

Abstract
Nutritional support has become a routine part of the care of the critically ill patient. It is an adjunctive therapy, the main goal of which is to attenuate the development of malnutrition, yet the effectiveness of nutritional support is often thwarted by an underlying hostile metabolic milieu. This requires that these metabolic changes be taken into consideration when designing nutritional regimens for such patients. There is also a need to conduct large, multi-center studies to acquire more knowledge of the cost-benefit and cost effectiveness of nutritional support in the critically ill.
Keywords: enteral nutrition, glucose, intensive care, metabolism, parenteral nutrition
Introduction
Nutritional support has become a routine part of the care of the critically ill patient. It is an adjunctive therapy, the main goal of which is to attenuate the development of malnutrition. The effectiveness of nutritional support is often stymied by an underlying hostile metabolic milieu. The design of a nutritional support regimen must, therefore, take into consideration this disordered metabolic situation.
The metabolic milieu
Acute stress caused by accidental or surgical injury, sepsis, burns or other serious illnesses, such as myocardial infarction, results in the outpouring of counter-regulatory endocrine hormones, cytokines and lymphokines. This results in changes in substrate utilization and substance synthesis rates, as well as catabolism and hypermetabolism. Consequently, there is loss of fat and lean body (muscle) mass, a problematic situation that has been dubbed 'auto-cannibalism'. Thus, it is not surprising that this abnormal metabolic milieu causes disordered utilization of exogenously administered nutrients. Conventional nutritional strategies, such as providing the equivalents of a usual human diet, frequently do not prevent or attenuate the loss of muscle and fat tissue. As a result, many investigational efforts have been directed at overcoming or circumventing the obstacles placed by this disordered milieu.
Glucose metabolism
Pathophysiology
The metabolic response to stress is characterized by major alterations in glucose metabolism. The increased secretion of the counter-regulatory (to insulin) hormones cortisol, catecholamines and glucagon [1] results in elevated endogenous glucose production that is secondary to accelerated hepatic gluconeogenesis (Fig. 1). This increased glucose production, coupled with peripheral tissue resistance to insulin, results in reduced glucose utilization and causes hyperglycaemia. Insulin levels are usually within the normal or mildly elevated range, but are not sufficiently elevated to prevent hyperglycaemia. Hyperglycaemia may also be due to reduced nonoxidative glucose disposal that results from decreased muscle glycogen synthetase activity [2,3]. The hyperglycaemia is thought to ensure a ready supply of glucose to predominantly glucose-consuming cells, such as the wound, inflammatory cells and immune cells [4].
Figure 1.
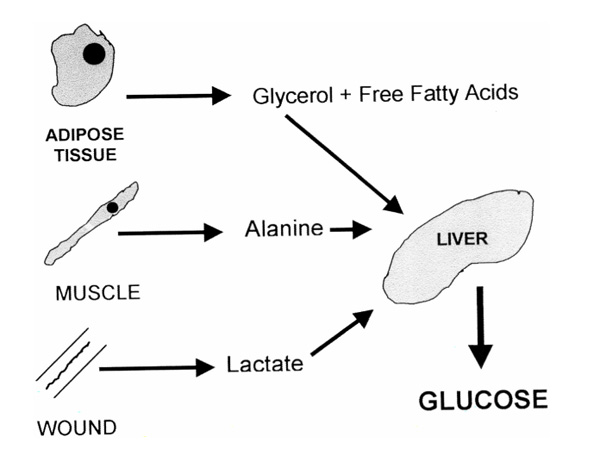
Hepatic gluconeogenesis is increased during stress. Sources of substrate include glycerol from lipolysis, alanine from proteolysis and lactate from anaerobic glycolysis.
Insulin resistance in peripheral tissues is thought to be due to a postreceptor defect, and is thought to hinder cellular glucose uptake. This defect is possibly caused by impaired activity of the intracellular glucose transport system and does not appear to be due to impaired signalling of phosphatidylinositol 3-kinase [5]. Insulin resistance occurred after both elective open and laparoscopic cholecystectomy, and persisted for at least 5 days [6,7]. The overall amount of glucose oxidized may be decreased, despite a glucose oxidization pathway that is thought to be intact, because of the reduced rate of glucose uptake into the cell [3]. The incremental response to increases in insulin concentration is maintained [8], but the ability to reduce blood glucose concentrations per insulin concentration is markedly diminished [9]. Despite the resistance to insulin, the overall peripheral uptake of glucose is often 'normal' or mildly elevated due to increased noninsulin-mediated glucose uptake. Relative to the blood glucose concentration the uptake is low, however [10]. Also, the 'normal' or mildly elevated peripheral glucose uptake may be fueled by the large amounts of glucose (due to hyperglycaemia) being presented to the partially resistant insulin pathway [1].
Glucose can be oxidized to produce adenosine triphosphate (ATP), water and carbon dioxide; converted to glycogen for storage in the liver and muscles; or converted to fat. The latter process is called lipogenesis and occurs in both liver and adipose tissue, although the latter appears to be the main site for lipogenesis [11]. Under normal circumstances carbohydrate intake inhibits fat oxidation, increases glucose oxidation and promotes fat storage [12]. Lipogenesis is usually quantitatively unimportant in humans because the rate of lipogenesis does not exceed the rate of lipid oxidation [13]. When carbohydrate intake exceeds total energy expenditure, however, lipogenesis becomes a more important pathway [14], and respiratory quotients may exceed 1.0, indicating net lipogenesis. This may be seen among patients receiving glucose infusions of > 4 mg/kg per min [15]. Critically ill patients administered intravenous glucose at a rate of 4 mg/kg per min as part of glucose-based total parenteral nutrition (TPN) exhibited some increased hepatic de-novo lipogenesis, despite achieving an respiratory quotient of only 0.90 [16]. This lipogenesis occurred despite the fact that during surgical or septic stress lipogenetic capability is thought to be reduced. This is possibly secondary to tumour necrosis factor (TNF), which, among other factors, can induce apoptosis of preadipocytes and adipocytes [17]. Interleukin (IL)-1β also appears to inhibit lipogenesis in human adipocytes [18].
Exogenous glucose and carbohydrate administration
The administration of exogenous glucose and carbohydrates to injured or septic patients either does not or only minimally diminishes the rate of gluconeogenesis [16]. This is in contradistinction to refeeding starved patients where carbohydrate administration reduces gluconeogenesis and lipolysis. Despite the reduced utilization of glucose, it is still important to administer carbohydrates, because some body tissues are unable to use other substrates readily. Furthermore, glucose and carbohydrate intakes stimulate the secretion of additional insulin, an anabolic hormone that promotes protein synthesis [19] and has an antilipolytic affect [20,21]. The appearance of hyperglycaemia limits the amount of glucose and carbohydrate that can be administered, however. The degree of TPN-induced hyperglycaemia is directly proportional to the rate of glucose infusion and the degree of injury. Elderly patients are more predisposed to develop hyperglycaemia [22].
Excessive glucose loads (> 4 mg/kg per min), especially when administered to acutely stressed patients receiving a total caloric intake greater than resting energy expenditure, results in a thermogenic response, further elevation of blood glucose concentrations and production of additional carbon dioxide [23]. If the patient is no longer stressed respiratory quotients may exceed 1.0, indicating net lipogenesis [24]. This additional carbon dioxide must be excreted via the lungs. A recent survey of US teaching hospitals by Schloerb et al [25] demonstrated that many were feeding their patients more than 4.5mg/kg per min glucose, thus increasing the possibility of glucose overload.
