

Abstract
Hypertrophied myocardium is associated with reductions in the transient outward K+ current (Ito) and expression of pore-forming Kv4.2/4.3 and auxiliary KChIP2 subunits. Here we show that KChIP2 mRNA and protein levels are dramatically decreased to 10% to 30% of control levels in the left ventricle of aorta-constricted rats in vivo and phenylephrine (PE)-treated myocytes in vitro. PE also markedly decreases Ito density. Inhibition of protein kinase Cs (PKCs) does not affect the PE-induced reduction in KChIP2 mRNA level, whereas activation of PKC with phorbol ester (phorbol myristate [PMA]) causes a marked reduction in KChIP2 mRNA level. Pharmacological inhibition of MEKs or overexpression of a dominant-negative MEK1 increases the basal KChIP2 mRNA expression and blocks the PMA-induced decrease in auxiliary subunit mRNA level. In addition, a constitutively active MEK1 decreases the basal KChIP2 mRNA level, and PMA causes no further reduction in auxiliary subunit mRNA level in active MEK1-expressing cells. Furthermore, pharmacological inhibition of JNKs or overexpression of a dominant-negative JNK1 prevents the PE-induced, but not PMA-induced, reduction in KChIP2 mRNA expression. These results suggest that downregulation of KChIP2 expression significantly contributes to the hypertrophy-associated reduction in Ito density. They also indicate that the expression of KChIP2 mRNA is controlled by the 2 branches of mitogen-activated protein kinase pathways: JNKs play a predominant role in mediating the PE-induced reduction, whereas the MEK-ERK pathway influences the basal expression and mediates the PKC-mediated downregulation.
Keywords: cardiac hypertrophy, voltage-gated K+ channel, signal transduction, gene regulation
Cardiac hypertrophy develops as a biological compensatory change in response to increased workload. However, sustained hypertrophy is a major risk factor for cardiac morbidity and mortality.1,2 One of the commonly observed changes in hypertrophied myocardium is a reduction in transient outward K+ current (Ito).3 Although it remains uncertain whether and how this reduction contributes to the increased cardiac dysfunction and sudden death seen with patients with hypertrophy, recent studies suggest that a marked reduction in Ito can be detrimental. For example, Ito is differentially expressed in distinct parts of the heart.4–11 Elimination of this differential expression may result in electrical instability by diminishing regional differences in action potential.12 Likewise, decreased Ito can facilitate the development of cardiac hypertrophy by prolonging action potential duration and a window for Ca2+ entry.13,14 Thus, an uncontrolled decline in Ito channel expression may directly or indirectly participate in the induction of hypertrophy-associated pathological changes of the heart.
Although Kv1.4-containing Kv1-family channels contribute to cardiac Ito, a large portion of the current is attributable to channels in the Kv4 family.15 These channels are multimeric complexes composed of pore-forming Kv4.2 and/or Kv4.316 –19 and auxiliary KChIP2 subunits.20 Many studies have shown that the expression of Kv4.2 and/or Kv4.3 subunits is reduced in hypertrophied myocardium.3,21 Our previous work indicates that transcriptional and posttranscriptional mechanisms are involved in the reduced pore-forming subunit expression.22 In contrast, little is known about the mechanisms controlling KChIP2 expression. Because deletion of KChIP2 gene leads to the loss of cardiac Ito,12 decreased expression of the auxiliary subunit may significantly contribute to hypertrophy-associated reduction in Ito.
Hence, we examined the contribution of decreased KChIP2 expression in hypertrophy-associated reduction in Ito. Using drugs and adenovirus-mediated expression, we also analyzed signaling mechanisms that regulate cardiac KChIP2 gene expression. In this article, we show that hypertrophy-inducing stimuli markedly reduce the expression of KChIP2 mRNA and protein in vivo and in vitro. This reduction in KChIP2 expression appears to be mediated mainly by the activation of JNKs, whereas MEKs influence its basal expression.
Materials and Methods
Animals and Neonatal Myocyte Culture
Abdominal aortic constriction or sham operation was performed on male Sprague-Dawley rats (11 to 12 weeks old). Two weeks after surgery, rats were euthanized and left ventricular free walls were obtained.
Neonatal cardiac myocytes were prepared from ventricular tissues of 1-day-old Sprague–Dawley rat pups by trypsinization, as described previously.22 Treatment with phenylephrine (PE) and/or other drugs was initiated on the third day after myocyte preparation, except that cells were maintained for 1additional day for experiments with virus infection.
Adenoviruses
Nucleotide substitutions were introduced in mouse MEK1 and JNK1 cDNAs using a 2-primer-based mutagenesis: constitutively active MEK1, MEK1S218,222E23; dominant-negative MEK1, MEK1K97M24; dominant-negative JNK1, JNK1 T183AY185F (JNK1APF).25 Adenoviruses carrying each of these constructs were generated with pShuttle-IRES-hrGFP-1 using a commercially available kit (Adeasy, Stratagene, La Jolla, Calif). These viruses express green fluorescent protein (GFP) and molecularly altered mitogen-activated protein (MAP) kinase as a single mRNA species in a cytomegalovirus promoter–driven manner.
Infection was performed at 1 × 109 total virus particles/mL in serum-free medium for 2 hours. Infected cells were cultured overnight in 5% calf serum–containing medium to enhance the expression of the virus-embedded gene, before experimental treatment. Fluorescent microscopy indicated that more than 85% cells were GFP positive under these conditions.
