

Introduction
The term 'agitation' describes a syndrome of excessive motor activity, usually nonpurposeful and associated with internal tension. For intensivists, agitation is not so much a diagnosis, but a consequence of more fundamental etiologies that, when expressed, result in disquietude. Agitation is important in the intensive care unit (ICU) because it can alter the diagnosis and course of medical treatment. It can cloud the etiology of underlying disease processes like a smoke screen, making effective diagnosis difficult or impossible. It may result in the inability of the patient to cooperate with monitoring and therapeutics that requires them to lie relatively still and quiet. Treatment of agitation without considering underlying causation gives the false impression of wellness when in reality end-organ damage is occurring either as a result of agitation itself, or as a result of exacerbation of underlying pathology.
Prior to the technological revolution in critical care medicine, agitation was a relatively minor issue. Little could be done for critically ill patients but make them as comfortable as possible and observe them for treatable decompensations. Modern ICUs now have the potential to return critically ill patients to productivity using technological advances in monitoring and closely titrated care, effectively pinning the patient firmly to the bed with tubes and appliances. As a result of our high-tech hemodynamic monitoring and support devices, we have conferred upon the already hemodynamically unstable patient new kinds of stress we never had to deal with before, and simplistic, symptomatic 'shotgun' sedation no longer applies.
Anatomy of the cognitive centers
The temporal lobes and the Hesh gyrus receive auditory information, modulate memory and language skills and relay information to the cortex where cognitive judgments are made and motor responses are integrated [1]. The thalamus and basal ganglia act as relay stations between lower centers and the cortex [2]. The brainstem enables endurance and survival capabilities, modulating heart rate, respiratory function and autonomic actions [3]. The pineal gland is thought to modulate sleep-wake cycles [4]. The hippocampal area including the mammillary bodies modulates spatial memory formation, declarative memory, working memory, memory indexing/storage, relating expectancy to reality, and internal inhibition. Memory is recorded in several parts of the brain at same time as 'memory molecules' for storage. These molecules are modulated by limbic system, especially the mammillary bodies. Bilateral hippocampal resection results in short-term anterograde amnesia [5]. The hippocampus has receptors for neurosteroids, both mineralocorticoid and glucocorticoid. The mineralocorticoid receptors (high affinity) are agonized by alderosterone, and antagonized by spironolactone. The glucocorticoid receptors (low affinity) are agonized by dexamethasone. There are no known antagonists to glucocorticoid receptors. The locus coeruleus is a small structure on the upper brainstem under the fourth ventricle and is involved in the regulation of wakefulness, attention and orientation [6] (Fig. 1).
Figure 1.
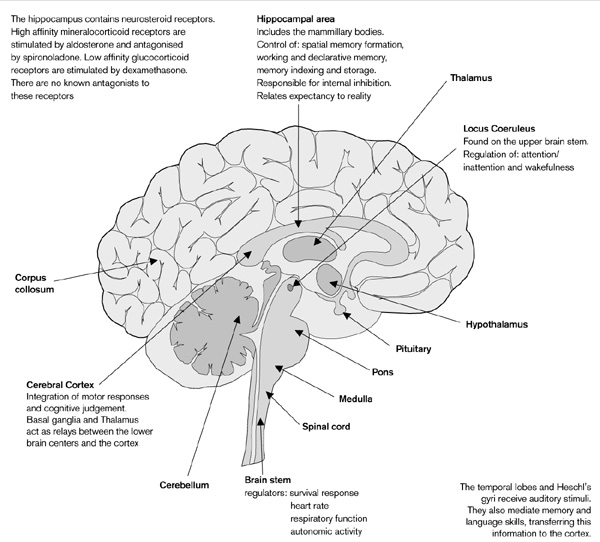
The anxiety producing centers of the brain.
Cerebral neuroreception
Receptors for neurotransmission are highly specialized and recognize only specific transmitter chemicals [7]. Neurotransmission is accomplished by at least three kinds of chemical transmitters (Table 1).
Table 1.
Neurotransmission and chemical transmitters
| Cholinergic | Biogenic | Amino acid |
| Acetylcholine | Dopamine | GABA |
| Norepinephrine | Glycine | |
| Serotonin | Glutamate | |
| Histamine | Aspartate |
GABA, γ-amino butyric acid.
Acetylcholine achieves high concentrations in the basal ganglia and account for the anticholinergic side effects of medications such as tricyclic antidepressants. The neurotransmitter dopamine is particularly active in the midbrain and limbic system, and the frontal lobes, modulating emotional responses. In its simplified form, schizophrenia is thought to be a result of increased dopamine neurotransmission activity. Norepinephrine exerts a diffuse modulating influence throughout the brain. Serotonin exerts a thalamic influence and has been implicated in clinical depression, sleep disorders, anxiety and pain. Other naturally occurring substances can exert neurotransmission activity. Branch chained and aromatic amino acids may act as false neurotransmitters during the encephalopathy of liver failure [8]. Glutamate has been implicated in the 'Chinese food syndrome', where food with high amounts of monosodium glutamate interfere with normal neurotransmission causing confusion [9]. Pain neurotransmission utilizes the opiate receptors found diffusely throughout the limbic system and basal ganglia, frontal and temporal cortex, thalamus, hypothalamus, midbrain and spinal cord. Kappa and Mu opiate receptors do not meet the typical criteria for neurotransmitters. They respond to endorphins and enkephalins which are peptides rather than hormones [10].
Neuroreceptors are responsible for highly selective signaling at synapses and also regulation. N-Methyl-D-aspartate (NMDA) receptors occur within the cerebral cortex, cerebellum and hippocampus [11]. Depolarization of the NMDA complex by glycine, the phencyclidine-like drugs and aspartate results in the elaboration of excitatory neurotransmitters that increase brain metabolic activity. The NMDA receptor is unique in that it is permeable to both sodium and calcium ions, so its electrical current is both agonist- and voltage-dependent, and relatively long lived. The γ -amino butyric acid (GABA) receptors are inhibitory complexes modulated by benzodiazepines, steroids and barbiturates [12]. The GABA-benzodiazepine complex hypothesis suggests that benzodiazepines attach to GABA-benzodiazepine complexes in the brain, enhancing chloride conduction resulting in the release of GABA. This opens a chloride channel leading to inhibition of neuronal excitation in the limbic system, resulting in anxiolysis, sedation or hypnosis, depending on dose. All neurotransmitter receptors can be 'desensitized' by a constant flux of agonists and become less responsive. This mechanism tends to prevent overstimulation at synapses by excess released neurotransmitter. Receptors may also be 'downregulated', the absolute number of receptors reduced as a response to the same kind of stimulus that results in desensitization [13].
Neurotransmitters and anxiety
Stress usually produces an elevated sense of fear and anxiety which causes increased norepinephrine turnover in the limbic regions (hypocampus, amygdala, locus coeruleus) and cerebral cortex [14]. Stress applied to laboratory animals results in a decreased density of α2-adrenergic autoreceptors in the hippocampus and amygdala, reflecting downregulation in response to elevated circulating endogenous circulating catecholamines, among other desensitizing actions [15]. This causes an increase in responsiveness of locus ceruleus neurons to excitatory stimulation that is associated with a reduction in α2-adrenergic autoreceptor sensitivity [16]. This phenomenon may be measured by assaying levels of platelet α2-adrenergic autoreceptors. Yohimbine activates noradrenergic neurons by blocking α2-adrenergic receptors and is thus anxiogenic. Clonidine, an α2-agonist, seems to diminish anxiety symptoms by decreasing norepinephrine transmission [17]. Yohimbine blocks the effects of clonidine, and panic attacks can be precipitated by its parenteral administration.
Behavioral sensitization to stress may also involve 'memory imprinting' alterations in noradrenergic function. This is thought to be the mechanism of the Post Traumatic Stress Disorder originally recognized in Vietnam veterans, but now recognized to be a sequelae to other prolonged inordinately stressful events [18]. This syndrome is not uncommon following extremely stressful ICU stays, especially if the patient experienced untreated pain, anxiety or delirium [19]. The limbic and cortical regions innervated by the locus coeruleus are those thought to be involved in the elaboration of adaptive responses to stress, eliciting increased responsiveness to excitatory stimuli when previously experienced stimuli occur again. Limited exposure to shock that does not increase noradrenergic activity in control animals increases norepinephrine release in animals previously exposed to the same kind of stress [20].