Critically ill patients often receive carbohydrates from a variety of sources other than the glucose in TPN or carbohydrates in enteral nutrition. They receive infusions of 5% dextrose and the glycerol in lipid emulsions also enters the gluconeogenic pathway. Glucose-containing solutions are also used for peritoneal dialysis and continuous veno-veno haemodialysis. The dialysate solution used during continuous veno-veno haemodialysis often contains glucose (usually 1.5%). A significant amount of the glucose (35-45%) can be absorbed, and is thus a source of carbohydrate calories [26]. Similarly, glucose-containing solutions used as replacement solution during haemofiltration can be a source of glucose calories, in one study [27] providing 300 g/day glucose.
Alternate fuels
Nonglucose carbohydrates have been tried experimentally in attempts to bypass the problems of decreased glucose disposal. Sorbitol, fructose and xylitol have been tried, but were found to be problematic [28]. Glycerol has been used with some success, although the amount that can be infused is limited [29].
Lipid metabolism
Pathophysiology
Various stresses, including injury, sepsis and congestive heart failure, cause alterations in lipid metabolism [30]. Lipolysis is accelerated secondary to increased β2-adrenergic stimulation [31,32]. Elevated concentrations of glucagon, TNF-α, IL-1, interferon-α and interferon-γ might also play a role in stimulating lipolysis [33,34,35]. Stimulation of the β2-receptors increases cyclic adenosine monophosphate concentrations, which in turn stimulates the activity of hormone-sensitive lipase [36]. The role of the newly discovered β3-adrenergic receptor in human lipolysis is still unclear [37]. The lipolytic response to β2-stimulation is greater in lean than in obese persons [38]. There are regional variations in the lipolytic rate, with visceral fat cells having the greatest rate due to increased activity of β2- and β3-receptors, and reduced activity of α2-adrenergic receptors. Subcutaneous fat has reduced lipolytic activity due to increased activity of insulin receptors and α2-adrenoreceptors [39].
Rapid glycerol and free fatty acid turnover rates reflect the accelerated lipolysis seen during stress [40]. The increase in lipolysis also results in an increased systemic supply of free fatty acids. The turnover rates are greater than those expected from the increases in the plasma concentrations of these substrates, however [40]. This indicates that there is both increased re-esterification of free fatty acids to triglycerides and increased lipolysis of triglycerides to free fatty acids. This increased activity of the triglycerides-fatty acid substrate cycle is thought to be one of the causes of the hypermetabolism during stress. β-Adrenergic receptor blockade with propranolol decreases lipid oxidation and resting metabolic rates of burn patients [41]. Glucose infusions further increase the lipolytic rate during abdominal surgery because they increase sympathetic nervous system activity [31].
In the stressed state the relative caloric contribution of fat oxidation to the resting energy expenditure is increased and the contribution of glucose oxidation is decreased [42]. The fatty acids released by lipolysis undergo β-oxidation, which in the stressed patient is the predominant ATP-producing pathway. This situation is reflected in observations made after oesophagectomy [43] in which there was a gradual decrease in the contribution of fat oxidation and increase in glucose oxidation as patients convalesced.
Exogenous lipid administration
Intravenous exogenous lipid is most frequently administered as an emulsion of long-chain triglycerides (LCT). In the blood the lipid emulsion is converted to triglyceride-rich particles, the size of which approximate those of chylomicrons (diameter 200-500 nm), and to phospholipid-rich particles called liposomes produced from the emulsifier [44]. The chylomicron-like particles are hydrolyzed by lipoprotein lipase and release fatty acids. The liposomes stimulate cholesterogenesis and accumulate in the blood as the long-lived abnormal lipoprotein-X. The athrogenic potential of lipoprotein-X is under study [45]. Patients with sepsis and multiple organ dysfunction efficiently metabolize intravenous lipid emulsion, even when chronic hepatic failure is present [46].
Exogenous lipid is needed to prevent essential free fatty acid deficiency, so it is recommended that patients receiving TPN receive lipid emulsion infusion (500 ml 10% LCT emulsion) two to three times a week. It is also used as an energy substrate, given that lipid oxidation is the predominant energy-producing pathway. There has been some debate as to whether exogenous lipids (LCT) are a useful energy substrate. This debate has been occasioned by observations that exogenous LCT failed to suppress glucose oxidation in the critically ill. In normal postabsorptive individuals the administration of LCT or LCT/medium-chain triglycerides (MCT) emulsions reduced glucose oxidation, but not tissue glucose uptake [47,48]. Others, however, have not observed suppression of glucose oxidation in normal persons when LCT emulsion was added to a glucose infusion [49]. Similarly, in critically ill patients the addition of LCT emulsion did not modify glucose metabolism so that 45% of the administered fat was stored [50]. Others observed that fat emulsions were well oxidized when administered to septic and trauma patients, even when glucose was administered [51]. Clarification is needed regarding the fate of administered fat emulsions and their effects on the metabolism of other substrates. Current practice is to consider lipid emulsions as an energy substrate and to adminster them as 30-40% of total calories.
Concern has been expressed over the possible immunosuppressive effects of lipid emulsions. Ex-vivo studies have demonstrated decreased neutrophil bacterial killing, depression of monokine expression and other immunodepressant effects [52]. A recent study in trauma patients [53] attributed an increased incidence of infections to lipid emulsion administration. This has led to recommendations to limit fat calories to 30% of total calories [54].
Alternate fuels
There has been interest in substituting MCT for some of the LCT. MCT do not require carnitine to enter the mitochondria and so may be advantageous in situations where carnitine is reduced, such as in some cases of sepsis. MCT are essentially an energy substrate; it is unclear whether they can be stored. Less lipoprotein-X is produced when emulsions containing both MCT and LCT are used. In an attempt to reduce the toxicity of the MCT, structured lipids have been developed. These are lipids with both LCT and MCT bonded to the glycerol backbone (Fig. 2). They are touted to have higher oxidation rates, faster clearance rates and lower reticuloendothelial system accumulation than MCT [55].
Figure 2.
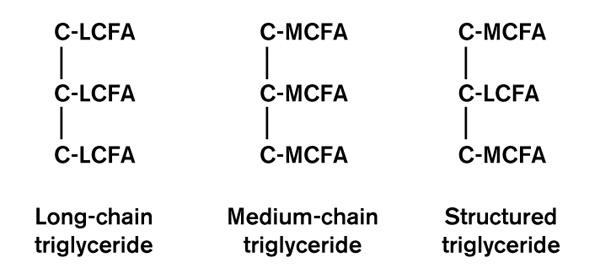
Various types triglycerides are used in nutritional support. They differ in the types of fatty acids attached to the glycerol backbone. Structured lipids are currently under investigation. LCFA, long chain fatty acid; MCFA, medium chain fatty acid.
The soybean-derived fat emulsions traditionally used for parenteral nutrition contain ω6 polyunsaturated fatty acids,specifically arachnodonic and linoleic acids. Arachnodonic acid is the precursor for prostaglandins such as thromboxane-A2 and prostaglandin-E1, which are associated with platelet aggregation and inflammation. Alternately, fish oils contain fatty acids (eg eicosapentaenoic and ω 3 linolenic acids). These are precursors of another class of prostaglandins that include thromboxane-A3 which have less platelet-aggregating activity and cause less inflammation. The platelets from postoperative patients infused with fish oil-enriched soybean oil lipid emulsions for 7 days after surgery aggregated less than those administered only soybean oil lipid emulsion [56]. ω3 Fatty acids also decreased the ex-vivo production of IL-1, IL-6, IL-2 and TNF by peripheral blood mononuclear cells [57]. Oral eicosapentaenoic acid intake after surgery improved lymphocyte proliferation and natural killer cell activity [58]. There has been some interest in using ω3 fatty acids in adult respiratory distress syndrome to reduce pulmonary microvascular permeability and alveolar macrophage prostaglandin and leukotriene synthesis [59,60].