RNA Measurement
RNA was extracted from the left ventricular free walls and cultured myocytes using a guanidium thiocyanate–based method following the instructions of the manufacturer (RNeasy, Qiagen, Valencia, Calif). Kv4 and KChIP2 subunit mRNA levels were determined by RNase protection and real-time PCR, respectively, according to the previously described methods.26,27 For the latter analysis, we used primers corresponding to the C-terminal portion of KChIP2 cDNA that was commonly present in all splicing variants.26 We also used standard RT-PCR for detection and quantitation of KChIP2 splicing variants.28 In using this method, we tested for the linearity of signals using various PCR cycles and amounts of template cDNA. PCR products were separated on a 2% agarose or 4% polyacrylamide gel and visualized by ethidium bromide staining. Stained gel images were captured, and band intensities were analyzed using a charge-coupled device camera–based system (BioChem II, UVP, Upland, Calif).
Immunoblot Analysis
A post–nuclear membrane fraction was prepared by homogenization of ventricular tissues, followed by differential centrifugation.29 The obtained membrane fraction was rehomogenized in Triton-containing lysis buffer (20 mmol/L Tris-HCl [pH 7.5], 0.2 mol/L NaCl, 1 mmol/L EDTA, and 1% Triton X-100) supplemented with protease inhibitors at ≈10 mg protein/mL using a Teflon-pestle homogenizer. Triton extract was then obtained by centrifugation at 100 000g for 30 minutes. Transfected HEK293 cells or cultured myocytes were collected with ice-cold PBS. Triton extract was prepared by suspending the cell pellet in the lysis buffer, followed by centrifugation at 10 000g for 10 minutes. Immunoblot analysis was performed following the separation of samples on an SDS gel.30 Immunoreactive KChIP2 proteins were detected with rabbit polyclonal antibody previously produced in our laboratory26 or monoclonal antibody31 supplied by Dr James Trimmer (University of California at Davis). The 2 antibodies provided indistinguishable patterns on immunoblots. Anti-Kv4.2 and -Kv4.3 antibodies were obtained from a commercial source (Alomone, Jerusalem, Israel). The blots for KChIP2 and Kv4.x were stripped and reprobed with anti-GAPDH (Novus Biologicals, Littleton, Colo). Immunoreactivity of channel subunits was determined using GAPDH level as a control.
Patch-Clamp Recording
Whole-cell voltage-clamp recording was performed on cultured neonatal myocytes with an Axopatch-1D patch-clamp amplifier, and data were acquired and analyzed by pClamp software. Patch pipettes were filled with a solution containing (in mmol/L): 135 KCl, 1 MgCl2, 10 EGTA, 5 glucose, and 10 HEPES (pH 7.3). The bath solution was (in mmol/L): 135 NaCl, 5 KCl, 2 MgCl2, 1 CaCl2, 5 CoCl2, 10 glucose, 0.02 tetrodotoxin, and 10 HEPES (pH 7.4). Leak currents were subtracted by a P/4 pulse protocol and the series resistance was compensated by 60% to 80%. Cell membrane capacitances were obtained by reading the value for whole-cell input capacitance neutralization directly from the amplifier.
Data Analysis
The mRNA and protein levels were normalized to the mean of control data in each set of experiments. Comparison between two groups with variances was performed by 2-tailed Student t test, whereas multiple comparisons were done by the layered Bonferroni test. Statistical analysis for data without variance was done by the Wilcoxon signed-rank test. Results are presented as the mean±SEM.
An expanded Materials and Methods section is available in the online data supplement at http://circres.ahajournals.org.
Results
Hypertrophy-Inducing Stimuli Markedly Reduce KChIP2 Expression
We first used abdominal aortic ligation to test for hypertrophy-associated changes in KChIP2 expression. The expression of KChIP2 mRNA in the left ventricular free wall was markedly reduced to 23.7±7.9% of the control sham-operated animal level (n=6 for each group; Figure 1A). Aortic constriction similarly decreased all three KChIP2 splicing variants.28 To detect KChIP2 proteins, we used polyclonal and monoclonal anti-KChIP2 antibodies.26,31 These antibodies specifically reacted with all three KChIP2 splicing variants (Figure 1C with monoclonal antibody and data with polyclonal antibody not shown). Rat brains contained immunoreactive KChIP2 proteins with distinct sizes: a doublet at ≈30 KDa and a band at ≈25 KDa (Figure 1C). KChIP2 proteins with distinct sizes were also seen in cardiac tissues of other species.32,33 The positions of the larger doublet and smaller band corresponded to those obtained with KChIP2a/2b- and KChIP2c-transfected HEK293 cells, respectively. In rat cardiac tissue, the smaller immunoreactive KChIP2 protein at ≈25 KDa was abundant, whereas very faint larger proteins were seen. Given the abundant expression of KChIP2c mRNA in rat heart, it is likely that the ≈25-KDa immunoreactive protein represents KChIP2c. The very low levels of KChIP2a/2b proteins detected under our experimental conditions might be, in part, attributable to the insolubility of these proteins. Importantly, aortic constriction significantly reduced the ≈25-KDa immunoreactive protein to 26.4±4.5% of sham-operated animal level (n=6 for each group; Figure 1C and 1D). In contrast to the dramatic reduction in KChIP2 expression, moderate reductions in the levels of Kv4.2 and Kv4.3 mRNAs and proteins (25% to 50%) were obtained (Figure 1B through 1D). Thus, aortic constriction leads to marked reductions in KChIP2 mRNA and protein levels.
Figure 1.

