

Abstract
Multiple organ failure is the major problem in intensive care patients. The failure of the organ 'immune system' is frequently overlooked, however. In this issue the article by Angele and Faist provides an excellent review of the topic. Deactivation of monocyte and lymphocyte functions seems to play a key role in post-traumatic immunodepression. To accompany that review we summarize our knowledge of the mechanisms of deactivation. Stress response, lipopolysaccharide translocalization and tissue injury contribute to 'immunoparalysis'. Recently developed, well standardized assays now allow us to monitor the immune system like other organ functions and opens new approaches for therapeutic interventions.
Keywords: interleukin-10, immunodepression, immunoparalysis, monocyte deactivation, stress response
Introduction
Sepsis and septic multiple organ failure are leading causes of morbidity and death in critically ill patients. Alterations to the patient's immune system, with an excessive systemic inflammatory response on one hand and paralysis of cell-mediated immunity on the other, appear to be the key elements in the pathogenesis of multiple organ failure and susceptibility to infection. Thus far, immune-based interventions have met with limited success, at least in phase III trials. A better understanding of the immunopathogenesis of multiple organ failure is needed if we are to develop new therapeutic strategies.
During the past few years our knowledge in the field of sepsis improved through the use of new animal models that are closer to the human situation than were their predecessors, and as a result of novel immune diagnostic techniques. The paper by Angele and Faist [1] focuses on the effects of blood loss and surgical injury on cell-mediated responses, and it provides a comprehensive review on this important issue. Those authors themselves significantly contributed to our current 'state of the art' knowledge.
The studies summarized in that review indicate that injury, trauma and blood loss result in a marked suppression in cell-mediated immunity that is associated with an increased susceptibility to wound infection and sepsis. In particular, the alterations in monocyte/macrophage and lymphocyte function are addressed. Monocytes from patients after blood loss, trauma and major surgery frequently exhibit depressed ex vivo secretion of tumour necrosis factor (TNF), IL-12 and other cytokines in response to lipopolysaccharide, and a downregulation in HLA-DR expression and antigen-presenting capacity. In addition, T cells, in particular type 1 cytokine-secreting T cells, show functional abnormalities. In some clinical situations an expansion of type 2 cytokine-secreting CD8+ T cells was observed. Moreover, B-cell deficiency was detected.
In addition, the review notes important differences between sexes in altered immune responsiveness following trauma, injury and blood loss. It is well established that male critically ill patients are more susceptible to infections and sepsis, and have a higher mortality than do female patients. By using murine models, it was demonstrated that sex hormones and dehydroepiandrosterone play important roles in sex-specific immune responsiveness following blood loss, trauma and injury.
On the basis of the well established observation of immunodeficiency following trauma, blood loss and major surgery, three questions arise. What are the mechanisms of immune deactivation? Do we have standardized and validated assays to detect the immunodeficiency? Finally, are there any therapeutic options to reverse the immune deactivation?
Mechanisms of immune deactivation
Activation of the stress response plays an important role in downregulating systemic immune responsiveness following trauma, injury and blood loss (Fig. 1). The regulatory role of the hypothalamic-pituitary-adrenal axis, which can stimulate corticosteroid release, is well established. Recent data [2,3] show that the sympathetic nerve system and the vagus are also involved in the regulation of immune responsiveness. The three systems downregulate monocyte/macrophage proinflammatory (e.g. TNF release) and antigen-presenting functions both directly and indirectly by induction of immunomodulatory cytokines (e.g. IL-10). IL-10 also deactivates T cells, in particular type 1 cytokine-secreting T cells.
Figure 1.
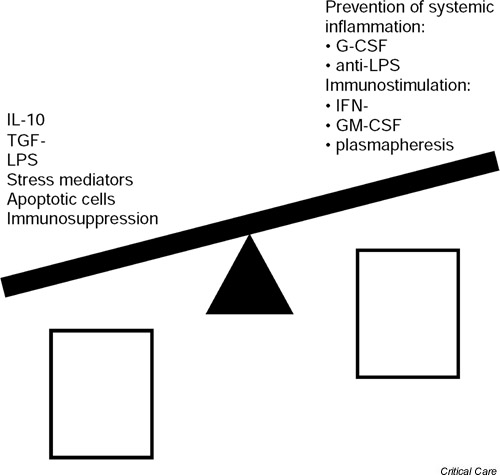
Regulation of monocytic HLA-DR, tumour necrosis factor (TNF) secretion and antigen-presenting cell (APC) activity. G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte/macrophage colony-stimulating factor; IFN, interferon; LPS, lipopolysaccharide; TGF, transforming growth factor.
This interaction between immune and central nervous systems is important for preventing excessive inflammatory reactions in intensive care unit patients. If the response is transient, then the beneficial effect dominates. However, extensive systemic inflammation (e.g. following endotoxin translocalization or severe infection) leads to a strong and long-lasting activation of the immune deactivating stress axis, which increases susceptibility to infection. In addition, high levels of corticosteroids and catecholamines induce apoptosis of lymphocytes, resulting in defects in the adaptive immune system. Moreover, a strong inflammatory response of monocyte/macrophages also directly deactivates these cells via negative feedback mechanisms (endotoxin desensitization, TNF-induced IL-10 and IL-1 receptor antagonist release, etc.). Inflammation- and hypoxia-related tissue injury results in release of apoptotic cells, which are taken up by monocyte/macrophages via a receptor that deactivates their inflammatory and antigen-presenting pathways.