Protein metabolism
Pathophysiology
One of the hallmarks of the metabolic response to injury is catabolism (negative nitrogen balance). There is accelerated proteolysis of skeletal muscle, which provides some of the substrate for increased hepatic gluconeogenesis. Reducing the rate of hepatic gluconeogenesis with somatostatin does not decrease the rate of peripheral protein breakdown, demonstrating that the accelerated rated of hepatic glucose production is not linked to the elevated level of peripheral protein breakdown [1]. The degree of nitrogen loss is proportional to the degree of stress, and abates as the patient convalesces [61]. The increased protein breakdown is thought to be modulated only partly by the endocrine stress hormones, such as cortisol [62]. Instead, other mediators such as the cytokines TNF-α, IL-1, IL-6 and interferon-γ are involved in mediating catabolic activity. It is the balance between these catabolic hormones and anabolic hormones such as insulin and insulin-like growth factors that determine the degree of catabolism. A number of metabolic pathways may be responsible for skeletal muscle proteolysis, including the lysosomal calcium-activated, ATP-ubiquitin-dependent proteolytic pathway [63]. The liver also contributes to catabolism through the increase in clearance of α-amino nitrogen (urea). After surgery the rate of this conversion is doubled [64]. Blockade of glucagon and cortisol secretions with a combination of etomidate, somatostatin and thoracic epidural anaesthesia decreased this clearance, thus indicating roles for glucagon and possibly cortisol and afferent neural reflexes in this process [65,66].
There is conflicting data regarding skeletal muscle protein synthetic activity during stress. Some studies indicate reduced (eg 31% decrease 24 h after open cholecystectomy) skeletal muscle protein synthesis, whether saline or TPN are infused [67,68]. Others claim that the net negative nitrogen balance is due to an accelerated rate of protein breakdown that is in excess of an increased protein synthetic rate [69]. Tissues with rapidly replicating cells, such as enterocytes, immune cells and keratinocytes, exhibit reduced protein synthesis. The derangement of protein metabolism is profound. Provision of TPN after open cholecystectomy does not prevent the decline in muscle protein synthesis observed 24 h after surgery [67].
During stress there is increased hepatic synthesis of the 'acute phase' proteins such as fibrinogen, complement, immunoglobulins and C-reactive protein. Increase in these proteins is thought to lead to increased ability to fight infection. Simultaneously, there is reduced synthesis of binding proteins, such as albumin prealbumin, and transferrin.
Exogenous protein administration
Amino acids and protein are basic components of nutritional support regimens. The aim of administering exogenous protein or amino acid is to attenuate the breakdown of endogenous proteins by providing an alternate source of amino acids for gluconeogenesis and protein synthesis. Unfortunately, in the stressed state exogenously administered amino acids and protein are not well utilized and nitrogen balance remains negative well into the convalescent stage. In the acutely stressed state proteolysis is relatively unresponsive to the usual feedback mechanisms, such as the administration of exogenous glucose, protein or amino acids. In the catabolic state an intake of 1.2-1.5 g/kg per day protein/amino acid is recommended and higher amounts do not promote further nitrogen retention. Instead the added protein/amino acid is metabolized to urea, so the blood urea nitrogen may rise. Situations associated with large external losses of protein, such as extensive burns and large draining abscesses, make it necessary to increase protein/amino acid intake.
One of the important consequences of glucose and carbohydrate administration is the stimulation of insulin secretion. At lower doses insulin decreases protein breakdown by inhibiting the major catabolic pathway, the ATP-ubiquitin proteasome proteolytic pathway [70]. At higher doses it is thought also to stimulate protein synthesis. Suppression of insulin secretion during stress hormone infusion (cortisol, glucagon, epinephrine) in normal individuals increased whole-body nitrogen losses and the forearm efflux of amino acids [71]. The administration of small doses of insulin to burn patients increased skeletal muscle protein synthesis [72] and improved wound matrix formation [73]. The provision of hypocaloric parenteral nutrition with high-dose insulin improved nitrogen balance in postoperative cancer patients [74]. The anabolic effects of insulin on protein metabolism were not evident in enterally fed trauma patients. Therefore, the route of nutrient intake may play a role in the anabolic effects of insulin [75].
Alternate approaches
The inability of the usual intake of protein/amino acids to attenuate nitrogen losses significantly has led investigators to examine ways of either decreasing proteolysis or increasing protein synthesis. An initial attempt was to provide branched-chain amino acid enriched solutions. This resulted in some improvement in nitrogen balance, but no improvements in outcome. More recent attempts have focused on administering anabolic substances to reduce protein oxidation and improve protein synthesis. During the flow phase of stress concentrations of growth hormone are reduced and there is resistance to its actions [76]. Because of its anabolic properties (mediated through insulin-like growth factor-1) there has been much study of growth hormone administration in catabolic patients. The aim of administering growth hormone is to increase nitrogen retention and promote wound healing. Growth hormone administration in critically ill patients receiving nutritional support has been observed to reduce nitrogen loss and improve phosphate retention. A disadvantage of growth hormone is its diabetogenic and lipolytic properties. In the studies performed thus far with growth hormone administration in the critically ill [77,78], no definitive improvement in patient outcome was observed. Insulin growth factors (IGFs) are a family of polypeptides that regulate cell growth. Their secretion is stimulated by growth hormone and their bioavailability is modified by IGF binding proteins. IGF-1 and its binding protein, IGF binding protein-3, are both reduced during stress [79]. Like growth hormone, IGF-1 stimulates protein synthesis at lower doses, and at higher doses it also reduces proteolysis [80]. The administration of IGF-1 for 3 days decreased protein oxidation and, when administered along with its binding protein, IGF binding protein-3, attenuated catabolism in burn patients [79,81]. More study of these substances is underway.
Renal replacement therapy
Continuous renal replacement therapies, especially continuous veno-veno haemodiafiltration, have improved the treatment of critically ill patients suffering from acute renal failure. These therapies have facilitated the provision of enteral and parenteral nutritional support by allowing patients to receive a full complement of calories and amino acids/protein. This is due to the ability of continuous therapies to efficiently remove excess fluid, urea and electrolytes. These therapies also cause loss of amino acids, however (10-15 g/day [26]). This loss is reported to be unrelated to amino acid intake and related to plasma amino acid concentration, volume of dialysate effluent and filter efficiency [82]. The concentrations of most amino acids are the same in the plasma and the dialysate [83]. Therefore, the amino acid/protein content of the nutritional support should be increased. With aggressive hemodiafiltration it was possible to feed patients as much as 2.5 g/kg per day protein and achieve a positive nitrogen balance [84].
Route of administration
Pathophysiology
There has been increased emphasis on using the enteral route for nutritional support occasioned by the desire to maintain gut integrity and so lessen the translocation of bacteria from the gut [85]. Lack of enteral intake, such as occurs among postoperative and critically ill patients receiving only parenteral nutrition, is associated with small intestinal villus atrophy, decreased villus cell count and reduced mucosal thickness, but not with changes in crypt cell count [86]. Intestinal permeability, measured using the urinary lactulose : mannitol ratio, is increased [86]. Also, crypt length is increased and mucosal surface patterns change from finger-like to leaf-like microvilli [87]. These changes were reversed with enteral feeding [86,88]. Similar changes, along with activation of the lamina propria mononuclear cells and enterocytes, were seen during nutritional depletion [89,90,91]. Interestingly, there was no evidence of mucosal atrophy among patients who had received 10 days of preoperative parenteral nutrition [92]. It is thought that these anatomic changes in and of themselves do not lead to bacterial translocation, but that further insult is needed. One such insult is ischaemiareperfusion injury to the small intestines after resuscitation from an episode of shock, which may also itself change gut integrity [93]. Furthermore, in the stressed patient there is the possibility that the constantly proliferating gut tissues (epithelial and lymphoid cells) may have an inadequate supply of nutrients, further compromising gut integrity [94].