Abdominal aortic constriction reduces KChIP2 expression. A, Two weeks after abdominal aortic constriction (AAC) or sham operation (Sham), total RNA was isolated from the left ventricular free wall. RT-PCR analyses for KChIP2 splicing variants and GAPDH were performed. Lines a, b, and c indicate the positions of KChIP2a, 2b, and 2c. B, Total KChIP2 mRNA level was determined by real-time PCR, whereas Kv4.2 and Kv4.3 mRNA levels were measured with RNase protection assays. * P<0.05 and **P<0.01 compared with Sham (2-tailed t-test, n=6 for each point). C, left, Monoclonal anti-KChIP2 antibody was tested with Triton extracts from adult brain and heart (12 weeks old) and neonatal heart (1 day old), as well as those from HEK293 cells transfected with indicated cDNAs. Arrows indicate the positions of KChIP2 variants. Bands at large sizes in HEK293 cell extracts (≈110 KDa) were attributable to a nonspecific reaction to endogenous cytosolic proteins. Right, Triton extracts were prepared from left ventricular free walls of aorta-constricted and sham-operated animals and subjected to immunoblot analysis with anti-KChIP2, anti-GAPDH, anti-Kv4.2, or anti-Kv4.3 antibody. D, KChIP2 and Kv4.x immunoreactivity was determined with GAPDH immunoreactivity as an internal control. * P<0.05 and **P<0.01 compared with Sham (2-tailed t test; n=6 for each group).
To further examine the relationship between KChIP2 level and functional channel expression, we used cultured neonatal ventricular myocytes. Recent studies have shown that treatment of these cells with PE, a robust inducer of hypertrophy, reduces Ito density.13,14 PE appeared to cause a marked decrease in KChIP2 mRNA expression: the level of auxiliary subunit message in cells treated with PE for 48 hours was 18.9±3.7% of that in control vehicle-treated cells (n≥6 for each group; Figure 2A through 2C). Again, the drug similarly reduced expression of the 3 KChIP2 splicing variants. The reduction in auxiliary subunit mRNAs occurred rapidly with concentrations of PE that were consistent with its action via α1-adrenergic receptors. Immunoblot analysis revealed that PE treatment for 48 hours caused a marked decrease in auxiliary subunit proteins to 17.9±4.4% of control level (n=6 for each group; Figure 2D and 2E). In contrast, only moderate decreases in Kv4.2 and 4.3 mRNA and protein levels (20% to 35%) were detected (Figure 2C through 2E). Finally, PE treatment for 48 hours significantly reduced Ito density: current density at +40 mV was 4.7±1.9 (n=11) and 19.8±5.8 (n=13) pA/pF in PE- and vehicle-treated cells, respectively (Figure 2F). No apparent changes in activation or inactivation kinetics were found. Thus, PE markedly decreases KChIP2 level and Ito density in neonatal myocytes. These findings suggest that the reduced availability of KChIP2 subunits limits formation of fully assembled functional channel complexes in PE-treated cells.
Figure 2.
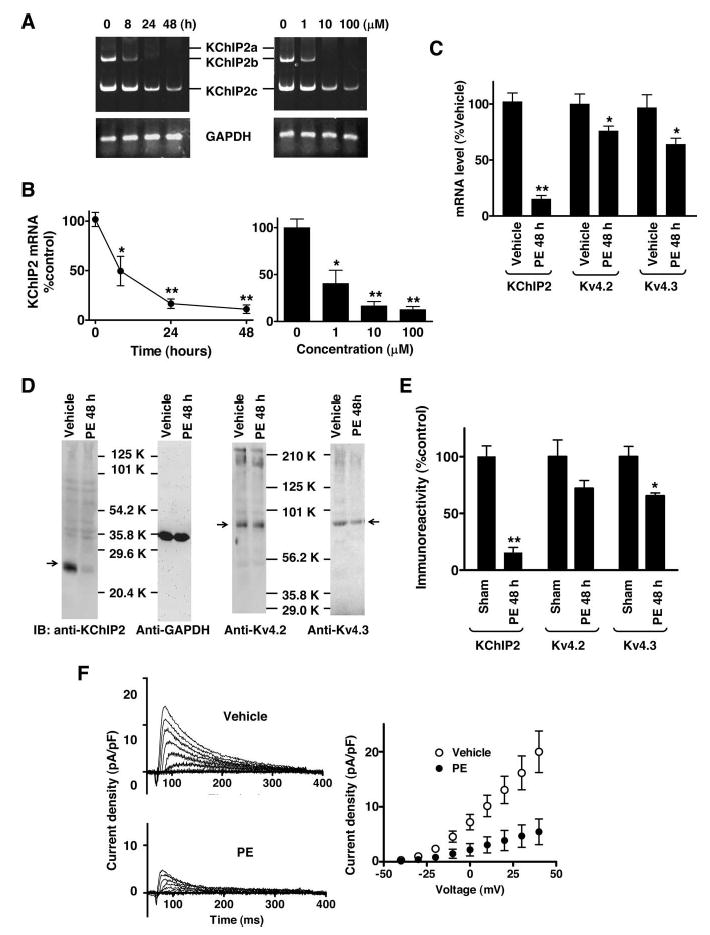
PE decreases KChIP2 expression and Ito density. A, Neonatal myocytes were treated with 100 μmol/L PE for various periods of time (left) or with various concentrations of PE for 48 hours (right). RT-PCR results for KChIP2 and GAPDH are shown. B, Total KChIP2 mRNA levels were determined by real-time PCR. *P<0.05 and **P<0.01 compared with control (Bonferroni test, n=4 for each condition). C, Neonatal myocytes were treated with 100 μmol/L PE or vehicle (0.1% ascorbic acid) for 48 hours. Total KChIP2 mRNA expression was determined by real-time PCR, whereas Kv4.2 and Kv4.3 mRNA levels were determined with RNase protection assays. *P<0.05 and **P<0.01 compared with control (2-tailed t-test, n=6 for each group). D, Triton extracts were prepared from myocytes treated with 100 μmol/L PE or vehicle for 48 hours. Immunoblots were obtained with monoclonal anti-KChIP2, anti-GAPDH, anti-Kv4.2, or anti-Kv4.3 antibody. Arrows indicate the position of channel subunit proteins. E, KChIP2 and Kv4.x protein levels were determined using GAPDH immunoreactivity as a control. *P<0.05 and **P<0.01 compared with vehicle (2-tailed t test, n=6 for each condition). F, Patch-clamp recording was performed with myocytes treated with 100 μmol/L PE or vehicle for 48 hours. Before recording, cells were washed 3 times with bath solution to eliminate potential acute effects by the drug. Currents were elicited by a 400-ms voltage pulse with 10-mV increment between −40 and +40 mV from holding potential of −80 mV. G, Peak current density-voltage relationship is shown (n=13 for vehicle and n=11 for PE). We eliminated the data from cells (≈10%) that exhibited very small outward current (≤0.1 pA/pF at ≤40 mV).