In summary, the greater the inflammation and injury, the greater the counter-regulation.
Measurement of immune responsiveness
During the past few decades several assays for measurement of immune responsiveness have been reported. A problem, however, is standardization of the assays. Many of the assays developed thus far do not fulfill the criteria of a modern diagnostic assay. As suggested in the review [1], measures of monocyte/macrophage and T-cell function play key roles in diagnosis. During the past few years some well standardized assays for detecting monocytic cytokine secretion (whole blood assay with semi-automatic cytokine measurement), monocytic HLA-DR and CD86 expression (quantitative flow cytometry, allowing counting of molecules per cell), and T-cell cytokine profiling (differentiation of type 1/2 cytokine patterns by intracellular cytokine staining and flow cytometric analysis) have been developed. Using standard operation procedures, interassay variation of less than 20% can be achieved [4,5]. Such assays are now available for use in multicentre trials.
Putative therapeutic options
What can we do to prevent/overcome excessive immunodepression? Following high-dose immunosuppression in transplant patients using corticosteroid bolus or pan-T-cell antibodies, we observed a temporary decrease both in monocyte and T-cell function [6]. Long-lasting 'immunoparalysis' is predictive of the occurrence and outcome of bacterial/fungal infectious complications. Reduction in immunosuppression improved immune function and antimicrobial defences. Those data suggest a direct relation between immune responsiveness and susceptibility to and course of infection.
However, what are the therapeutic options in patients who are not immunosuppressed? As suggested above, a strong perioperative systemic inflammation contributes to long-lasting immune depression. Therefore, it makes sense to attempt to prevent extensive counter-regulation by blocking systemic inflammation. Angele and Faist [1] reported on a pilot trial of granulocyte colony-stimulating factor (G-CSF) in surgical patients. Interestingly, G-CSF, which cannot directly stimulate monocytes and T cells, prevented immune deactivation (monocytic TNF secretion, HLA-DR expression, IL-10 release, etc.). The mechanisms underlying this action are not well understood. However, it is likely that, in addition to the advanced antimicrobial capacity of G-CSF-activated neutrophils, the anti-inflammatory capacity of G-CSF contributes to the effect [7].
Endotoxin-neutralizing approaches may also help to prevent inflammation-related immunodepression. In animal models, targeting stress mediators (e.g. steroid receptor or β2-adrenergic receptor antagonists) is very successful in preventing immune depression [3], but transfer of this approach from animal house to bedside is difficult because of the short therapeutic window.
If 'immunoparalysis' becomes established, then immunostimulatory approaches may be considered. In pilot trials [8], treatment with plasmapheresis (removal of inhibitory factors), interferon-γ and GM-CSF showed promise.
None of the approaches described above has yet been tested in phase III trials. The availability of well standardized immunoassays will now permit enrollment of patients with 'immunoparalysis' into multicentre trials.
Competing interests
None declared.
Abbreviations
G-CSF = granulocyte colony-stimulating factor; IL = interleukin; TNF = tumour necrosis factor.
See related Review: http://ccforum.com/content/6/4/298
References
- Angele MK, Faist E. Clinical review: Immunodepression in the surgical patient and increased susceptibility to infection. Crit Care. 2002;6:298–305. doi: 10.1186/cc1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- Woiciechowsky C, Asadullah K, Nestler D, Eberhardt B, Platzer C, Schoning B, Glockner F, Lanksch WR, Volk HD, Docke WD. Sympathetic activation triggers systemic IL-10 release in immunodepression induced by brain injury. Nat Med. 1998;4:808–813. doi: 10.1038/nm0798-808. [DOI] [PubMed] [Google Scholar]
- Reinke P, Docke WD, Kox W, Zuckermann H, Volk HD. New developments in the immunodiagnosis of patients in intensive care [in German]. Dtsch Med Wochenschr. 1999;124:1527–1529. doi: 10.1055/s-2007-1023903. [DOI] [PubMed] [Google Scholar]
- Volk HD, Zuckermann H, Kox W, Woicieczowsky CH, Hoeflich C, Buhl K, Graeta G, Doeckel I, Reinke P. Immune monitoring and strategies for immune modulation. In: Linden P, Doughty LA, editor. In Molecular and Cellular Biology of Critical Care Medicine, Immunology and Infectious Disease Chapter 6. Kluwer Academic Publishers; 2002. [Google Scholar]
- Volk HD, Reinke P, Falck P, Staffer G, v Backer R. Diagnostic value of an immune monitoring program for the clinical management of immunosuppressed patients with septic complications. Clin Transplant. 1989;3:246–252. [Google Scholar]
- Hartung T, Doecke WD, Bundschuh D, Foote MA, Gantner F, Hermann C, Lenz A, Milwee S, Rich B, Simon B, Volk HD, von Aulock S, Wendel A. Effect of filgrastim treatment on inflammatory cytokines and lymphocyte functions. Clin Pharmacol Ther. 1999;66:415–424. doi: 10.1053/cp.1999.v66.a101210. [DOI] [PubMed] [Google Scholar]
- Volk HD, Reinke P, Docke WD. Clinical aspects: from systemic inflammation to 'immunoparalysis'. Chem Immunol. 2000;74:162–177. doi: 10.1159/000058753. [DOI] [PubMed] [Google Scholar]