These changes in gut integrity have led many to consider whether after a systemic insult intestinal translocation of bacteria occurs in critically ill humans. This concern is based on studies of animal models that describe the translocation of gut organisms across intestinal mucosa with enhanced permeability. This bacterial translocation leads to local activation of the gut's immune inflammatory system (Peyer's patches and hepatic Kuppfer cells [95]). The released cytokines and other mediators then exacerbate the already existing systemic inflammatory response syndrome leading to multiple organ failure [96]. There is no overwhelming evidence of translocation in humans [97]. Intestinal serosal and mesenteric lymph nodes showed evidence of bacteria in 10.3% of 269 general surgery patients and in only 5% when those with inflammatory bowel disease and intestinal obstruction were excluded. These observations led to the conclusions that translocation can spontaneously occur in humans [98]. Indirect evidence comes from patients with intra-abdominal sepsis who showed reduced numbers of immunoglobulin A and M plasma cells. It was speculated that the reduced expression of these immunoglobulins might favour bacterial adhesion to enterocytes [99].
Nutritional support
This 'gut hypothesis of multiple organ failure' has led to the recommendation that enteral nutrition be started as soon as possible after surgery or in nonsurgical patients after admission to the intensive care unit. It is hoped that enteral nutrition will preserve splanchnic flow and prevent mucosal breakdown [100]. Further impetus to begin enteral nutrition early has been lower stress hormone concentrations, lower infection rates, shorter hospital stays and better survivals in some, but not all [101,102] studies in which nutrition was started within 4-24 h, and not more than 24h after admission [103,104,105]. Other studies that compared parenteral with enteral nutrition in pancreatitis, postoperative states, burns and the critically ill have indicated fewer infections and even shorter hospital stays, although many of these studies contained only a small number of patients.
A review of the literature by Lipman [106] revealed little absolute evidence that enteral nutrition is better than parenteral nutrition, with the exception of lower cost and possible reduced septic morbidity in acute trauma patients [107,108]. It is intuitive, however, that the eventual goal in critically ill patients is to feed them enterally as soon as they are able to tolerate such intake. The problem is that enteral feeding is fraught with both technical and physiological challenges. These often result in inadequate caloric intake, especially upon starting the nutrition. Therefore, it may be necessary to supplement the enteral intake with parenteral nutrition.
Alternate therapies
Glutamine has been the focus of much study because it may be important in the maintenance of gut and immunological integrity during critical illness. It is the most abundant free amino acid in the circulation and is a primary fuel for rapidly dividing cells such as enterocytes and immunocytes [108]. It is also involved in the inter-organ transport of nitrogen. Evidence from animal studies has indicated that glutamine may be an essential amino acid during critical illness and that parenteral and enteral glutamine-supplemented nutrition may prevent bacterial translocation. The data in humans are less compelling [109]. Glutamine is rather insoluble and thus is difficult to administer. Some success has been achieved infusing it as the L-amino acid, glutamine-dipeptides or alanyl-glutamate. Initial reports [110,111] indicate that in some catabolic patients glutamine-containing nutrition may improve gut structure and function, exert an anabolic effect and reduce morbidity, hospital costs, infection rates and duration of hospital stay. More studies are needed to ascertain whether glutamine administration improves outcome [112].
Access for enteral nutrition is a problematic issue due to the problems of securing a reliable route. Placement of the tip of the tube into the jejunum is considered ideal so as to avoid the often present gastric ileus and possibly prevent aspiration. There has been some disagreement as to whether it is mandatory to place the tube into the jejunum. The ideal route is a jejunostomy catheter placed during laparotomy, because feeding may begin within hours of surgery. Complication rates are comparatively low (1.5%) and most commonly involve catheter occlusion or dislodgment. Very rarely bowel necrosis or intra-abdominal leaks may occur [113]. Alternatively, a nasoenteric tube can be passed during surgery and manually manipulated into the jejunum. In patients without surgical placement, nasoenteric tubes may be placed. It is often difficult to pass such tubes from the stomach into the jejunum. Right lateral decubitus positioning and prokinetic drugs may be tried, but often placement must be performed under radiological or endoscopic guidance [114].
Pancreatitis
The traditional treatment of both acute pancreatitis and acute exacerbation of chronic pancreatitis has been exclusion of all oral intake and institution of nasogastric suction. Nutritional support therefore has been delivered via the parenteral route. Recently, there have been reports of successful jejunal feedings with concomitant nasogastric decompression among patients with moderate pancreatitis (≥3, Ranson criteria; APACHE II score of ≥10, major organ failure and/or pancreatic necrosis [115,116]). A small randomized trial [117] found fewer total and septic complications among patients who received parenteral nutrition. Further study using larger randomized trials is needed to confirm these findings.
The bottom line
The ultimate question is whether nutritional support improves patient outcome, and, specifically, whether nutritional support lowers mortality and mobility and reduces intensive care unit and hospital durations of stay. Unfortunately, there are no definitive large multicenter studies that have examined this issue. There are a few meta-analyses and large studies that have examined selected aspects of nutritional support. A recent study [118] claimed that starting enteral nutrition within 3 days of intensive care unit admission reduces duration of mechanical ventilation and improves outcome. A meta-analysis using 26 studies [119] examined the relationship between TPN and standard care (usual oral diet plus intravenous dextrose), and complications and mortality rates in surgical and critically ill patients. TPN had no effect on mortality and only lowered complication rates in malnourished patients. Studies published before 1988 had significant treatment effect, whereas those published after 1989 did not. Complication rates, but not mortality, were lower among patients who did not receive lipids. Another meta-analysis of 11 randomized trials of both critically ill and gastrointestinal malignancy patients [120] examined the issue of immunonutrition (ie enteral nutrition supplemented with arginine, ω3 fatty acids and purines). This formulation is purported to improve immune function and showed significant reductions in the risk of developing infectious complications and in hospital stay. No effect on mortality was observed.
Conclusion
There is a need to acquire more knowledge through large multicenter studies of the cost-benefit and cost-effectiveness of nutritional support in the critically ill, and specifically to clarify the issues of TPN versus enteral nutrition and the use of specialized nutrients. Until such studies are performed and the results analyzed, nutritional support should continue to be viewed as an important adjunctive therapy. It should be used in a manner that maximizes positive effects and minimizes detrimental ones.