MEKs Are Involved in Basal and Protein Kinase C–Regulated KChIP2 mRNA Expression
To identify signaling mechanisms underlying the PE-induced decrease in KChIP2 mRNA expression, we used various inhibitors for kinases and phosphatases. Inhibitors of protein kinase Cs (1 μmol/L bisindolmaleimide), tyrosine kinases (5 μmol/L genestein), Ca2+-calmodulin–dependent kinases (1 μmol/L KN93), calcineurin (5 μmol/L cyclosporin A), or Akt-phosphatidyl inositol 3-kinase (100 nmol/L rapamycin) did not significantly affect the basal or PE-induced decrease in KChIP2 mRNA expression (data not shown). On the other hand, direct activation of protein kinase Cs (PKCs) with phorbol ester (100 nmol/L phorbol 12-myristate 13-acetate [PMA]) dramatically decreased KChIP2 mRNA level (Figure 3A and 3B). This PMA-induced reduction was completely prevented with the inhibitor of PKCs. Moreover, the inactive homologue 4α- phorbol ester (4α-PMA) did not affect the auxiliary subunit mRNA level. Thus, activation of PKCs inhibits KChIP2 mRNA expression, whereas the PE-induced decrease in auxiliary subunit mRNA level does not require PKCs.
Figure 3.

Activation of protein kinase Cs reduces KChIP2 mRNA level. A, Neonatal myocytes were treated with 100 nmol/L PMA, 4α-PMA, or vehicle (0.1% DMSO [None]) for 8 hours in the presence of 1 μmol/L bisindolmaleimide (BIM) or vehicle (0.1% DMSO). RT-PCR results for KChIP2 variants and GAPDH are shown. Note that PMA similarly decreased 3 splicing variant mRNA levels. B, Total KChIP2 mRNA level was determined by real-time PCR. **P<0.01 (Bonferroni test, n≥6). PE-induced decrease in KChIP2 mRNA level was not significantly different between the presence and absence of BIM.
The MEK-ERK branch of MAP kinase pathway is involved in compensatory hypertrophy.34 In addition, PKCs are known to phosphorylate and activate MEKs. We also observed robust phosphorylation of ERKs, downstream targets of MEKs, by aortic banding in vivo and PE treatment in vitro (supplemental Figure I). Activation of PKC modestly increased their phosphorylation. Thus, the observed PMA-induced decrease in KChIP2 mRNA expression might be mediated by activation of the MEK-ERK pathway. To test this possibility, we first used pharmacological inhibition of MEKs. Treatment with U0126 (10 μmol/L), a selective inhibitor of MEKs, caused a significant increase in the basal level of KChIP2 mRNA (Figure 4A and 4B). This increase was caused by inhibition of MEKs, because the inactive homologue U0124 did not affect auxiliary subunit mRNA level. Furthermore, the MEK inhibitor potently blocked the PMA-induced decrease in KChIP2 mRNA level (Figure 4A, 4B, and 4D). In contrast, inhibition of MEKs produced only a moderate effect on the PE-induced reduction.
Figure 4.
