production and stable-isotope labeling of milligram quantities of proteins. FEBS Lett., 442, 15–19. [DOI] [PubMed] [Google Scholar]
- 36.Madin K., Sawasaki,T., Ogasawara,R. and Endo,Y. (2000) A highly efficient and robust cell-free protein synthesis system prepared from wheat embryos: plants apparently contain a suicide system directed at ribosomes. Proc. Natl Acad. Sci. USA, 97, 559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans T.C. Jr, Martin,D., Lolly,R., Panne,D., Sun,L., Ghosh,I., Chen,L., Benner,J., Liu,X.Q. and Xu,M.Q. (2000) Protein trans-splicing and cyclization by a naturally split intein from the dnaE gene of Synechocystis species PCC6803. J. Biol. Chem., 275, 9091–9094. [DOI] [PubMed] [Google Scholar]
- 38.Mills K.V., Lew,B.W., Jiang,S. and Paulus,H. (1998) Protein splicing in trans by purified N- and C-terminal fragments of the Mycobacterium tuberculosis RecA intein. Proc. Natl Acad. Sci. USA, 95, 3543–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]