References
- Wolfe RR. Herman Award Lecture 1996: relation of metabolic studies to clinical nutrition: the example of burn injury. Am J Clin Nutr. 1996;64:800–808. doi: 10.1093/ajcn/64.5.800. [DOI] [PubMed] [Google Scholar]
- Thorell A, Nygren J, Hirshman MF, et al. Surgery-induced insulin resistance in human patients: relation to glucose transport and utilization. Am J Physiol. 1999;276:E754–E761. doi: 10.1152/ajpendo.1999.276.4.E754. [DOI] [PubMed] [Google Scholar]
- Green CJ, Campbell IT, O'Sullivan E, et al. Septic patients in multiple organ failure can oxidize infused glucose, but non-oxidative disposal (storage) is impaired. Clin Sci (Colch) 1995;89:601–609. doi: 10.1042/cs0890601. [DOI] [PubMed] [Google Scholar]
- Chiolero R, Revelly JP, Tappy L. Energy metabolism in sepsis and injury. Nutrition. 1997;13 (suppl 9):45S–51S. doi: 10.1016/s0899-9007(97)00205-0. [DOI] [PubMed] [Google Scholar]
- Strommer L, Permert J, Arnelo U, et al. Skeletal muscle insulin resistance after trauma: insulin signaling and glucose transport. . Am J Physiol. 1998;275:E351–E358. doi: 10.1152/ajpendo.1998.275.2.E351. [DOI] [PubMed] [Google Scholar]
- Thorell A, Efendic S, Gutniak M, Haggmark T, Ljungvist O. Insulin resistance after abdominal surgery. Br J Surg. 1994;81:59–63. doi: 10.1002/bjs.1800810120. [DOI] [PubMed] [Google Scholar]
- Hawthorne GC, Ashworth L, Albertic KG. The effect of laparoscopic cholecystectomy on insulin sensitivity. Horm Metab Res. 1994;26:474–477. doi: 10.1055/s-2007-1001736. [DOI] [PubMed] [Google Scholar]
- Brandi LS, Santoro D, Natali A, et al. Insulin resistance of stress: sites and mechanisms. Clin Sci. 1993;85:525–535. doi: 10.1042/cs0850525. [DOI] [PubMed] [Google Scholar]
- Wolfe RR, Durkot MJ, Allsop JR, Burke JF. Glucose metabolism in severely burned patients. Metabolism. 1979;28:1031–1039. doi: 10.1016/0026-0495(79)90007-6. [DOI] [PubMed] [Google Scholar]
- Nelson KM, Long CL, Bailey R, Smith RJ, Laws HL, Blakemeore WS. Regulation of glucose kinetics in trauma patients by insulin and glucagon. Metabolism. 1992;41:68–75. doi: 10.1016/0026-0495(92)90193-e. [DOI] [PubMed] [Google Scholar]
- Aarsland A, Chinkes D, Wolfe RR. Hepatic and whole body fat synthesis in humans during carbohydrate overfeeding. Am J Clin Nutr. 1997;65:1774–1782. doi: 10.1093/ajcn/65.6.1774. [DOI] [PubMed] [Google Scholar]
- Stubbs RJ, Prentice AM, James WP. Carbohydrates andenergy balance. Ann NY Acad Sci. 1997;819:44–69. doi: 10.1111/j.1749-6632.1997.tb51798.x. [DOI] [PubMed] [Google Scholar]
- Jequier E. Carbohydrates as a source of energy. Am J Clin Nutr. 1994;59 (suppl):682S–685S. doi: 10.1093/ajcn/59.3.682S. [DOI] [PubMed] [Google Scholar]
- Hellerstein MK. De novo lipogenesis in humans: metabolic and regulatory aspects. Eur J Clin Nutr. 1999;53 (suppl):S52–S65. doi: 10.1038/sj.ejcn.1600744. [DOI] [PubMed] [Google Scholar]
- Guenst JM, Nelson LD. Predictors of total parenteral nutrition-induced lipogenesis. Chest. 1994;105:553–559. doi: 10.1378/chest.105.2.553. [DOI] [PubMed] [Google Scholar]
- Tappy L, Schwarz JM, Schneiter P, et al. Effects of isoenergetic glucose-basedor lipid-based parenteral nutrition on glucose metabolism, de novo lipogenesis, and respiratory gas exchanges in critically ill patients. Crit Care Med. 1998;26:860–867. doi: 10.1097/00003246-199805000-00018. [DOI] [PubMed] [Google Scholar]
- Prins JB, Niesler CU, Winterford CM, et al. Tumor necrosis factor-alpha induces apoptosis of human adipose cells. . Diabetes. 1997;46:1939–1944. doi: 10.2337/diab.46.12.1939. [DOI] [PubMed] [Google Scholar]
- Delikat SE, Galvani DW, Zuzel M. The metabolic effects of interleukin 1 beta on human bone marrow adipocytes. Cytokine . 1995;7:3387–3343. doi: 10.1006/cyto.1995.0043. [DOI] [PubMed] [Google Scholar]
- Wolfe RR. Substrate utilization/insulin resistance in sepsis/trauma. Ballieres Clin Endocrinol Metab. 1997;11:645–657. doi: 10.1016/s0950-351x(97)80926-3. [DOI] [PubMed] [Google Scholar]
- Fellander G, Nordenstrom J, Ungerstedt U, Arner P, Bolinder J. Influence of operation on glucose metabolism and lipolysis in human adipose tissue: a microdialysis study. Eur J Surg. 1994;160:87–95. [PubMed] [Google Scholar]
- Jeevanandam M, Shamos RF, Petersen SR. Substrate efficacy in early nutrition support of the critically ill multiple trauma victims. . J Parenterol Enterol Nutr. 1992;16:511–520. doi: 10.1177/0148607192016006511. [DOI] [PubMed] [Google Scholar]
- Watters JM, Kirkpatrick SM, Hopbach D, Norris SB. Aging exaggerates the blood glucose response to total parenteral nutrition. . Can J Surg. 1996;39:481–485. [PMC free article] [PubMed] [Google Scholar]
- Rosemarin DK, Wardlaw GM, Mirtallo J. Hyperglycemia associated with high, continuous infusion rates of total parenteral nutrition dextrose. . Nutr Clin Pract. 1996;11:151–156. doi: 10.1177/0115426596011004151. [DOI] [PubMed] [Google Scholar]
- Guenst JM, Nelson LD. Predictors of total parenteral nutrition-induced lipogenesis. Chest. 1994;105:553–559. doi: 10.1378/chest.105.2.553. [DOI] [PubMed] [Google Scholar]
- Schloerb PR, Henning JF. Patterns and problems of adult total parenteral nutrition use in US academic medical centers. Arch Surg. 1998;133:7–12. doi: 10.1001/archsurg.133.1.7. [DOI] [PubMed] [Google Scholar]
- Frankenfield DC, Reynolds HN. Nutritional effect of continuous hemofiltration. Nutrition. 1995;11:388–393. [PubMed] [Google Scholar]
- Monaghan R, Watters JM, Clancey SM, Moulton SB, Rabin EZ. Uptake of glucose during continuous arteriovenous hemofiltration. . Crit Care Med. 1993;21:1159–1163. doi: 10.1097/00003246-199308000-00014. [DOI] [PubMed] [Google Scholar]
- Schricker T, Gross G, Wolfel R, Georgieff M. Enhancement of fatty acid mobilization and oxidation by glucose-xylitol compared to glucose alone in posttraumatic and septic patients. Nutr Hosp. 1995;10:13–18. [PubMed] [Google Scholar]
- Singer P, Bursztein S, Kirvela O, et al. Hypercaloric glycerol in injured patients. Surgery. 1992;112:509–514. [PubMed] [Google Scholar]
- Kupari LJ, Yki-Javinen H. Free fatty acid kinetics and oxidation in congestive heart failure. Am J Cardiol. 1998;81:45–50. doi: 10.1016/s0002-9149(97)00804-7. [DOI] [PubMed] [Google Scholar]
- Fellander G, Nordenstrom J, Tjader I, Bolinder J, Arner P. Lipolysis during abdominal surgery. J Clin Endocrinol Metab . 1994;78:150–155. doi: 10.1210/jcem.78.1.8288698. [DOI] [PubMed] [Google Scholar]