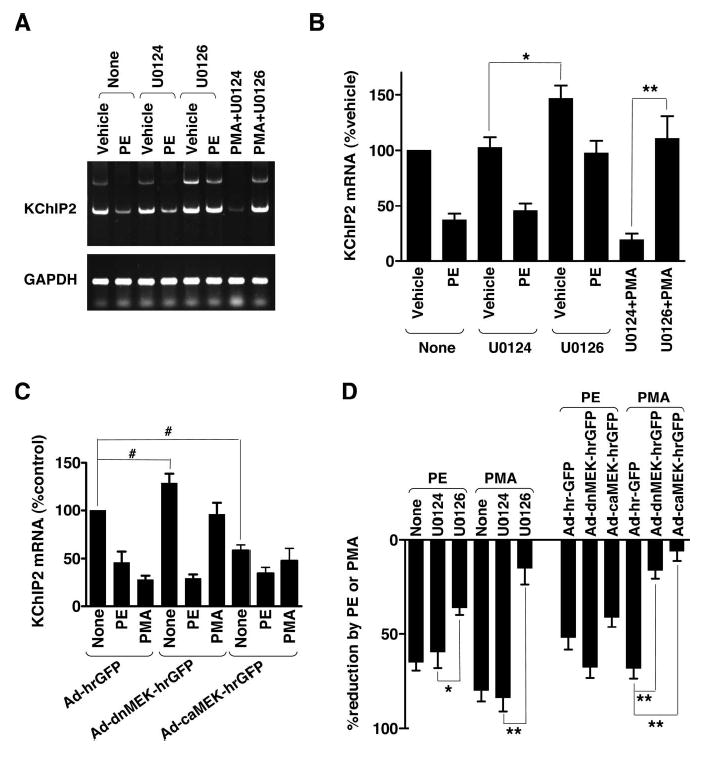
MEKs influence basal KChIP2 mRNA expression and mediate protein kinase C-induced downregulation. A, Neonatal myocytes were treated with 100 μmol/L PE, its corresponding vehicle (0.1 mmol/L ascorbic acid), or 100 nmol/L PMA for 8 hours in the presence or absence of 10 μmol/L U0126 or U0124. RT-PCR data for KChIP2 variants and GAPDH are shown. B, Total KChIP2 mRNA level was determined by real-time PCR. *P<0.05 and **P<0.01 (2-tailed t test, n≥6 for each condition). C, Myo-cytes were infected with adenoviruses carrying a dominant-negative (Ad-dnMEK-hrGFP) or constitutively active (Ad-caMEK-hrGFP) MEK1 or no additional insert (Ad-hrGFP). Infected cells were then untreated (None) or treated with 100 μmol/L PE or 100 nmol/L PMA for 8 hours. #P<0.05 (Wilcoxon signed-rank test, n≥6 for each condition). D, Relative reductions in total KChIP2 mRNA level were obtained from the data in B and C. *P<0.05 and **P<0.01 (Bonferroni test, n≥6 for each condition).
To further examine the involvement of MEKs in KChIP2 mRNA expression, we used adenovirus-mediated overexpression of dominant-negative (K97M, dnMEK) and constitutively active (S218,222E, caMEK) MEK1s (Figure 4C). Similarly to the pharmacological inhibition, overexpression of the dominant-negative MEK1 increased the basal KChIP2 mRNA level (Figure 4C and 4D). It also caused a substantial reduction in the PMA-induced decrease in KChIP2 mRNA level. Moreover, expression of the constitutively active MEK1 decreased the basal KChIP2 mRNA level. In active MEK1-expression cells, PMA was unable to produce a further decrease in KChIP2 mRNA expression. In contrast, PE was still capable of reducing KChIP2 mRNA level in these cells. Taken together, MEKs control basal KChIP2 mRNA expression and mediate the PKC-regulated decrease in auxiliary subunit message. Inhibition of MEKs may increase the basal KChIP2 mRNA level by eliminating basal activity or spontaneous activation of these enzymes in untreated myocytes. This PKC-MEK–mediated signaling pathway plays less important role in the PE-induced decrease in KChIP2 mRNA expression.
JNKs Mediate PE-Induced Decrease in KChIP2 mRNA Expression
Because the JNK and p38 branches of the MAP kinase cascade are implicated in mediating hypertrophic responses in neonatal myocytes,35,36 we also examined whether these kinases might be involved in the PE-induced downregulation of KChIP2 mRNA expression. Inhibition of p38 with 20 μmol/L SB20219 did not affect the PE- or PMA-induced decreases in KChIP2 mRNA level (data not shown). On the contrary, inhibition of JNKs with 20 μmol/L SP00125 appeared to prevent the PE-induced reduction in auxiliary subunit mRNA expression (Figure 5A through 5C). Furthermore, adenovirus-mediated overexpression of a dominant-negative JNK1 (JNK1APF, dnJNK) significantly blocked the PE-induced decrease in KChIP2 mRNA expression (Figure 5B and 5C). In contrast to the marked prevention of the PE effect, pharmacological or genetic inhibition of JNKs did not influence the PMA-induced downregulation. Finally, we found that aortic banding and PE treatment caused increases in phosphorylation of p55 JNK1 in vivo and in vitro (supplemental Figure I). Thus, JNKs play a predominant role in mediating the PE-induced downregulation of KChIP2 mRNA expression, whereas these kinases are not required for the PKC-mediated decrease.
Figure 5.

JNKs are required for PE-induced decrease in KChIP2 mRNA expression. A, Neonatal myocytes were treated with 100 μmol/L PE, 100 nmol/L PMA, or untreated (None) for 8 hours in the presence of 10 μmol/L SP00125 or vehicle (0.1% DMSO). RT-PCR data for KChIP2 splicing variants and GAPDH are shown. B, Total KChIP2 mRNA level was determined by real-time PCR. *The PE-induced reduction in KChIP2 mRNA level was significantly smaller with SP00125 than that with vehicle (P<0.05, 2-tailed t test, n≥6 for each condition). C, Myocytes were infected with adenoviruses carrying a dominant-negative JNK1 (Ad-dnJNK-hrGFP) or no additional insert (Ad-hrGFP). Infected cells were treated with 100 μmol/L PE, 100 nmol/L PMA, or untreated (None) for 8 hours. Total KChIP2 mRNA level was determined by real-time PCR. *The PE-induced reduction in cells infected with Ad-dnJNK-hrGFP was significantly smaller than that with Ad-hrGFP (P<0.05, 2-tailed t test, n≥6 for each condition).
Discussion
A reduced Ito density is commonly observed in hypertrophied myocardium.3,21 Associated decreases in the pore-forming Kv4.2/4.33,21 and auxiliary KChIP212,32 subunits have been seen in some cases. Yet the mechanisms underlying these reductions largely remain unknown. In this article, we have shown that a decrease in KChIP2 is more dramatic than those in the pore-forming subunits using abdominal aortic constriction in vivo and PE treatment in vitro. Current measurement in vitro further demonstrated that a reduction in KChIP2 is correlated with that in Ito density. Furthermore, our experiments revealed that 2 branches of the MAP kinase pathway regulate cardiac KChIP2 mRNA expression. Activation of JNKs is mainly responsible for the PE-induced downregulation, whereas MEKs are involved in controlling the basal level and protein kinase C (PKC)-mediated decrease (Figure 6). Thus, KChIP2 gene expression may be a key event in controlling cardiac Ito channel expression under physiological and pathological conditions
Figure 6.