Noradrenergic hyperfunction and agitation
Panic attacks, insomnia, accentuated startle, autonomic hyperarousal and hypervigilence are characteristics of noradrenergic hyperfunction [21]. Conditioned fear and recollection of immobilization stress may be experienced by patients who have experienced traumatic emergency endotracheal intubation and mechanical ventilation in the past. In ICU patients the sensitizing factor may result from hemodynamic and metabolic decompensations as a result of multisystem insufficiency [22]. In a panic attack, the main symptom is dyspnea; the patient feels like he or she wants to breathe but cannot. There are two kinds of panic [23]: type one includes typical symptoms of diaphoresis and tachycardia, and is effectively treated by anxiolytic drugs including the benzodiazepines, especially low dose continuous infusions of titratable ones like midazolam and lorazepam; type two panic attacks are characterized by subjective dyspnea and hyperventilation. For this variant of the syndrome, imiprimine tends to downregulate the brain's 'suffocation alarm system' that promotes the subjective sensation of dyspnea. It is thought that this center is modulated by serotonin uptake neuroreceptors [24]. Imiprimine, however, is not useful in the ICU because of the long therapeutic lag period and because of its many difficult-to-control side effects.
Symptoms of panic attacks are easily confused with those of chronic heart failure, which a large population of ICU patients are predisposed to. In addition, metabolic imbalances associated with left heart failure and respiratory failure may precipitate anxiety de novo. Patients controlled on mechanical ventilation and chronic lung failure patients may manifest acute increases in pCO2 resulting in further catecholamine release, increasing agitation [25]. Agitated patients tend to increase peripheral musculoskeletal metabolism, increasing lactate and carbon dioxide production [26]. Both lactate and increased CO2 are evolutionary signals that danger is coming, prompting a responsive response to stress and potential danger. Hypercapnea stimulates the sympathetic centers resulting in tachycardia and mild hypertension, and possibly promoting panic. Increased levels of lactate and CO2 enter the brain quickly and can precipitate panic attacks [27]. During hyperventilation, pCO2 declines, causing cerebral vessels to constrict reflexly, further limiting blood flow and O2 transport to the brain which can result in mental confusion.
The treatment for heart failure radically differs from that of panic attacks and the exact etiology of each disorder must be accurately identified before treatment is begun. If the panic episode is secondary to pump failure resulting in tissue hypoxia and hypercarbia, establishing an airway and ventilation will rapidly ameliorate hemodynamic instability, resolving the agitation episode. However, if the patient's agitation results from a panic attack, aggressive airway, ventilation and hemodynamic support will only make them more agitated. In addition, effective treatment for the two different kinds of panic are different in effectiveness and complications. For type 1 panic, the 'cost' of benzodiazepines is sedation with possible ventilation impairment, the potential for tolerance, rebound and withdrawal on removal, all of which interfere with titration of ICU ventilation and hemodynamic support. For type 2 panic, the 'cost' of imiprimine therapy is the drug's side effects, usually worse than placebo for the first 4weeks of therapy, then results improve. This period of time is too long to benefit most ICU patients who suffer from relatively short duration ailments, rapidly corrected by titrated life support.
Dopaminergic neurotransmitters and agitation
Acute stress increases dopamine release and metabolism in a number of brain areas [28]. Dopaminergic innervation of the medial and dorsolateral prefrontal cortex appears to be particularly vulnerable to stress and relatively low intensity levels of stress are capable of promoting significant responses. The prefrontal dopaminergic neurons have a number of higher functions including attention and 'working' memory, and the acquisition of coping patterns in response to stress [29]. Amphetamines and cocaine agonize these receptors and have a similar effect as stress, resulting in symptoms such as anxiety, panic, hypervigilence, exaggerated startle reflexes and paranoia [30]. NMDA and opiate receptors are plentiful in this area and stress-induced innervation of the fronto-cortical neurons is prevented if these receptors are selectively blocked. This increase of dopamine from the dendrites of dopamine neurons may be due to an alteration in GABA regulation of the dopamine neurons. As with noradrenergic systems, single or repeated exposures to stress potentiates the capacity of a subsequent stressor to increase dopamine function in the forebrain without altering basal dopamine turnover, suggesting that the receptors have been hypersensitized [31].
Sensory and cognitive dissociations resulting from dopaminergic hyperfunction produce a state of fear and anxiety via direct anatomic connections from cortical brain structures to the limbic system principally through mesolimbic pathways [32]. It is thought that this disinhibition of mesolimbic dopamine neurons causes the bizarre behavioral and cognitive symptoms experienced by patients in schizophrenia and, by extension, with delirium [33]. Delirium resulting from dopaminergic hyperfunction is characterized by global disorders of cognition and wakefulness and by impairment of psychomotor behavior [34]. Major cognitive functions such as perception, deductive reasoning, memory, attention and orientation are all globally disordered. Excessive motor activity frequently accompanies severe cases of delirium and, when this occurs, the resulting constellation of symptoms is called 'agitated delirium' [35]. Integrative brain failure in the ICU environment is almost always associated with a hemodynamic or metabolic decompensation, either intra-or extracranial. The ICU environment provides a repository of typical predisposing factors of a hemodynamic or metabolic nature, including acute or chronic organic brain vascular insufficiency, endocrine insufficiency, acute or chronic cardiopulmonary decompensations, multiple organ-system insufficiency, relative hypoxia, poor tissue perfusion, multiple medications, and finally sleep–wake cycle disruption caused by immobilization, anxiety and pain [36].
If excessive responses to dopaminergic systems contribute to the aforementioned manifestations, the neuroleptic drugs that decrease neurodopamine activity such as haloperidol should alleviate some of the symptoms, particularly hypervigilence and paranoia. Haloperidol is a butryphenone structurally similar to droperidol with mechanisms of action similar to piperazine-based phenothiazines [37]. Haloperidol is a dopamine antagonist; benzodiazepines are GABA agonists. Theoretically, there should be a synergistic relationship between the two when used in a conjoined fashion. In addition, butryphenones such as haloperidol suppress spontaneous movements and complex behavior patterns which result from disharmonious brain function, with minimal central nervous system (CNS) depressive effect [38]. There is little or no ataxia, incoordination or dysarthria at ordinary doses. Haloperidol appears to exert a diffuse depressive effect by inhibiting dopaminergic receptors and reuptake of neurodopamine in the subcortical, midbrain and brainstem reticular formation [39]. A unique effect of haloperidol is a relatively strong suppression of spontaneous musculoskeletal hyperactivity and behavior that results from hyperdopaminergic brain function without pronounced sedation or hypotension [40]. Haloperidol produces less sedation than other phenothiazines, with very little effect on heart rate, blood pressure and respiration, and it appears to have a very wide range between therapeutic doses and the dose which precipitates extrapyramidal reactions [41]. It is thought that haloperidol's molecular structure is changed in some fashion when given orally, increasing the possibility of extrapyramidal reactions [42].
Opiate neurotransmitters and agitation
One of the fundamental behavioral effects of intense stress is analgesia, resulting from the release of endogenous opiates in the brain stem [43]. This analgesic effect can be blocked by naloxone and shows cross tolerance to morphine [44]. It is unknown whether the effects of stress on endogenous opiates are related to the core clinical symptoms associated with anxiety and panic. The recent development of novel drugs (termed peptoids) that mimic or block neuropeptide function have opened up new clinical approaches to a number of conditions [45]. Thus high efficacy κ opioid-receptor agonists such as CI-977 (enadoline) have more potential for the treatment of pain-related anxiety because the hemodynamic and ventilatory side effects are fewer [46]. Peptoid antagonists appear to be relatively free of side effects possibly because neuropeptide systems are only activated under very selective conditions. Peptoid agonists, on the other hand, can exert extremely powerful actions on brain function and this may be related to the key position neuropeptide receptors occupy in the hierarchy of chemical communication in the brain.
Much evidence now exists that very complex neural connections involving diverse areas of the nervous system play a part in pain modulation. Pain signals may be edited at the spinal cord level, in the periaqueductal gray matter and brain stem raphe nuclei prior to reaching relays and gating mechanisms in the thalamus on the way to the cerebral cortex [47]. The perception of noxious stimuli may depend not only on peripheral stimulation and transmission, but also on modulation occurring in spinal cord and higher structures. Accordingly, the subjective sensation of pain can be effectively blocked at the brain level by narcotic analgesics and also at the inflow tract level, explaining the efficacy of spinal or epidural anesthesia [48].
Perceptions of pain from peripheral nocieceptors are integrated and relayed via integrated afferent pathways from the hypothalamus to the reticular activating system via the reticular formation, beginning in the medulla and extending to the midbrain. This pathway links the brain with perception of external events. Pain is a very potent activator of this system, and this explains the importance of the elicitation of pain in the evaluation of consciousness [49]. Modulated signals ultimately reach the medulla oblongata and the sympathetic outflow tracts of the spinal cord leading to the pupils, heart, blood vessels, gastrointestinal tract, pancreas, and adrenal medulla. Norepinephrine is released from the postganglionic fibers into the target organ and both epinephrine and norepinephrine are released into the blood stream from the adrenal medulla. The more intense the stimuli, the more pronounced the response. The levels of norepinephrine generally increase about that of epinephrine and the levels of 11- and 17-hydroxycorticosteroids also increase. Painful stress has two separate components: psychic and somatic; both of these usually combine to stimulate the hypothalamus via a common pathway [50]. In addition to promoting the psychic symptom of anxiety, increased catecholamine levels increase heart rate and myocardial contractility to bolster cardiac output [51].