- Herndon DN, Nguyen TT, Wollfe RR, et al. Lipolysis in burned patients is stimulated by the beta-2-receptor for catecholamines. Arch Surg. 1994;129:1301–1305. doi: 10.1001/archsurg.1994.01420360091012. [DOI] [PubMed] [Google Scholar]
- Perea A, Clemente F, Martinell J, et al. Physiological effect of glucagon in human isolated adipocytes. Horm Metab Res . 1995;27:372–375. doi: 10.1055/s-2007-979981. [DOI] [PubMed] [Google Scholar]
- Miles JM. Lipid fuel metabolism in health and disease. . Curr Opin Gen Surg. 1993:78–84. [PubMed] [Google Scholar]
- Doerrler W, Feingold KR, Grunfeld C. Cytokines induce catabolic effects in cultured adipocytes by multiple mechanisms. Cytokine. 1994;6:478–484. doi: 10.1016/1043-4666(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Carey GB. Mechanisms regulating adipocyte lipolysis. . Adv Exp Med Biol. 1998;441:157–170. doi: 10.1007/978-1-4899-1928-1_15. [DOI] [PubMed] [Google Scholar]
- Granneman JG. Why do adipocytes make the beta 3 adrenergic receptor? Cell Signal. 1995;7:9–15. doi: 10.1016/0898-6568(94)00066-k. [DOI] [PubMed] [Google Scholar]
- Townsend RR, Klein S, Wolfe RR. Changes in lipolytic sensitivity following repeated epinephrine infusion in humans. Am J Physiol. 1994;266:E155–E160. doi: 10.1152/ajpendo.1994.266.2.E155. [DOI] [PubMed] [Google Scholar]
- Arner P. Differences in lipolysis between human subcutaneous and omental tissues. Ann Med. 1995;27:435–438. doi: 10.3109/07853899709002451. [DOI] [PubMed] [Google Scholar]
- Schricker T, Berroth A, Pfeiffer U, et al. Assessment of perioperative glycerol metabolism by stable isotope tracer technique. . Nutrition. 1997;13:191–195. doi: 10.1016/s0899-9007(96)00400-5. [DOI] [PubMed] [Google Scholar]
- Breitenstein E, Chiolero RL, Jequier E, et al. Effects of beta-blockade on energy metabolism following burns. Burns. 1990;16:259–264. doi: 10.1016/0305-4179(90)90136-k. [DOI] [PubMed] [Google Scholar]
- Goldstein SA, Elwyn DH. The effects of injury and sepsis on fuel utilization. Ann Rev Nutr. 1989;9:445–473. doi: 10.1146/annurev.nu.09.070189.002305. [DOI] [PubMed] [Google Scholar]
- Sato N, Oyamatsu M, Tsukada K, et al. Serial changes in contribution of substrates to energy expenditure after thoracic esophagectomy for cancer. Nutrition. 1997;13:100–103. doi: 10.1016/s0899-9007(96)00382-6. [DOI] [PubMed] [Google Scholar]
- Ferezou J, Bach AC. Structure and metabolic fate of triacylglycerol and phospholipid-rich particles of commercial parenteral fat emulsions. Nutrition. 1999;15:44–50. doi: 10.1016/S0899-9007(98)00130-0. [DOI] [PubMed] [Google Scholar]
- Abe M, Kawano M, Tishiro T, et al. Catabolism of lipoprotein-x induced by infusion of 10% fat emulsion. Nutrition . 1997;13:417–421. doi: 10.1016/s0899-9007(97)91279-x. [DOI] [PubMed] [Google Scholar]
- Druml W, Fischer M, Ratheiser K. Use of intravenous lipids in critically ill patients with sepsis without and with hepatic failure. . J Parenterol Enterol Nutr. 1998;22:217–223. doi: 10.1177/0148607198022004217. [DOI] [PubMed] [Google Scholar]
- Stouthard JM, Endert E, Romiju Jam Sauerwein HP. Infusion of long-chain or medium-chain triglycerides inhibits peripheral glucose metabolism in men. J Parenterol Enterol Nutr. 1994;18:436–441. doi: 10.1177/0148607194018005436. [DOI] [PubMed] [Google Scholar]
- Haesler E, Schneiter P, Temler E, Jequier E, Tappy L. Effects of infused amino acids on glucose metabolism in healthy lean humans. . Int J Obes Relat Metab Dis. 1994;18:307–312. [PubMed] [Google Scholar]
- Wolfe BM, Klein S, Peters EJ, Schmdt BF, Wolfe RR. Effects of elevated free fatty acids on glucose oxidation in normal humans. Metabolism. 1988;37:323–329. doi: 10.1016/0026-0495(88)90131-x. [DOI] [PubMed] [Google Scholar]
- Tissot S, Normand S, Khalfallah Y, et al. Effects of a continuous lipid infusion on glucose metabolism in critically ill. . Am J Physiol. 1995;269:E753–E758. doi: 10.1152/ajpendo.1995.269.4.E753. [DOI] [PubMed] [Google Scholar]
- Nordenstrom J, Carpentier YA, Askanazi J, et al. Metabolic utilization of intravenous fat emulsion during total parenteral nutrition. Ann Surg. 1982;196:221–231. doi: 10.1097/00000658-198208000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waitzberg DL, Bellinati-Pres R, Salgado MM, et al. Effect of total parenteral nutrition with different lipid emulsions on human monocyte and neutrophil functions. Nutrition. 1997;13:128–132. doi: 10.1016/s0899-9007(96)00386-3. [DOI] [PubMed] [Google Scholar]
- Battistella FD, Widergren JT, Anderson JT, et al. A prospective, randomized trial of intravenous fat emulsion administration in trauma victims requiring total parenteral nutrition. J Trauma . 1997;43:52–60. doi: 10.1097/00005373-199707000-00013. [DOI] [PubMed] [Google Scholar]
- Chan S, McCowen KC, Blackburn GL. Nutritional management in the ICU. Chest. 1999;115 (suppl):145S–148S. doi: 10.1378/chest.115.suppl_2.145s. [DOI] [PubMed] [Google Scholar]
- Bellantone R, Bossola M, Carriero C, et al. Structured versus long-chain triglycerides: a safety, tolerance, and efficacy randomized study in colorectal surgical patients. J Parenterol Enterol Nutr. 1999;23:123–127. doi: 10.1177/0148607199023003123. [DOI] [PubMed] [Google Scholar]
- Roulet M, Frascarolo P, Pilet M, Chapuis G. Effects of intravenously infused fish oil on platelet fatty acid phospholipid composition and on platelet function in postoperative trauma. J Parenterol Enterol Nutr. 1997;21:296–301. doi: 10.1177/0148607197021005296. [DOI] [PubMed] [Google Scholar]
- Calder PC. n-3 polunsaturated fatty acids and cytokine production in health and disease. Ann Nutr Metab. 1997;41:203–234. doi: 10.1159/000177997. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Tashiro T, Yamamori H, Takagi K, Morishima Y. Effects of soybean oil emulsion and eicosapentanoic acid on stress response and immune function after a severely stressful operation. . Ann Surg. 1999;229:255–261. doi: 10.1097/00000658-199902000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso P, Whelan J, DeMichele SJ, et al. Effects of eicosapentaenoic and gamma-linolenic acid on lung permeability and alveolar macrophage eicosanoid synthesis in endotoxic rats. Crit Care Med . 1997;25:523–532. doi: 10.1097/00003246-199703000-00024. [DOI] [PubMed] [Google Scholar]
- Planas M, Masclans JR, Iglesia R, et al. Eicosanoids and fat emulsions in acute respiratory distress syndrome patients. . Nutrition. 1997;13:202–205. doi: 10.1016/s0899-9007(96)00402-9. [DOI] [PubMed] [Google Scholar]
- Tashiro T, Yamamori H, Takagi K, Morishima Y, Nakajima N. Effect of severity of stress on whole-body protein kinetics in surgical patients receiving parenteral nutrition. Nutrition. 1996;12:763–765. doi: 10.1016/s0899-9007(96)00214-6. [DOI] [PubMed] [Google Scholar]