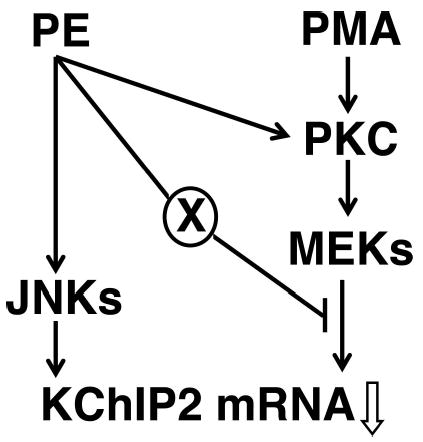
Signaling mechanisms controlling cardiac KChIP2 mRNA expression. The results in this study indicate that there are 2 signaling pathways leading to downregulation of KChIP2 mRNA expression: the JNK and MEK MAP kinase pathways. Whereas stimulation of α1-adrenergic receptors with phenylephrine (PE) downregulates KChIP2 mRNA expression mainly by activating the former, direct activation of PKCs with PMA uses the latter. Our data also suggest that there are 2 routes of the crosstalk between these 2 pathways. One point of the crosstalk is activation of PKCs, originated from the well-established Gq-coupled receptor-phospholipase C–mediated release of intracellular Ca2+ and diacyl glycerol. The other unidentified signaling mechanism (X) suppresses the MEK-mediated reduction.
We found good correlation of the PE-induced reduction in KChIP2 level to that in Ito density in neonatal myocytes. Our data also showed that KChIP2 is dramatically decreased in vivo by aortic constriction. These findings suggest that the reduced availability of KChIP2 subunits may limit formation of fully assembled functional channels in not only neonatal, but also adult, hypertrophied myocytes. However, the levels of pore-forming and auxiliary subunits differ between neonatal and adult myocytes. For example, cardiac Ito density increases during development in many species. In rats, expression of KChIP2 is increased in the early postnatal period,37 whereas a gradual increase in Kv4.2 expression is seen during development.38 We also found that KChIP2 protein level, when normalized to GAPDH immunoreactivity, is approximately double in left ventricle of adult rats compared with that of 1-day-old neonatal animals. Therefore, the contribution of reduced KChIP2 expression to hypertrophy-associated decrease in Ito density may significantly differ between neonatal and adult myocytes. In addition to the developmental changes, there are significant species differences in the expression of Ito-forming subunits. Whereas Kv4.2 is the major pore-forming subunit in cardiac tissues of rodents, larger animals including humans contain a large amount of Kv4.3 but very little Kv4.2.18,39 Moreover, the transmural gradient of Ito density across the left ventricular wall is generated by differential expression of Kv4.2 in rodents and KChIP2 in larger animals.39–41 These findings suggest that the limited amount of the pore-forming and auxiliary subunits determine the level of functional Ito channel complexes in adult myocytes in rodents and larger animals, respectively. Thus, the impact of hypertrophy-associated reduction in KChIP2 on Ito channel expression may significantly differ in heart regions and animal species.
Stimulation of α1-adrenergic receptors with PE and direct activation of PKCs with PMA each resulted in a marked reduction in KChIP2 mRNA level. Although α1-adrenergic receptors are known to couple to Gq proteins leading to activation of PKCs,42,43 our data indicate that the two stimuli use independent signaling pathways to decrease the expression of auxiliary subunit mRNA (see Figure 6). Specifically, inhibition of PKCs or MEKs nearly completely abolished the PMA-induced reduction in KChIP2 mRNA expression but produced less apparent effects on the PE-induced decrease. We also observed that PMA resulted in a relatively moderate increase in the phosphorylation of ERKs, whereas PE led to a marked elevation in their phosphorylation. Therefore, the PKC-MEK–mediated reduction seemed to be suppressed in the context of PE treatment. This apparent reduction in the contribution of the MEK-mediated effect might be attributable to the fact that PE stimulates various signaling molecules, in addition to classical and novel PKCs that are activated by PMA. For example, PE activates various kinases and phosphatases, such as p38, tyrosine kinases, Ca2+-cal-modulin– dependent kinases, calcineurin and Akt-phosphatidyl inositol 3-kinase. Therefore, PE-induced activation of any of these enzymes might suppress the MEK-mediated reduction in KChIP2 mRNA expression. However, we found that inhibitors of these enzymes did not unmask the MEK-mediated effect in the context of PE treatment isolated with the blocker of JNK SP00125 (data not shown). In addition, the adrenergic agonist is known to increase the production of cAMP in neonatal myocytes. Again, inhibitor of protein kinase A or cAMP analogue appeared to produce no effects on the contribution of the MEK-mediated reduction in the context of PE treatment or the PMA-induced decrease in KChIP2 mRNA expression. Other possible mediators of this PE-induced suppression of the MEK-mediated effect are atypical PKCs, such as PKCζ and PKCL/λ.44,45 Whereas a myristoylated pseudosubstrate of PKCζ did not affect the contribution of the MEK-mediated reduction in the context of PE treatment, we were unable to test the involvement of the latter enzyme because of the lack of available inhibitors. Hence, it remains uncertain at this moment what signaling molecules may mediate the PE-induced suppression of the PKC-MEK-ERK effect on KChIP2 mRNA expression.