The analgesia and blunted emotional response to trauma produced by release of endogenous opioids increase the chances of survival after injury [52]. However, the emotional response to opioids has been described as euphoric [53]. The difference between suicide and adventure is that the adventurer leaves a margin of safety. The narrower the margin the more the adventure. It has been suggested that precipitous increases in endogenous opiates secondary to short-lived stress may explain the joys of elective risk-taking behavior [54]. Opiates such as morphine decrease stress-induced norepinephrine release in the hippocampus, hypothalamus, thalamus and midbrain, promoting anxiolysis and sedation as well as analgesia. In addition to their analgesic and sedative effect, opiates decrease the sympathetic discharge associated with the pain of myocardial ischemia and pulmonary edema, and thus exert a mild negative inotropic and chronotropic effect. Opiates also exert a direct depressive effect on the medullary respiration center. However, humoral responses of patients in pain, such as hypertension and tachypnea tend to counterbalance the side effects of narcotic analgesics, such as hypotension from histamine release and medullary ventilation center depression [55].
The hypothalamic–pituitary–adrenal axis and agitation
Acute multifactorial stress increases corticotropin (ACTH) and corticosterone levels. Stress-induced corticosterone release may be involved in the central processing of stress-related phenomena and subsequent learned behavior responses [56]. The mechanism responsible for transient stress induced hyperadrenocorticism and feedback resistance may be a downregulation of glucocorticoid receptors as a result of high circulating glucocorticoid levels elicited by stress. This results in increased corticosterone secretion and feedback resistance. Following termination of stress and decreased circulating levels of glucocorticoids, the number of receptors gradually return to normal and feedback sensitivity normalizes [57]. The effect of chronic stress may also result in augmented corticosterone responses to a previous exposure to stress [58,59]. Adrenalectomy has been shown to increase the frequency of behavioral deficits induced by intense stress [60]. This effect is reversed by the administration of corticosteroids. Learning deficits produced by intense stress may be related to neurotoxic levels of glucocorticoids to hippocampal neurons [61].
Corticotropin-releasing factor (CRF) is the hypothalamic hypophysiotropic hormone that activates the pituitary-adrenal axis [62]. CRF can also be a neurotransmitter in other areas of the brain. CRF is anxiogenic when injected intravenously, probably interacting with noradrenergic neurons in these areas, increasing activity [63]. CRF may also exhibit activity in the dopaminergic areas within the frontal cortex as well, a reaction similar to that promoted by stress. Severe stress also produces increased levels of CRF in the locus coeruleus, hippocampus and amygdala, increasing emotional lability [64].
Autonomic hyperarousal and hypervigilence facilitate appropriate rapid behavioral reaction to threat. Stress-induced levels of cortisol may promote metabolic activation necessary for sustained physical demands necessary to avoid further injury. Elevated catecholamine levels increase heart rate and myocardial contractility to bolster cardiac output as potential compensation for injury during 'fight or flight'. Painful stimulation of somatic afferent nerves is a potent activator of neuroendocrine changes. Immediate inhibition of insulin production occurs coincidentally with an increase in glucogon production, resulting in increased blood sugar (hyperglycemia of stress), free fatty acids, triglycerides and cholesterol to fuel possible 'fight or flight' [65]. Growth hormone and cortisol secretion increase, providing an anti-inflammatory response for potential trauma. Aldosterone production acts to conserve salt and water, bolstering intravascular volume in case of potential blood loss.
Previous studies with receptor antagonists suggested that α1-adrenergic receptors were involved in defensive withdrawal in rats [66]. However, β -adrenoreceptor antagonists may also be involved in stress-related responses [67]. Propranolol pretreatment prevents the restraint-induced changes in the behavior of mice after stressful maze testing [68]. These results suggest the involvement of CNS β -adrenergic receptors in stress-related behavioral changes and suggest that β -adrenergic agonists exert anxiolytic effects that differ from those of the benzodiazepines. The activation of β -adrenoceptors may be an important mechanism in the behavioral inhibition induced by CRF, and that the neurochemical mechanisms that underlie the 'anxiogenic' and the 'activating' behavioral effects of CRF are neuropharmacologically distinct. The anxiolytic benzodiazepine alprazolam seems to selectively decrease CRF concentration in the locus coeruleus [69].
Clinical implications of neurotransmission and agitation in the ICU
The acute behavioral responses brought about by the activation of neurotransmission-modulated humoral responses by psychological and physical trauma represent evolutionary adaptive responses critical for survival in an uncertain and potentially dangerous environment. These compensatory responses were presumably created at a time in the universe when there were no high-tech surrogates for naturally induced environmental stress. Patients in the hybrid ICU environment undergo stress but no natural environmental threat. The highly stressful environment of the ICU may lead to a loss of orientation to time and place. Monotonous sensory input such as repetitive and noisy monitoring equipment, prolonged immobilization, especially with indwelling life support hardware, frequently interrupted sleep patterns and social isolation eventually contribute to the onset of brain dysfunction. However, this high-tech habitat is capable of reversing multiple organ-system insufficiency if the patient is able to tolerate the inherent stress of the environment. Therefore, normally beneficial responses act to the patient's detriment in the artificial ICU environment and it is necessary to block them as selectively as possible.
Ameliorating neurotramsission dysfunction in the ICU
Blocking deleterious pain responses
The pain reflex is normally beneficial, allowing those affected to recognize and avoid impending peril quickly. However, when the pain cannot be avoided, the reflex promotes decompensatory hemodynamic and metabolic changes. An ideal treatment for pain would stop the pain-induced reflex, calming the resultant numeral response, with a minimum effect on other organ-system functions (Table 2). This is not always possible with currently utilized sedatives. Hypnotics such as the benzodiazepines do not resolve pain, they merely superimpose a layer of CNS depression which makes it harder to diagnose where the pain is coming from and what effect it is having on other organ systems. Antipsychotics such as haloperidol do not have any analgesic effect, and their side effects will predominate if given for analgesia, adding more bizarre CNS symptoms to the already agitated patient.
Table 2.
Treatment choices for anxiety in the intensive care unit
| Benzodiazapines | Central nervous system depressants with anterograde amnestic musculoskeletal relaxation and anxiolytic action. |
| Blunts the patient's perception of distress. No analgesic activity. | |
| Lorezapam | Mild anxiolytic, slow acting, long acting, not titratable. Accumulates quickly when used in continuous infusion. Low |
| performance-high safety factor. | |
| Midazolam | Potent, titratable for 48 h can be titrated to siut the sedation requirements of the individual. Moderate |
| performance-moderate safety. | |
| Propofol | Very potent, very titratable (up to 1 week). Facilitates control of life threatening agitation. High performance, low |
| safety. | |
| Neuroleptics | Not sedatives. Treatment for true delerium, not anxiety or discomfort. Reorganizes brain chemistry at level of |
| dopamine. | |
| Haloperidol | Always used intravenously. Step up dosing required. Continuous infusion useful in selected patients. |
| Droperidol | Similar to haloperidol except associated with frightening dreams that may require benzodiazepines for relief (thus |
| limiting its action). | |
| Analgesics | Stops pain reflex and offers comfort and mild anxiolysis. |
| Morphine sulfate | Gold standard of analgesia/sedation. Multiple routes of delivery. Reversible. May cause hemodynamic respiratory |
| supression in patients with little reserve. | |
| Fentanyl | As effective as morphine but titratable in real time for 48 h. No histamine release. Effectively titrates analgesia for |
| unstable patients. | |
| Meperidine | Not titratable. Causes hypotension, tachycardia, seizures and mental status changes in critically ill patients. |
| Ketorolac | Pure analgesia without sedation. Effective in stopping pain reflex for hemodynamically unstable patients. |
| Combination therapy | Effective real time titration of both analgesia and sedation at the same time in the same patient. |
| Midazolam and fentanyl | When separation of theraputic effect is desired, start with one and then add the other. The doses of both must be |
| reduced. | |
| Speciality sedation agents | Usually used as adjuncts to treatment for complicated patients. |
| Clonidine | Offers analgesia, decreases adrenergic response. Side effects of bradycardia and dry mouth. Intravenous |
| formulation if possible. | |
| Dexmetomidine | In trial. A purer α2 action. More beneficial effects, fewer side effects. Will be useful in treating substance |
| withdrawal. | |
| Reversal agents | Titrating the effect of sedation or analgesia at the level of brain receptors. |
| Naloxone | Rapid reversal of narcotics. Short acting. Should be used in continuous infusion to avoid complications of sudden |
| awakening. | |
| Flumazenil | Rapid reversal of benzodiazepines. Short acting. Should be used in continuous infusion to avoid complications of |
| sudden awakening. |
Morphine sulfate is the most widely used of all narcotic analgesic/sedatives [70]. The drug is easily titrated by multiple routes and reversible with narcotic antagonists. However, in addition to its sedative action, morphine has profound effects on cardiac hemodynamics. Doses as small as 0.1–0.2 mg/kg can produce orthostatic hypotension in normal subjects due to vasodilatation in the splanchnic beds, decreasing preload and right heart filling pressures [71]. This vasodilatory effect has been attributed to both histamine release and direct effects from neural mediators [72]. The respiratory depressive effect can be profound and unpredictable. Fentanyl is a synthetic opiate 75–200-times more potent than morphine, significantly more rapid acting (1–2 min) and with a shorter duration (30–40 min) [73]. The most used opiate, morphine, frequently promotes hypotension by a histamine vasodilating effect. Compared to morphine, fentanyl promotes minimal histamine release and exhibits significantly less effect on cardiac dynamics than morphine. However, fentanyl's affinity for fat can lead to its accumulation during prolonged use, ultimately 'leaching out' after discontinuation of the drug, limiting its long-term use as a continuous infusion [74].