- Brown JA, Gore DC, Jahoor F. Catabolic hormones alone fail to reproduce the stress-induced efflux of amino acids. Arch Surg. 1994;129:819–824. doi: 10.1001/archsurg.1994.01420320041007. [DOI] [PubMed] [Google Scholar]
- Mansoor O, Beaufrere B, Boirie Y, et al. Increased mRNA levels for components of the lysosomal, Ca++ activated, and ATP-ubiquitin-dependent proteolytic pathways in skeletal muscle from head trauma patients. Proc Natl Acad Sci USA. 1996;493:2714–2718. doi: 10.1073/pnas.93.7.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindorff HA. The hepatic catabolic stress response: hormonal regulation of urea synthesis after surgery. Dan Med Bull. 1993;40:224–234. [PubMed] [Google Scholar]
- Heindorff H, Schulze S, Mogensen T, et al. Hormonal and neural blockade prevent the postoperative increase in amino acid clearance and urea synthesis. Surgery. 1992;111:543–550. [PubMed] [Google Scholar]
- Heindorff H, Billesbolle P, Pedersen SL, Hansen R, Vistrup H. Somatostatin prevents the postoperative increases in plasma amino acid clearance and urea synthesis after elective cholecystectomy. . Gut. 1995;36:766–770. doi: 10.1136/gut.36.5.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essen P, McNurlan MA, Sonnenfeld T, et al. Muscle protein synthesis after operation: effects of intravenous nutrition. . Eur J Surg. 1993;159:195–200. [PubMed] [Google Scholar]
- Tjader I, Essen P, Thorne A, et al. Muscle protein synthesis rate decreases 24 hours after abdominal surgery irrespective of total parenteral nutrition. J Parenterol Enterol Nutr. 1996;20:135–138. doi: 10.1177/0148607196020002135. [DOI] [PubMed] [Google Scholar]
- Gore DC, Jahoor F, Wolfe RR, Herndon DN. Acute response of human muscle protein to catabolic hormones. Ann Surg. 1993;218:679–684. doi: 10.1097/00000658-199321850-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grizard J, Dardevet D, Balage M, et al. Insulin action on skeletal muscle protein metabolism during catabolic states. . Reprod Nutr Dev. 1999;39:61–74. doi: 10.1051/rnd:19990104. [DOI] [PubMed] [Google Scholar]
- Bessey PQ, Lowe KA. Early hormonal changes affect the catabolic response to trauma. Ann Surg. 1993;218:476–491. doi: 10.1097/00000658-199310000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrando AA, Chinkes DL, Wolfe SE, et al. A submaximal dose of insulin promotes net skeletal muscle protein synthesis in patients with severe burns. Ann Surg. 1999;229:11–18. doi: 10.1097/00000658-199901000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre EJ, Barrow RE, Hawkins HK, et al. Effects of insulin on wound healing. J Trauma. 1998;44:342–345. doi: 10.1097/00005373-199802000-00019. [DOI] [PubMed] [Google Scholar]
- Pearlstone DB, Wolf RF, Berman RS, Burt M, Brennan MF. Effect of systemic insulin on protein kinetics in postoperative cancer patients. . Ann Surg Oncol. 1994;1:321–332. doi: 10.1007/BF02303571. [DOI] [PubMed] [Google Scholar]
- Clements RH, Hayes CA, Gibbs ER, et al. Insulin's anabolic effect is influenced by route of administration of nutrients. . Arch Surg. 1999;134:274–277. doi: 10.1001/archsurg.134.3.274. [DOI] [PubMed] [Google Scholar]
- Jenkins RC, Ross RJ. Acquired growth hormone resistance in catabolic states. Ballieres Clin Endocrinol Metab. 1996;10:411–419. doi: 10.1016/s0950-351x(96)80545-3. [DOI] [PubMed] [Google Scholar]
- Koea JB, Breier BH, Douglas RG, Gluckman PD, Shaw JH. Anabolic and cardiovascular effects of recombinant human growth hormone in surgical patients with sepsis. Br J Surg. 1996;83:196–202. [PubMed] [Google Scholar]
- Voerman BJ, Srack van Schijndel RJ, de Boer H, et al. Effects of human growth hormone on fuel utilization and mineral balance in critically ill patients on full intravenous nutritional support. . J Crit Care. 1994;9:143–150. doi: 10.1016/0883-9441(94)90010-8. [DOI] [PubMed] [Google Scholar]
- Herndon DN, Ramzy PI, DebRoy MA, et al. Muscle protein catabolism after severe burn: effects of IGF-1/IGFBP-3 treatment. . Ann Surg. 1999;229:713–722. doi: 10.1097/00000658-199905000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miers WR, Barrett EJ. The role of insulin and other hormones in the regulation of amino acid and protein metabolism in humans. J Basic Clin Physiol Pharmacol. 1998;9:235–253. doi: 10.1515/jbcpp.1998.9.2-4.235. [DOI] [PubMed] [Google Scholar]
- Cioffi WG, Gore DC, Rue LW, III, et al. Insulin-like growth factor-1 lowers protein oxidation in patients with thermal injury. . Ann Surg. 1994;220:310–319. doi: 10.1097/00000658-199409000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankenfield DC, Badellino MM, Reynolds HN, et al. Amino acid loss and plasma concentration during continuous hemofiltration. J Parenterol Enterol Nutr. 1993;17:551–561. doi: 10.1177/0148607193017006551. [DOI] [PubMed] [Google Scholar]
- Novak I, Sramek V, Pittrova H, et al. Glutamine and other amino acid losses during continuous venoveno hemofiltration. . Artif Organs. 1997;21:359–363. doi: 10.1111/j.1525-1594.1997.tb00731.x. [DOI] [PubMed] [Google Scholar]
- Bellomo R, Seacombe J, Daskalakis M, et al. A prospective comparative study of moderate versus high protein intake for critically ill patients with acute renal failure. Ren Fail. 1997;19:111–120. doi: 10.3109/08860229709026265. [DOI] [PubMed] [Google Scholar]
- Archer SB. Current uses and abuses of total parenteral nutrition. Adv Surg. 1996;26:165–189. [PubMed] [Google Scholar]
- Buchman AL, Moukarzel AA, Bhuta S, et al. Parenteral nutrition is associated with intestinal morphologic and functional changes in humans. J Parenterol Enterol Nutr. 1995;19:453–460. doi: 10.1177/0148607195019006453. [DOI] [PubMed] [Google Scholar]
- Sedman PC, MacFie J, Palmer MD, Mitchell CJ, Sagar PM. Preoperative total parenteral nutrition is not associated with mucosal atrophy or bacterial translocation in humans. Br J Surg . 1995;82:1663–1667. doi: 10.1002/bjs.1800821226. [DOI] [PubMed] [Google Scholar]
- Hadfield RJ, Sinclair DG, Houldsworth PE, Evans TW. Effects of enteral and parenteral nutrition on gut mucosal permeability in the critically ill. Am J Respir Crit Care Med. 1995;152:1545–1548. doi: 10.1164/ajrccm.152.5.7582291. [DOI] [PubMed] [Google Scholar]
- Van der Hulst RR, von Meyenfeldt MF, van Kreel BK, et al. Gut permeability, intestinal morphology, and nutritional depletion. Nutrition. 1998;14:1–6. doi: 10.1016/S0899-9007(97)00385-7. [DOI] [PubMed] [Google Scholar]