MAP kinases play important roles in the development of hypertrophy and other pathological changes of the heart. Three branches of the MAP kinase pathway, each involving MEK-ERK, JNK, or p38, appear to control distinct aspects of cardiac myocyte hypertrophy and apoptosis.35,36,46–48 Although activation of JNK and p38 leads to hypertrophy in cultured neonatal myocytes, studies with animal models revealed that these kinases are antihypertrophic. Instead, they may mediate cardiac myocyte apoptosis and other pathological alterations, leading to dilated cardiomyopathy and heart failure. In contrast, the MEK-ERK pathway has been shown to mediate compensatory hypertrophy without pathological features. In this study, we identified JNK and MEK as important regulators of cardiac KChIP2 gene expression. Thus, the 2 signaling pathways may independently contribute to adaptive and maladaptive changes in Ito expression. For example, regulation of cardiac Ito density can act as a biological adaptive response to compensate for required work force. Our data indicate that MEK affects basal KChIP2 mRNA expression. Therefore, the MEK-mediated regulation of KChIP2 mRNA expression may represent adaptive regulation of Ito expression. On the other hand, the JNK-mediated marked reduction by PE may be a part of pathological alterations associated with hypertrophied myocardium. In this regard, it is important to note that a reduction in Ito density is seen in myocytes from failing hearts, regardless of the presence of hypertrophy, including myocytes from dilated cardiomyopathic hearts.49 Taken together, KChIP2 gene expression may play a pivotal role in the physiological regulation and pathological alterations of the heart.
Supplementary Material
Acknowledgments
Support for this study provided by NIH grant HL074111. We thank Dr James Trimmer for monoclonal anti-KChIP2 antibody.
References
- 1.Kannel WB, Gordon T, Offutt D. Left ventricular hypertrophy by electrocardiogram. Prevalence, incidence, and mortality in the Framingham study. Ann Intern Med. 1969;71:89–105. doi: 10.7326/0003-4819-71-1-89. [DOI] [PubMed] [Google Scholar]
- 2.Kannel WB, Levy D, Cupples LA. Left ventricular hypertrophy and risk of cardiac failure: insights from the Framingham Study. J Cardiovasc Pharmacol. 1987;10(suppl 6):S135–S140. [PubMed] [Google Scholar]
- 3.Nabauer M, Kaab S. Potassium channel down-regulation in heart failure. Cardiovasc Res. 1998;37:324–334. doi: 10.1016/s0008-6363(97)00274-5. [DOI] [PubMed] [Google Scholar]
- 4.Casis O, Iriarte M, Gallego M, Sanchez-Chapula JA. Differences in regional distribution of K+ current densities in rat ventricle. Life Sci. 1998;63:391–400. doi: 10.1016/s0024-3205(98)00287-2. [DOI] [PubMed] [Google Scholar]
- 5.Li GR, Feng J, Yue L, Carrier M. Transmural heterogeneity of action potentials and Ito1 in myocytes isolated from the human right ventricle. Am J Physiol. 1998;275:H369–H377. doi: 10.1152/ajpheart.1998.275.2.H369. [DOI] [PubMed] [Google Scholar]
- 6.Litovsky SH, Antzelevitch C. Transient outward current prominent in canine ventricular epicardium but not endocardium. Circ Res. 1988;62:116–126. doi: 10.1161/01.res.62.1.116. [DOI] [PubMed] [Google Scholar]
- 7.Liu DW, Gintant GA, Antzelevitch C. Ionic bases for electrophysiological distinctions among epicardial, midmyocardial, and endocardial myocytes from the free wall of the canine left ventricle. Circ Res. 1993;72:671–687. doi: 10.1161/01.res.72.3.671. [DOI] [PubMed] [Google Scholar]
- 8.Nabauer M, Beuckelmann DJ, Uberfuhr P, Steinbeck G. Regional differences in current density and rate-dependent properties of the transient outward current in subepicardial and subendocardial myocytes of human left ventricle. Circulation. 1996;93:168–177. doi: 10.1161/01.cir.93.1.168. [DOI] [PubMed] [Google Scholar]
- 9.Shimoni Y, Severson D, Giles W. Thyroid status and diabetes modulate regional differences in potassium currents in rat ventricle. J Physiol. 1995;488(pt 3):673–688. doi: 10.1113/jphysiol.1995.sp020999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang ZG, Fermini B, Nattel S. Repolarization differences between guinea pig atrial endocardium and epicardium: evidence for a role of Ito. Am J Physiol. 1991;260:H1501–H1506. doi: 10.1152/ajpheart.1991.260.5.H1501. [DOI] [PubMed] [Google Scholar]
- 11.Wettwer E, Amos GJ, Posival H, Ravens U. Transient outward current in human ventricular myocytes of subepicardial and subendocardial origin. Circ Res. 1994;75:473–482. doi: 10.1161/01.res.75.3.473. [DOI] [PubMed] [Google Scholar]
- 12.Kuo HC, Cheng CF, Clark RB, Lin JJ, Lin JL, Hoshijima M, Nguyen-Tran VT, Gu Y, Ikeda Y, Chu PH, Ross J, Giles WR, Chien KR. A defect in the Kv channel-interacting protein 2 (KChIP2) gene leads to a complete loss of I(to) and confers susceptibility to ventricular tachycardia. Cell. 2001;107:801–813. doi: 10.1016/s0092-8674(01)00588-8. [DOI] [PubMed] [Google Scholar]