Ketorolac is a parenteral nonsteroidal anti-inflammatory agent that has almost pure analgesic activity. Sixty milligrams of ketorolac intramuscularly is 800-times as potent as aspirin and approximately equal in analgesic effect to 10 mg morphine sulfate [75]. However, ketorolac has significantly less respiratory and hemodynamic effects than morphine [76]. Ketorolac is useful in blocking the pain reflex, and therefore the increased catecholamine response in patients with marginal hemodynamic reserve. Incisional pain prevents postoperative patients with upper midline abdominal incisions from coughing effectively, causing a significant decrease in forced expiratory volume, and compromising clearance of tracheal secretions. A 'pure' analgesic may decrease risk of nosocomial pneumonia by allowing patients to clear their secretions more effectively with less risk of respiratory depression. ICU patients in heart failure who must undergo painful procedures such as invasive vascular catheterization, chest tube thoracostomy or intra-aortic balloon placement also tolerate narcotic side effects poorly and in whom the hemodynamic effects of a catecholamine release would be decompensatory.
Anxiety and discomfort
The benzodiazepines have been the mainstay of ICU anxiety treatment for a number of years because they offer a relatively wide margin of safety from unwanted side effects [77]. Benzodiazepines with short half lives are especially useful when hour-to-hour titration is required in unstable hemodynamics and for patients with coincident liver disease [78]. Benzodiazepines attenuate stress-induced increases in norepinephrine release in the hippocampus, cortex amygdaloid and locus coeruleus region, effectively reducing conditioned fear and generalized anxiety. Anterograde amnesia occurs almost immediately after intravenous administration and usually persists for 20–40 min after a single dose [79]. However, during intense stress, these drugs may not be able to exert an effective anxiolytic action except in hypnotic doses [80]. All of the benzodiazepines reduce ventilatory responses to hypoxia when administered rapidly or in large doses [81]. Benzodiazepine toxicity usually results in an amplification of their therapeutic effects, but rarely cardiac arrest unless other cardioactive drugs have been given concurrently.
α2-Adrenoreceptors are located both centrally and peripherally. Their function is to inhibit norepinephrine release from presynaptic junctions by several negative feedback mechanisms, effectively suppressing neuronal firing and norepinephrine secretion in all target effecter organs containing α2-receptors, including the central sympathetic nervous system [82]. As a result, α2-adrenergic agonists potently inhibit sympathoadrenal outflow, as evidenced by the decreased levels of circulating norepinephrine and the diminution of catecholamine metabolites in the urine. α2-Agonists, which long ago established themselves as antihypertensives, have also been found to possess intrinsic anxiolytic, sedative, analgesic, and antiemetic properties [83]. These attributes make them attractive for use in the treatment of agitation and delirium associated with noradrenergic hyperfunction [84]. α2-Agonists administered concurrently with benzodiazepines or opiate analgesics permit significantly decreased doses of these sedative narcotics, minimizing side effects while maintaining effective levels of sedation and analgesia [85].
Among clinically available α2-agonists, clonidine seems to be the most selective. Clonidine is thought to act by competitively binding opiate catecholaminergic receptors, decreasing the amount of opiates required to get the same sedative effect. As a consequence, respiratory depression, hypotension, and other side effects of narcotic sedatives are significantly attenuated, especially in hemodynamically unstable patients [86]. Clonidine has been shown to decrease the amount of anesthesia required to obtain operative analgesia [87]. Clonidine has been effectively used intrathecally for analgesia in terminal cancer patients who had become tolerant to intrathecal morphine [88]. Clonidine has been extensively used on psychiatry wards to attenuate drug withdrawal syndrome after chronic benzodiazepine and alcohol use [89]. Clonidine has also been proved to be effective in patients with panic disorders due to its anxiolytic action and its ability to decrease the brain noradrenergic neuronal hyperactivity.
Clonidine is almost completely absorbed after oral administration, but takes 60-90min to reach peak plasma concentration [90]. A large number of ICU patients are not able to take the medication enterally, or, because of concurrent hemodynamic or metabolic instabilities, it is absorbed erratically by that route. Drug delivery through a transdermal patch takes much longer to reach effective blood levels and a minimum of 2days to achieve a steady state concentration, making this route ponderous for acute care use [91]. Unfortunately, clonidine is not yet approved for intravenous use in the USA, but intravenous administration is common in Europe [92]. Careful titration of intravenous clonidine as a supplement to analgesics or sedatives in severe agitation syndromes in the critical care patients is a new area of clinical investigation. Other α2-agonists not currently used in clinical practice havepractical potential in the treatment of severe agitation and delirium. The highly selective α2-agonist dexmedetomidine has been shown to produce anxiolytic effects comparable to benzodiazepines, but fewer negative effects on hemodynamics. Dexmedetomidine is not yet available for clinical use [93,94].
New horizons: selective serotonin and cholecystokinin antagonists
Because of the clinical side effects and shortcomings of the previous list of therapeutic medications, new agents that act selectively on sensory pathways afferent to the limbic system by decreasing the secretion of neurotransmitters could be more selective in conditioned responses without concurrent sedation and hypnosis. The serotonin reuptake antagonists show promise as superselective anxiolytics, potentially valuable in the ICU where sedative side effects directly affect hemodynamic, ventilatory and metabolic stability during life support management [95].
Serotonin (5-hydroxytryptamine; 5-HT) is involved in numerous physiological processes such as appetite, sleep, pain, sexual behavior and temperature regulation. 5-HT reuptake antagonists have been found effective in the alleviation of depression and panic attacks, and are at varying stages of clinical evaluation in the treatment of obsessive-compulsive disorder, chronic pain, and bulimia nervosa. Selective 5-HT receptor agonists and antagonists show promise in the treatment of migraine, nausea and vomiting, schizophrenia, anxiety, hypertension, and Raynaud's disease [96]. 5-HT interacts with multiple brain 5-HT receptor subtypes to influence a wide range of behaviors. Three main families of 5-HT receptors (5-HT1, 5-HT2 and 5-HT3) have been described. Several different 5-HT receptor subtypes and (5-HT1A, 5-HT1C, 5-HT2 and 5-HT3) may produce anxiolytic effects; 5-HT1A 5-HT2 receptors may be involved in the etiology of major depression and the therapeutic effects of antidepressant treatment; and 5-HT3 receptors have been linked to reward mechanisms and cognitive processes [97]. Serotonin1A agonists seem to be promising for anxiety and also mixed anxiety-depression [98].
Buspirone, with a pharmacologic profile which distinguishes it from the benzodiazepines, appears to hold future promise [99]. Buspirone does not act on the GABA receptor; rather, its most salient interaction with neurotransmitter receptors occurs at the 5-HT1A receptor. Because it lacks the anticonvulsant, sedative, and muscle-relaxant properties associated with other anxiolytics, buspirone has been termed 'anxioselective' [100]. Buspirone does not have a euphoric effect and therefore has a low potential for abuse. Pharmacologic studies on the molecular level indicate that buspirone interacts with dopamine and 5-HT receptors. This action is supported by studies focused on receptor binding, anatomical localization, biochemistry, neurophysiology, and animal behavior. However, the lengthy 'lag period' before buspirone begins to show pharmacologic activity limits its use in acute care areas like the ICU.
The recognition that action at 5-HT1A receptors may be aviable approach to the pharmacotherapy of anxiety is evidenced by the number of other agents of this class under development by a number of pharmaceutical companies. The cyclopyrrolone zopiclone functions as a selective hypnotic, extending the duration of slow wave sleep and concomitantly shortening the awake periods [101]. This slow wave sleep inducing effect of zopiclone did not depress rapid eye movement (REM) sleep and shows no rebound of activity in wakefulness or REM sleep after treatment. At the cortical level in rats, zopiclone increases the spectral energy in the Δ band (0.5–4 Hz). This rise in energy can also reach the fast frequencies (β band: 12–16 Hz). This power spectrum is characteristic of a compound having tranquilizing-hypnotic potential. The relatively short duration of action of zopiclone minimizes the residual effects seen upon waking (drowsiness, impairment of psychomotor performance). Binding is thought to occur at the benzodiazepine receptor complex, or to a site closely linked to this complex [102]. Although zopiclone exhibits anticonvulsant, muscle relaxant and anxiolytic properties in animals, its hypnotic effects are the most useful in humans. In clinical trials, zopiclone improved sleep in chronic insomniacs similarly to flurazepam [103]. Minimal impairment of psychomotor skills and mental acuity have been reported in the relatively small number of patients studied to date [104].