- Welsh FK, Farmery SM, MacLennan K, et al. Gut barrier function in malnourished patients. Gut. 1998;42:396–401. doi: 10.1136/gut.42.3.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds JV, O'Farrelly C, Feighery C, et al. Impaired gut barrier function in malnourished patients. Br J Surg. 1996;83:1288–1291. [PubMed] [Google Scholar]
- Groos S, Hunefeld G, Luciano L. Parenteral versus enteral nutrition: morphological changes in human adult intestinal mucosa. . J Submicrosc Cytol Pathol. 1996;26:61–74. [PubMed] [Google Scholar]
- Kong SE, Blennerhassett LR, Heel KA, McCauley RD, Hall JC. Ischaemia-reperfusion injury in the intestine. Aust NZ J Surg. 1998;68:554–561. doi: 10.1111/j.1445-2197.1998.tb02099.x. [DOI] [PubMed] [Google Scholar]
- McBurney MI. The gut: central organ in nutrient requirements and metabolism. Can J Physiol Pharmacol. 1994;72:260–265. doi: 10.1139/y94-040. [DOI] [PubMed] [Google Scholar]
- Hartung T, Sauer A, Hermann C, Brockhaus F, Wendel A. Overactivation of the immune system by translocated bacteria and bacterial products. Scand J Gastroenterol Suppl. 1997;222:98–99. doi: 10.1080/00365521.1997.11720729. [DOI] [PubMed] [Google Scholar]
- Swank GM, Deitch EA. Role of the gut in multiple organ failure: bacterial translocation and permeability changes. World J Surg. 1996;20:411–417. doi: 10.1007/s002689900065. [DOI] [PubMed] [Google Scholar]
- Lipman TO. Bacterial translocation and enteral nutrition in humans: an outsider looks in. J Parenterol Enterol Nutr. 1995;19:156–165. doi: 10.1177/0148607195019002156. [DOI] [PubMed] [Google Scholar]
- Sedman PC, MacFie J, Sagar P, et al. The prevalence of gut translocation in humans. Gastroenterology. 1994;107:643–649. doi: 10.1016/0016-5085(94)90110-4. [DOI] [PubMed] [Google Scholar]
- Coutinho HB, Robalinho TI, Coutinho VB. Intra-abdominal sepsis: an immunocytochemical study of the small intestine mucosa. J Clin Pathol. 1997;50:294–298. doi: 10.1136/jcp.50.4.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost P, Bihari D. The route of nutritional support in the critically ill: physiological and economic considerations. . Nutrition. 1997;13 (suppl):58S–63S. doi: 10.1016/s0899-9007(97)00207-4. [DOI] [PubMed] [Google Scholar]
- Eyer SD, Micon LT, Konstantinides FN, et al. Early enteral feeding does not attenuate metabolic response after blunt trauma. . J Trauma. 1993;34:639–644. doi: 10.1097/00005373-199305000-00005. [DOI] [PubMed] [Google Scholar]
- Heslin MJ, Latkany L, Brooks AD, et al. A prospective, randomized trial of early enteral feeding after resection of upper gastrointestinal malignancy. Ann Surg. 1997;226:567–577. doi: 10.1097/00000658-199710000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TW, Zadrozny DB, Harrington T. The benefits of early jejunal hyperalimentation in the head-injured patient. . Neurosurgery. 1989;25:729–725. doi: 10.1097/00006123-198911000-00007. [DOI] [PubMed] [Google Scholar]
- Beiei-Halgersen R, Boesby S. Influence of postoperative enteral nutrition on postsurgical infections. Gut. 1996;39:833–835. doi: 10.1136/gut.39.6.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr CS, Ling KDE, Boulos P, et al. Randomized trial of safety of efficacy of intermediate postoperative enteral feeding in patients undergoing gastrointestinal resection. . BMJ. 1996;312:869–871. doi: 10.1136/bmj.312.7035.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipman TO. Grains or veins: is enteral nutrition realy better than parenteral nutrition? A look at the evidence. J Parenterol Enterol Nutr. 1998;22:167–182. doi: 10.1177/0148607198022003167. [DOI] [PubMed] [Google Scholar]
- Moore FA, Feliciano DV, Andrassy RJ, et al. Early enteral feeding, compared with parenteral, reduces postoperative septic complications. The results of a meta-analysis. Ann Surg. 1992;216:172–183. doi: 10.1097/00000658-199208000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JC, Heel K, McCauley R. Glutamine. Br J Surg . 1996;83:305–312. doi: 10.1002/bjs.1800830306. [DOI] [PubMed] [Google Scholar]
- Buchman AL. Glutamine: is it a conditionally required nutrient for the human gastrointestinal system? J Am Coll Nutr. 1996;15:199–205. doi: 10.1080/07315724.1996.10718590. [DOI] [PubMed] [Google Scholar]
- Ziegler TR, Szeszycki EE, Estivariz CF, Puckett AB, Leader LM. Glutamine: from basic science to clinical applications. . Nutrition. 1996;12 (suppl):S68–S70. doi: 10.1016/s0899-9007(96)00019-6. [DOI] [PubMed] [Google Scholar]
- Jones C, Palmer TE, Griffiths RD. Randomized clinical outcome study of critically ill patients given glutamine-supplemented enteral nutrition. Nutrition. 1999;15:108–115. doi: 10.1016/S0899-9007(98)00172-5. [DOI] [PubMed] [Google Scholar]
- Sacks GS. Glutamine supplementation in catabolic patients. . Ann Pharmacother. 1999;33:348–354. doi: 10.1345/aph.18200. [DOI] [PubMed] [Google Scholar]
- Myers JG, Page CP, Stewart RM, et al. Complications of needle catheter jejunostomy in 2,022 consecutive applications. Am J Surg. 1995;170:547–551. doi: 10.1016/s0002-9610(99)80013-0. [DOI] [PubMed] [Google Scholar]
- Coates NE, MacFadyen BV., Jr Endoscopic jejunal access for enteral feeding. Am J Surg. 1995;169:627–628. doi: 10.1016/s0002-9610(99)80235-9. [DOI] [PubMed] [Google Scholar]
- McClave SA, Snider H, Owens N, Sexton LK. Clinical nutrition in pancreatitis. Dig Dis Sci. 1997;42:2035–2044. doi: 10.1023/a:1018806131924. [DOI] [PubMed] [Google Scholar]
- Simpson WG, Marsano L, Gates L. Enteral nutritional support in acute alcoholic pancreatitis. J Am Coll Nutr. 1995;14:662–665. doi: 10.1080/07315724.1995.10718557. [DOI] [PubMed] [Google Scholar]
- Kalfarentzos F, Kehagias J, Mead N, Nokkinis K, Gogos CA. Enteral nutrition is superior to parenteral nutrition in severe acute pancreatitis: results of a randomized prospective trial. Br J Surg. 1997;87:1665–1669. [PubMed] [Google Scholar]
- Nutrition intervention in ICU improves outcomes. Health Benchmarks. 1998;5:175–176. [PubMed] [Google Scholar]
- Heyland DK, MacDonald S, Keele L, Drover JW. Total parenteral nutrition in the critically ill patient. JAMA. 1998;280:2013–2019. doi: 10.1001/jama.280.23.2013. [DOI] [PubMed] [Google Scholar]
- Heys SD, Walker LG, Smith I, Eremin O. Enteral nutritional supplementation with key nutrients in patients with critical illness and cancer: a meta-analysis of randomized controlled clinical trials. Ann Surg. 1999;229:467–477. doi: 10.1097/00000658-199904000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]