Unlike zopiclone which exhibits a hypnotic action, suriclone is a novel benzodiazepine receptor ligand with enhanced anxiolytic properties [105]. Although chemically entirely different from the benzodiazepines, it acts as a functional benzodiazepine agonist with very high affinity for the benzodiazepine receptors. In studies, suriclone and diazepam had a different side-effect profile; suriclone produced mainly dizziness, while diazepam caused sedation [106]. This may reflect the fact that suriclone and benzodiazepines bind to distinct sites or different allosteric conformations of the benzodiazepine receptors. The drug, when effective, has a duration of action between 6 and 8h. There was no evidence of a rebound phenomenon. There was, however, a rapid return to pretreatment level of anxiety, which makes its use as a continuous, titratable infusion attractive.
The role of cholecystokinin (CCK) receptors in the development of anxiety is a new field of investigation [107]. CCK is an octapeptide normally synthesized in the gut, but also with large concentrations in the brain where it acts as a neurotransmitter. Central CCKergic neurotransmission has been implicated in the genesis of negative emotions, feeding disorders such as anorexia, nociception alterations, movement disorders, schizophrenia, anxiety and panic disorders [108]. The interaction of CCK with GABAergic inhibitory neurotransmission, mediated probably through CCK-B receptors, could be the neurochemical substrate for anxious type of exploratory behavior. The brain cholecystokinin-B/gastrin receptor (CCK-B/gastrin) has been implicated in mediating anxiety, panic attacks, satiety, and the perception of pain [109]. The isolation of rats for 7days produced anxiogenic-like effect on their behavior and increased the number of CCK receptors in the frontal cortex without affecting benzodiazepine receptors [110].
CCK and benzodiazepine receptor binding characteristics were analyzed in the brain tissue samples from 13 suicide victims and 23 control cases. In the frontal cortex, significantly higher apparent number of CCK receptors and affinity constants were found in the series of suicide victims. The results of this investigation suggest that CCKergic neurotransmission is linked to self-destructive behavior, probably through its impact on anxiety and adaptational deficits [111]. The behavioral effects of an experimental selective CCK-B receptor antagonist CI-988 were investigated in rodents. In three rodent tests of anxiety (rat elevated X-maze, rat social interaction test and mouse light-dark box), CI-988 produced an anxiolytic-like action over a wide dose range. The magnitude of this effect was similar to that of chlordiazepoxide [112]. In contrast, the selective CCK-A receptor antagonist devazepide was inactive [113].
Central but not peripheral administration of the selective CCK-B receptor agonist, pentagastrin, produced an anxiogenic-like action [114]. The pentagastrin-induced anxiety was dose-dependently antagonized by CI-988, whereas devazepide was inactive. CI-988 did not interact with alcohol or barbiturates. Thus, CI-988 appears to be an anxioselective compound unlike benzodiazepines but the anxiolytic-like action was dose-dependently antagonized by flumazenil. The possible involvement of endogenous CRF in the anxiogenic and pituitary-adrenal axis activating effects of CCK octapeptide sulfate ester (CCK8) was investigated in rats [115]. The results strongly suggest that the anxiogenic and hypothalamo-pituitary-adrenal activating effects of CCK8 are mediated via CRF [116].
Tetronothiodin is a novel CCK-B receptor antagonist produced from the fermentation broth of the NR0489a Streptomyces species [117]. Tetronothiodin inhibited the binding of CCK8 (C-terminal octapeptide of CCK) to rat cerebral cortex membranes (CCK-B receptors), but did not inhibit CCK8 binding to rat pancreatic membranes (CCK-A receptors). This finding indicated tetronothiodin was an antagonist of CCK-B receptors and may have use as a superselective antianxiety agent. It is a useful tool for investigating the pharmacological and physiological roles of CCK-B receptors and has no clinical role as yet.
References
- Davidson RJ, Irwin W. The functional neuroanatomy of emotion and affective style. Trends Cogn Sci. 1999;3:11–21. doi: 10.1016/s1364-6613(98)01265-0. [DOI] [PubMed] [Google Scholar]
- Kropotov JD, Etlinger SC. Selection of actions in the basal gangliathalamocortical circuits: review and model. Int J Psychophysiol. 1999;31:197–217. doi: 10.1016/s0167-8760(98)00051-8. [DOI] [PubMed] [Google Scholar]
- Reid SG, Milsom WK. Respiratory pattern formation in the isolated bullfrog (Rana catesbeiana) brainstem-spinal cord. Respir Physiol. 1998;114:239–255. doi: 10.1016/s0034-5687(98)00091-7. [DOI] [PubMed] [Google Scholar]
- Barrera-Mera B, Barrera-Calva E. The Cartesian clock metaphor for pineal gland operation pervades the origin of modern chronobiology. . Neurosci Biobehav Rev. 1998;23:1–4. doi: 10.1016/s0149-7634(97)00062-6. [DOI] [PubMed] [Google Scholar]
- Wise SP, Murray EA. Role of the hippocampal system in conditional motor learning: mapping antecedents to action. Hippocampus. 1999;9:101–117. doi: 10.1002/(SICI)1098-1063(1999)9:2<101::AID-HIPO3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Smythies J. The functional neuroanatomy of awareness: with a focus on the role of various anatomical systems in the control of intermodal attention. Conscious Cogn. 1997;6:455–458. doi: 10.1006/ccog.1997.0315. [DOI] [PubMed] [Google Scholar]
- Philip Strange. Brain Biochemistry and Brain Disorders Oxford: Oxford University Press; 1992.
- Fischer JE, Rosen HM, Ebeid AM, et al. The effect of normalization of plasma amino acids on hepatic encephalopathy in man. . Surgery. 1976;80:77–91. [PubMed] [Google Scholar]
- Settipane GA. The restaurant syndromes. N Engl Reg Allergy Proc. 1987;8:39–46. doi: 10.2500/108854187779045330. [DOI] [PubMed] [Google Scholar]
- McConalogue K, Grady EF, Minnis J, et al. Activation and internalization of the mu-opioid receptor by the newly discovered endogenous agonists, endomorphin-1 and endomorphin-2. Neuroscience. 1999;90:1051–1059. doi: 10.1016/S0306-4522(98)00514-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaud LL, Morrow AL. Gender-selective effects of ethanol dependence on NMDA receptor subunit expression in cerebral cortex, hippocampus and hypothalamus. Eur J Pharmacol. 1999;369:331–334. doi: 10.1016/s0014-2999(99)00103-x. [DOI] [PubMed] [Google Scholar]
- Bonci A, Malenka RC. Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. . Neurosci. 1999;19:3723–3730. doi: 10.1523/JNEUROSCI.19-10-03723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt RA, Bateson AN, Martin IL. Decreased GABA enhancement of benzodiazepine binding after a single dose of diazepam. J Neurochem. 1999;72:2219–2222. doi: 10.1046/j.1471-4159.1999.0722219.x. [DOI] [PubMed] [Google Scholar]
- Crippen DW, Ermakov S. Stress, agitation and brain failure in critical care medicine. Crit Care Q. 1992;15:52–74. doi: 10.1097/00002727-199208000-00005. [DOI] [PubMed] [Google Scholar]
- Rusnak M, Zorad S, Buckendahl P, et al. Tyrosine hydroxylase mRNA levels in locus ceruleus of rats during adaptation to long-term immobilization stress exposure. Mol Chem Neuropathol. 1998;33:249–258. doi: 10.1007/BF02815186. [DOI] [PubMed] [Google Scholar]
- Dunn AJ. Stress-related changes in cerebral catecholamine and indoleamine metabolism: lack of effect of adrenalectomy and corticosterone. J Neurochem. 1988;51:406–412. doi: 10.1111/j.1471-4159.1988.tb01053.x. [DOI] [PubMed] [Google Scholar]
- Schafers RF, Nurnberger J, Herrmann B, et al. Adrenoceptors mediating the cardiovascular and metabolic effects of α -methylnoradrenaline in humans. J Pharmacol Exp Ther. 1999;289:918–925. [PubMed] [Google Scholar]
- Fontana A, Rosenheck R. A model of war zone stressors and post-traumatic stress disorder. J Trauma Stress . 1999;12:111–126. doi: 10.1023/A:1024750417154. [DOI] [PubMed] [Google Scholar]
- Crippen DW. The role of sedation in the ICU patient with pain and agitation. Crit Care Clin. 1990;6:369–392. [PubMed] [Google Scholar]
- Bagley J, Moghaddam B. Temporal dynamics of glutamate efflux in the prefrontal cortex and in the hippocampus following repeated stress: effects of pretreatment with saline or diazepam. Neuroscience . 1997;77:65–73. doi: 10.1016/S0306-4522(96)00435-6. [DOI] [PubMed] [Google Scholar]
- Middleton HC. Panic disorder: a theoretical synthesis of medical and psychological approaches. J Psychosom Res . 1998;44:121–132. doi: 10.1016/s0022-3999(97)00208-0. [DOI] [PubMed] [Google Scholar]
- Crippen DW. Pharmacologic treatment of brain failure and delirium. Crit Care Clin. 1994;10:733–766. [PubMed] [Google Scholar]
- Leaman TL. Anxiety disorders. Primary Care. 1999;26:197–210. doi: 10.1016/s0095-4543(08)70002-2. [DOI] [PubMed] [Google Scholar]
- Szabo ST, de Montigny C, Blier P. Modulation of noradrenergic neuronal firing by selective serotonin reuptake blockers. Br J Pharmacol. 1999;126:568–571. doi: 10.1038/sj.bjp.0702343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buerk DG, Osanai S, Mokashi A, Lahiri S. Dopamine, sensory discharge, and stimulus interaction with CO2 and O2 in cat carotidbody. Appl Physiol. 1998;85:1719–1726. doi: 10.1152/jappl.1998.85.5.1719. [DOI] [PubMed] [Google Scholar]
- Langston P, Gorman D, Runciman W, et al. The effect of carbon monoxide on oxygen metabolism in the brains of awake sheep. Toxicology. 1996;114:223–232. doi: 10.1016/S0300-483X(96)03513-5. [DOI] [PubMed] [Google Scholar]
- Griez E, Schruers K. Experimental pathophysiology of panic. Psychosom Res. 1998;45:493–503. doi: 10.1016/s0022-3999(98)00027-0. [DOI] [PubMed] [Google Scholar]
- Zangen A, Overstreet DH, Yadid G. Increased catecholamine levels in specific brain regions of a rat model of depression: normalization by chronic antidepressant treatment. Brain Res. 1999;824:243–250. doi: 10.1016/s0006-8993(99)01214-7. [DOI] [PubMed] [Google Scholar]
- Castellano C, Cabib S, Puglisi-Allegra S, et al. Strain-dependent involvement of D1 and D2 dopamine receptors in muscarinic cholinergic influences on memory storage. Behav Brain Res . 1999;98:17–26. doi: 10.1016/s0166-4328(98)00046-1. [DOI] [PubMed] [Google Scholar]
- Horger BA, Iyasere CA, Berhow MT, et al. Enhancement of locomotor activity and conditioned reward to cocaine by brain-derived neurotrophic factor. J Neurosci. 1999;19:4110–4122. doi: 10.1523/JNEUROSCI.19-10-04110.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso AM, Gioino G, Molina VA, et al. Chronic amphetamine facilitates immunosuppression in response to a novel aversive stimulus: reversal by haloperidol pretreatment. Pharmacol Biochem Behav. 1999;62:307–314. doi: 10.1016/s0091-3057(98)00166-x. [DOI] [PubMed] [Google Scholar]
- Iturriaga R, Alcayaga J, Zapata P. Dissociation of hypoxia-induced chemosensory responses and catecholamine efflux in cat carotid body superfused in vitro. J Physiol (Lond) 1996;497:551–564. doi: 10.1113/jphysiol.1996.sp021788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PJ. The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain. 1999;122:593–624. doi: 10.1093/brain/122.4.593. [DOI] [PubMed] [Google Scholar]
- Miller NE, Lipowski JZ, Lebowitz BD. Delirium: Advances in research and Clinical Practice New York: Springer; 1991.
- Crippen DW. Neurologic monitoring in the intensive care unit. New Horizons. 1994;2:107–120. [PubMed] [Google Scholar]
- Crippen D. Understanding neurohumoral causes of anxiety in the ICU. J Crit Ill. 1995;10:550–560. [PubMed] [Google Scholar]
- Settle EC, Ayd FJ. Haloperidol: a quarter century of experience. J Clin Psychiatry. 1983;44:440–448. [PubMed] [Google Scholar]
- Tesar GE, Murray GB, Cassem NH. Use of haloperidol for acute delirium in intensive care setting. J Clin Psychopharmacol. 1985;5:344–347. [PubMed] [Google Scholar]
- Todd CL, Grace AA. Interaction of benztropine and haloperidol actions on rat substantia nigra dopamine cell electrophysiological activity in vivo. Brain Res Bull. 1999;48:219–222. doi: 10.1016/s0361-9230(98)00166-x. [DOI] [PubMed] [Google Scholar]
- Wagner BK, O'Hara DA, Hammond JS. Drugs for amnesia in the ICU. . Am J Crit Care. 1997;6:192–203. [PubMed] [Google Scholar]
- Gerlach J, Larsen EB. Subjective experience and mental side-effects of antipsychotic treatment. Acta Psychiatr Scand Suppl. 1999;395:113–117. doi: 10.1111/j.1600-0447.1999.tb05990.x. [DOI] [PubMed] [Google Scholar]
- Rosebush PI, Mazurek MF. Neurologic side effects in neuroleptic-naive patients treated with haloperidol or risperidone. . Neurology. 1999;52:782–785. doi: 10.1212/wnl.52.4.782. [DOI] [PubMed] [Google Scholar]
- Cannon JT, Lewis JW, Weinberg VE, et al. Evidence for the independence of brainstem mechanisms mediating analgesia induced by morphine and two forms of stress. Brain Res. 1983;269:231–236. doi: 10.1016/0006-8993(83)90132-4. [DOI] [PubMed] [Google Scholar]
- Rosow C. Agonist-antagonist opioids: theory and clinical practice. Can J Anaesth. 1989;36:S5–8. doi: 10.1007/BF03005319. [DOI] [PubMed] [Google Scholar]
- Hughes J, Woodruff GN. Neuropeptides. Function and clinical applications. Arzneimittelforschung. 1992;42:250–255. [PubMed] [Google Scholar]
- Field MJ, Carnell AJ, Gonzalez MI, et al. Enadoline, a selective kappa-opioid receptor agonist shows potent antihyperalgesic and antiallodynic actions in a rat model of surgical pain. Pain. 1999;80:383–389. doi: 10.1016/S0304-3959(98)00237-1. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Wiertelak EP, McGorry M, et al. Neurocircuitry of conditioned inhibition of analgesia: effects of amygdala, dorsal raphe, ventral medullary, and spinal cord lesions on antianalgesia in the rat. Behav Neurosci. 1998;112:360–378. doi: 10.1037//0735-7044.112.2.360. [DOI] [PubMed] [Google Scholar]
- Furst S. Transmitters involved in antinociception in the spinal cord. Brain Res Bull. 1999;48:129–141. doi: 10.1016/s0361-9230(98)00159-2. [DOI] [PubMed] [Google Scholar]
- Sahni R, Menegazzi JJ, Mosesso VN., Jr Paramedic evaluation of clinical indicators of cervical spinal injury. Prehosp Emerg Care . 1997;1:16–18. doi: 10.1080/10903129708958778. [DOI] [PubMed] [Google Scholar]
- Riedel W, Layka H, Neeck G. Secretory pattern of GH, TSH, thyroid hormones, ACTH, cortisol, FSH, and LH in patients with fibromyalgia syndrome following systemic injection of the relevant hypothalamic-releasing hormones. Rheumatol. 1998;57 (suppl 2):81–87. doi: 10.1007/s003930050242. [DOI] [PubMed] [Google Scholar]
- King SM. Escape-related behaviours in an unstable, elevated and exposed environment. II. Long-term sensitization after repetitive electrical stimulation of the rodent midbrain defence system. Behav Brain Res. 1999;98:127–142. doi: 10.1016/s0166-4328(98)00061-8. [DOI] [PubMed] [Google Scholar]
- Wang TL, Chang H, Hung CR, et al. Morphine preconditioning attenuates neutrophil activation in rat models of myocardial infarction. Cardiovasc Res. 1998;40:557–563. doi: 10.1016/s0008-6363(98)00192-8. [DOI] [PubMed] [Google Scholar]
- Schlaepfer TE, Strain EC, Greenberg BD, et al. Site of opioid action in the human brain: mu and kappa agonists' subjective and cerebral blood flow effects. Am J Psychiatry. 1998;155:470–473. doi: 10.1176/ajp.155.4.470. [DOI] [PubMed] [Google Scholar]
- Janal MN, Colt EW, Clark WC, Glusman M. Pain sensitivity, mood and plasma endocrine levels in man following long-distance running: effects of naloxone. Pain. 1984;19:13–25. doi: 10.1016/0304-3959(84)90061-7. [DOI] [PubMed] [Google Scholar]
- Visi ES, et al. Neurochemistry and pharmacology of the major hippocampal transmitter systems: synaptic and nonsynaptic interactions [review]. Hippocampus. 1998;8:566–607. doi: 10.1002/(SICI)1098-1063(1998)8:6<566::AID-HIPO2>3.3.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Arief AI, Griggs RC. Metabolic Brain Dysfunction in Systemic DisordersBoston: Little, Brown Co; 1992.
- Casadevall M, Saperas E, Panes J, et al. Mechanisms underlying the anti-inflammatory actions of central corticotropin-releasing factor. Am J Physiol. 1999;276:G1016–G1026. doi: 10.1152/ajpgi.1999.276.4.G1016. [DOI] [PubMed] [Google Scholar]
- Ignar DM, Kuhn CM. Effects of specific mu and kappa opiate tolerance and abstinence on hypothalamo-pituitary-adrenal axis secretion in the rat. Pharmacol Exp Ther. 1990;255:1287–1295. [PubMed] [Google Scholar]
- Marti O, Harbuz MS, Andres R, et al. Activation of the hypothalamic–pituitary axis in adrenalectomised rats: potentiation by chronic stress. Brian Res. 1999;821:1–7. doi: 10.1016/s0006-8993(98)01212-8. [DOI] [PubMed] [Google Scholar]
- Marti O, Harbuz MS, Andres R, et al. Activation of the hypothalamic-pituitary axis in adrenalectomised rats: potentiation by chronic stress. Brain Res. 1999;821:1–7. doi: 10.1016/s0006-8993(98)01212-8. [DOI] [PubMed] [Google Scholar]
- Bremner JD. Does stress damage the brain? Biol Psychiatry. 1999;45:797–805. doi: 10.1016/s0006-3223(99)00009-8. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Suemaru S, Takao T, et al. Corticotropin-releasing hormone and pituitary–adrenocortical responses in chronically stressed rats. Regul Pept. 1988;23:117–126. doi: 10.1016/0167-0115(88)90019-5. [DOI] [PubMed] [Google Scholar]
- To CT, Bagdy G. Anxiogenic effect of central CCK administration is attenuated by chronic fluoxetine or ipsapirone treatment. . Neuropharmacology. 1999;38:279–282. doi: 10.1016/S0028-3908(98)00176-2. [DOI] [PubMed] [Google Scholar]
- Hotta M, Shibasaki T, Arai K, et al. Corticotropin-releasing factor receptor type 1 mediates emotional stress-induced inhibition of food intake and behavioral changes in rats. . Brain Res. 1999;823:221–225. doi: 10.1016/s0006-8993(99)01177-4. [DOI] [PubMed] [Google Scholar]
- Marshall SB, Marshall LF, et al. Neuroscience Critical Care: Pathophysiology and Patient Management Philadelphia: WB Saunders; 1990.
- Arnsten AF. Catecholamine regulation of the prefrontal cortex. Psychopharmacology. 1997;11:151–162. doi: 10.1177/026988119701100208. [DOI] [PubMed] [Google Scholar]
- Gorman AL, Dunn AJ. β-Adrenergic receptors are involved in stress-related behavioral changes. Pharmacol Biochem Behav. 1993;45:1–7. doi: 10.1016/0091-3057(93)90078-8. [DOI] [PubMed] [Google Scholar]
- Khasar SG, McCarter G, Levine JD. Epinephrine produces a β-adrenergic receptor-mediated mechanical hyperalgesia and in vitro sensitization of rat nociceptors. J Neurophysiol. 1999;81:1104–1112. doi: 10.1152/jn.1999.81.3.1104. [DOI] [PubMed] [Google Scholar]
- Gray AM. Effect of alprazolam on opiate withdrawal: a combined behavioural and microdialysis study. Eur J Pharmacol. 1996;313:73–77. doi: 10.1016/0014-2999(96)00599-7. [DOI] [PubMed] [Google Scholar]
- Sidebotham D, Dijkhuizen MR, Schug SA. The safety and utilization of patient-controlled analgesia. Pain Symptom Manage . 1997;14:202–209. doi: 10.1016/s0885-3924(97)00182-6. [DOI] [PubMed] [Google Scholar]
- Gan TJ, Ginsberg B, Glass PS, et al. Opioid-sparing effects of a low-dose infusion of naloxone in patient-administered morphine sulfate. Anesthesiology. 1997;87:1075–1081. doi: 10.1097/00000542-199711000-00011. [DOI] [PubMed] [Google Scholar]
- Katcher J, Walsh D. Opioid-induced itching: morphine sulfate and hydromorphone hydrochloride. Pain Symptom Manage. 1999;17:70–72. doi: 10.1016/s0885-3924(98)00115-8. [DOI] [PubMed] [Google Scholar]
- Fleischmann E, Akca O, Wallner T, et al. Onset time, recovery duration, and drug cost with four different methods of inducing general anesthesia. Anesth Analg. 1999;88:930–935. doi: 10.1097/00000539-199904000-00046. [DOI] [PubMed] [Google Scholar]
- Byun MY, Fine NA, Lee JY, et al. The clinical outcome of abdominoplasty performed under conscious sedation: increased use of fentanyl correlated with longer stay in outpatient unit. Plast Reconstr Surg. 1999;103:1260–1266. doi: 10.1097/00006534-199904040-00026. [DOI] [PubMed] [Google Scholar]
- Jung D, Mroszczak E, Bynum L. Pharmacokinetics of ketorolac in humans after intravenous, intramuscular and oral administration. Eur J Clin Pharmacol. 1988;35:423–425. doi: 10.1007/BF00561376. [DOI] [PubMed] [Google Scholar]
- Bravo LJ, Mattie H, Spierdijk J. The effects on ventilation of ketorolac in comparison with morphine. Eur J Clin Pharmacol. 1988;35:491–494. doi: 10.1007/BF00558243. [DOI] [PubMed] [Google Scholar]
- Barvais L, Dejonckheere M, Dernovoi B, et al. Continuous infusion of midazolam or bolus of diazepam for postoperative sedation cardiac surgical patients. Acta Anaesthesiol Belgica. 1988;39::239–245. [PubMed] [Google Scholar]
- Behne M, Asskali F, Steuer , et al. Continuous midazolam infusion for sedation of respirator patients. Anesthetist. 1987;36:228–232. [PubMed] [Google Scholar]
- Coenen AM, van Luijtelaar EL. Effects of benzodiazepines, sleep and sleep deprivation on vigilance and memory. Acta Neurol Belg. 1997;97:123–129. [PubMed] [Google Scholar]
- Shafer A. Complications of sedation with midazolam in the intensive care unit and a comparison with other sedative regimens. . Crit Care Med. 1998;26:947–956. doi: 10.1097/00003246-199805000-00034. [DOI] [PubMed] [Google Scholar]
- Nordt SP, Clark RF. Midazolam: a review of therapeutic uses and toxicity. Emerg Med. 1997;15:357–365. doi: 10.1016/s0736-4679(97)00022-x. [DOI] [PubMed] [Google Scholar]
- Sperlagh B, Sershen H, Lajtha A, et al. Co-release of endogenous ATP and [3 H]noradrenaline from rat hypothalamic slices: origin and modulation by α2- adrenoceptors. Neuroscience. 1998;82:511–520. doi: 10.1016/S0306-4522(97)00306-0. [DOI] [PubMed] [Google Scholar]
- Boher H, Bach A, Layer M, et al. Clonidine as a sedative adjunct. Intensive Care Med. 1990;16:265–266. doi: 10.1007/BF01705163. [DOI] [PubMed] [Google Scholar]
- Kulkarni SK, Parale MP, Kulkarni GK. Clonidine in alcohol withdrawal: a clinical report. Methods Find Exp Clin Pharmacol . 1987;9:697–698. [PubMed] [Google Scholar]
- Nutt D, Glue P, Molyneux S, et al. α-2-Adrenoceptor function in alcohol withdrawal: a pilot study of the effects of iv. clonidine in alcoholics and normals. Alcohol Clin Exp Res. 1988;12:14–18. doi: 10.1111/j.1530-0277.1988.tb00125.x. [DOI] [PubMed] [Google Scholar]
- Wing LM, Reid JL, Hamilton CA, et al. Effects of clonidine on biochemical indices of sympathetic function in normotensive man. Clin Sci Mol Med. 1977;53:45–53. doi: 10.1042/cs0530045. [DOI] [PubMed] [Google Scholar]
- Ghignone M, Quintin L, Kehler CH, et al. Effects of clonidine on narcotic requirements and hemodynamic response during indduction of fentanyl anesthesia and endotracheal intubation. . Anesthesiology. 1986;64:36–42. doi: 10.1097/00000542-198601000-00007. [DOI] [PubMed] [Google Scholar]
- Coombs DW, Saunders RL, Fratkin JD, et al. Continuous intrathecal hydromorphone and clonidine for tractable cancer pain. . J Neurosurg. 1986;64:890–894. doi: 10.3171/jns.1986.64.6.0890. [DOI] [PubMed] [Google Scholar]
- Cushman PJ, Sowers JR. Alcohol withdrawal syndrome: clinical and hormonal responses to α-2 adrenergic treatment. . Alcoholism. 1989;13:361–364. doi: 10.1111/j.1530-0277.1989.tb00336.x. [DOI] [PubMed] [Google Scholar]
- Hokfelt B, Hedeland H, Hasson BG. The effect of clonidine and penbutol, respectively on catecholamines in blood and urine, plasma renin activity and urinary aldosterone in hypertensive patients. Arch Int Pharmacodyn. 1975;213:307–321. [PubMed] [Google Scholar]
- Toon S, Hopkins KJ, Aarons L, Rowland M. Rate and extent of absorption of clonidine from a transdermal therapeutic system. J Pharm Pharmacol. 1989;41:17–21. doi: 10.1111/j.2042-7158.1989.tb06321.x. [DOI] [PubMed] [Google Scholar]
- Bernard JM, Hommeril JL, Passuti N, Pinaud M. Postoperative analgesia by intravenous clonidine. Anesthesiology. 1991;75:577–582. doi: 10.1097/00000542-199110000-00006. [DOI] [PubMed] [Google Scholar]
- Aantaa RE, Kanto JH, Scheinin M. Dexmedetomidine premedication for minor gynecologic surgery. Anesth Analg. 1990;4:407–413. doi: 10.1213/00000539-199004000-00011. [DOI] [PubMed] [Google Scholar]
- Aantaa RE, Kanto JH. Dexmedotomidine, an α2-adrenoceptor agonist, reduces anesthetic requirements for patients undergoing minor gynecologic surgery. Anesthesiology. 1990;73:230–235. doi: 10.1097/00000542-199008000-00007. [DOI] [PubMed] [Google Scholar]
- Lane R, Baldwin D. Selective serotonin reuptake inhibitor-induced serotonin syndrome [review]. J Clin Psychopharmacol. 1997;17:208–221. doi: 10.1097/00004714-199706000-00012. [DOI] [PubMed] [Google Scholar]
- Silver H, Shmugliakov N. Augmentation with fluvoxamine but not maprotiline improves negative symptoms in treated schizophrenia: evidence for a specific serotonergic effect from a double-blind study. J Clin Psychopharmacol. 1998;18:208–211. doi: 10.1097/00004714-199806000-00005. [DOI] [PubMed] [Google Scholar]
- Davidson C, Stamford JA. Evidence that 5-hydroxytryptamine release in rat dorsal raphe nucleus is controlled by 5-HT1A, 5-HT1B and 5-HT1D autoreceptors. Br J Pharmacol. 1995;114:1107–1109. doi: 10.1111/j.1476-5381.1995.tb13321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hery F, Boulenguez P, Semont A, et al. Identification and role of serotonin 5-HT1A and 5-HT1B receptors in primary cultures of rat embryonic rostral raphe nucleus neurons. Neurochem. 1999;72:1791–1801. doi: 10.1046/j.1471-4159.1999.0721791.x. [DOI] [PubMed] [Google Scholar]
- Yocca FD. Neurochemistry and neurophysiology of buspirone and gepirone: interactions at presynaptic and postsynaptic 5-HT1A receptors. Clin Psychopharmacol. 1990;10 (suppl 3):6S–12S. doi: 10.1097/00004714-199006001-00003. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Hamada C, Wada T, et al. Comparative study on the behavioral and EEG changes induced by diazepam, buspirone and a novel anxioselective anxiolytic, DN- in the cat. Neuropsychobiology. 1992;26:89–99. doi: 10.1159/000118901. [DOI] [PubMed] [Google Scholar]
- Kato M, Kajimura N, Okuma T, et al. Association between delta waves during sleep and negative symptoms in schizophrenia.Pharmaco-EEG studies by using structurally different hypnotics. Neuropsychobiology. 1999;39:165–172. doi: 10.1159/000026577. [DOI] [PubMed] [Google Scholar]
- Yoshimoto M, Higuchi H, Kamata M, et al. The effects of benzodiazepine (triazolam), cyclopyrrolone (zopiclone) and imidazopyridine (zolpidem) hypnotics on the frequency of hippocampal theta activity and sleep structure in rats. Eur Neuropsychopharmacol. 1999;9:29–35. doi: 10.1016/s0924-977x(97)00102-8. [DOI] [PubMed] [Google Scholar]
- Lancel M. Role of GABAA receptors in the regulation of sleep: initial sleep responses to peripherally administered modulators and agonists. Sleep. 1999;22:33–42. doi: 10.1093/sleep/22.1.33. [DOI] [PubMed] [Google Scholar]
- Strohle A, Antonijevic IA, Steiger A, et al. Dependency of non-benzodiazepine hypnotics. Two case reports. . Nervenarzt. 1999;70:72–75. doi: 10.1007/s001150050403. [DOI] [PubMed] [Google Scholar]
- Concas A, Serra M, Santoro G, et al. The effect of cyclopyrrolones on GABAA receptor function is different from that of benzodiazepines. Naunyn Schmiedebergs Arch Pharmacol. 1994;350:294–300. doi: 10.1007/BF00175035. [DOI] [PubMed] [Google Scholar]
- Wong G, Uusi-Oukari M, Hansen HC, et al. Characterization of novel ligands for wild-type and natural mutant diazepam-insensitive benzodiazepine receptors. Eur J Pharmacol. 1995;289:335–342. doi: 10.1016/0922-4106(95)90111-6. [DOI] [PubMed] [Google Scholar]
- Revel L, Mennuni L, Garofalo P, et al. CR 2945: a novel CCKB receptor antagonist with anxiolytic-like activity. Behav Pharmacol. 1998;9:183–194. [PubMed] [Google Scholar]
- Maletinska L, Pirkova J, Hlavacek J, et al. Cholecystokinin analogs with suppressed central activities. . Peptides. 1998;19:301–308. doi: 10.1016/S0196-9781(97)00371-9. [DOI] [PubMed] [Google Scholar]
- Dethloff LA, Barr BM, Bestervelt LL. Inhibition of gastrin-stimulated cell proliferation by the CCK-B/gastrin receptor ligand CI-988. Food Chem Toxicol. 1999;37:105–110. doi: 10.1016/s0278-6915(98)00119-7. [DOI] [PubMed] [Google Scholar]
- Acosta GB. Administration of cholecystokinin sulphated octapeptide (CCK-8S) induces changes on rat amino acid tissue levels and on a behavioral test for anxiety. Gen Pharmacol. 1998;31:637–641. doi: 10.1016/s0306-3623(98)00075-5. [DOI] [PubMed] [Google Scholar]
- Lofberg C, Agren H, Harro J, et al. Cholecystokinin in CSF from depressed patients: possible relations to severity of depression and suicidal behaviour. Eur Neuropsychopharmacol. 1998;8:153–157. doi: 10.1016/s0924-977x(97)00046-1. [DOI] [PubMed] [Google Scholar]
- Crespi F. Cholecystokinin-B (CCK-B) receptor antagonists improve 'aged' sleep: a new class of sleep modulators? Methods Find Exp Clin Pharmacol. 1999;21:31–38. doi: 10.1358/mf.1999.21.1.527016. [DOI] [PubMed] [Google Scholar]
- Singh L, Field MJ, Hughes J, et al. The behavioural properties of CI-988, a selective cholecystokininB receptor antagonist. . Br J Pharmacol. 1991;104:239–245. doi: 10.1111/j.1476-5381.1991.tb12413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawman-Mintzer O, Lydiard RB, Bradwejn J, et al. Effects of the cholecystokinin agonist pentagastrin in patients with generalized anxiety disorder. Am J Psychiatry. 1997;154:700–702. doi: 10.1176/ajp.154.5.700. [DOI] [PubMed] [Google Scholar]
- Biro E, Sarnyai Z, Penke B, et al. Role of endogenous corticotropin-releasing factor in mediation of neuroendocrine and behavioral responses to cholecystokinin octapeptide sulfate ester in rats. Neuroendocrinology. 1993;57:340–345. doi: 10.1159/000126377. [DOI] [PubMed] [Google Scholar]
- Singh L, Field MJ, Hill DR, et al. Peptoid CCK receptor antagonists: pharmacological evaluation of CCKA, CCKB and mixed CCKA/B receptor antagonists. Eur J Pharmacol. 1995;286:185–191. doi: 10.1016/0014-2999(95)00445-q. [DOI] [PubMed] [Google Scholar]
- Ohtsuka T, Nakayama N, Itezono Y, et al. Tetronothiodin, a novel cholecystokinin type-B receptor antagonist produced by Streptomyces sp. NR0489. III. Structural elucidation. . J Antibiot (Tokyo) 1993;46:18–24. doi: 10.7164/antibiotics.46.18. [DOI] [PubMed] [Google Scholar]