- 13.Kassiri Z, Zobel C, Nguyen TT, Molkentin JD, Backx PH. Reduction of I(to) causes hypertrophy in neonatal rat ventricular myocytes. Circ Res. 2002;90:578–585. doi: 10.1161/01.res.0000012223.86441.a1. [DOI] [PubMed] [Google Scholar]
- 14.Zobel C, Kassiri Z, Nguyen TT, Meng Y, Backx PH. Prevention of hypertrophy by overexpression of Kv4.2 in cultured neonatal cardiomyocytes. Circulation. 2002;106:2385–2391. doi: 10.1161/01.cir.0000033970.22130.93. [DOI] [PubMed] [Google Scholar]
- 15.Barry DM, Nerbonne JM. Myocardial potassium channels: electrophysiological and molecular diversity. Annu Rev Physiol. 1996;58:363–394. doi: 10.1146/annurev.ph.58.030196.002051. [DOI] [PubMed] [Google Scholar]
- 16.Fiset C, Clark RB, Shimoni Y, Giles WR. Shal-type channels contribute to the Ca2+-independent transient outward Kω current in rat ventricle. J Physiol. 1997;500(pt 1):51–64. doi: 10.1113/jphysiol.1997.sp021998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johns DC, Nuss HB, Marban E. Suppression of neuronal and cardiac transient outward currents by viral gene transfer of dominant-negative Kv4.2 constructs. J Biol Chem. 1997;272:31598–31603. doi: 10.1074/jbc.272.50.31598. [DOI] [PubMed] [Google Scholar]
- 18.Dixon JE, Shi W, Wang HS, McDonald C, Yu H, Wymore RS, Cohen IS, McKinnon D. Role of the Kv4.3 K+ channel in ventricular muscle. A molecular correlate for the transient outward current. Circ Res. 1996;79:659–668. doi: 10.1161/01.res.79.4.659. [DOI] [PubMed] [Google Scholar]
- 19.Barry DM, Xu H, Schuessler RB, Nerbonne JM. Functional knockout of the transient outward current, long-QT syndrome, and cardiac remodeling in mice expressing a dominant-negative Kv4 alpha subunit. Circ Res. 1998;83:560–567. doi: 10.1161/01.res.83.5.560. [DOI] [PubMed] [Google Scholar]
- 20.An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KI, Strassle BW, Trimmer JS, Rhodes KJ. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 2000;403:553–556. doi: 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- 21.Takimoto K, Levitan ES. Altered K+ channel expression in the hypertrophied and failing heart. In: Archer SL, Rusch NJ, eds. Potassium Channels in Cardiovascular Disease Kluwer Academic/Plenum Publishers; 2005:773–783.
- 22.Zhang TT, Takimoto K, Stewart AF, Zhu C, Levitan ES. Independent regulation of cardiac Kv4.3 potassium channel expression by angiotensin II and phenylephrine. Circ Res. 2001;88:476–482. doi: 10.1161/01.res.88.5.476. [DOI] [PubMed] [Google Scholar]
- 23.Janssen RA, Veenstra KG, Jonasch P, Jonasch E, Mier JW. Ras- and Raf-induced down-modulation of nonmuscle tropomyosin are MEK-independent. J Biol Chem. 1998;273:32182–32186. doi: 10.1074/jbc.273.48.32182. [DOI] [PubMed] [Google Scholar]
- 24.Allen MP, Xu M, Linseman DA, Pawlowski JE, Bokoch GM, Heidenreich KA, Wierman ME. Adhesion-related kinase repression of gonadotropin-releasing hormone gene expression requires Rac activation of the extra-cellular signal-regulated kinase pathway. J Biol Chem. 2002;277:38133–38140. doi: 10.1074/jbc.M200826200. [DOI] [PubMed] [Google Scholar]
- 25.Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki T, Takimoto K. Differential expression of Kv4 pore-forming and KChIP auxiliary subunits in rat uterus during pregnancy. Am J Physiol Endocrinol Metab. 2005;288:E335–E341. doi: 10.1152/ajpendo.00250.2004. [DOI] [PubMed] [Google Scholar]
- 27.Takimoto K, Li D, Hershman KM, Li P, Jackson EK, Levitan ES. Decreased expression of Kv4.2 and novel Kv4.3 K+ channel subunit mRNAs in ventricles of renovascular hypertensive rats. Circ Res. 1997;81:533–539. doi: 10.1161/01.res.81.4.533. [DOI] [PubMed] [Google Scholar]
- 28.Takimoto K, Yangi EK, Conforti L. Palmitoylation of KChIP splicing variants is required for efficient cell surface expression of Kv4.3 channels. J Biol Chem. 2002;277:26904–26911. doi: 10.1074/jbc.M203651200. [DOI] [PubMed] [Google Scholar]
- 29.Takimoto K, Levitan ES. Glucocorticoid induction of Kv1.5 K+ channel gene expression in ventricle of rat heart. Circ Res. 1994;75:1006–1013. doi: 10.1161/01.res.75.6.1006. [DOI] [PubMed] [Google Scholar]
- 30.Ren X, Shand SH, Takimoto K. Effective association of KChIPs with Kv4 channel is mediated with their unique core peptide. J Biol Chem. 2003;278:43564–43570. doi: 10.1074/jbc.M302337200. [DOI] [PubMed] [Google Scholar]
- 31.Rhodes KJ, Carroll KI, Sung MA, Doliveira LC, Monaghan MM, Burke SL, Strassle BW, Buchwalder L, Menegola M, Cao J, An WF, Trimmer JS. KChIPs and Kv4 alpha subunits as integral components of A-type potassium channels in mammalian brain. J Neurosci. 2004;24:7903–7915. doi: 10.1523/JNEUROSCI.0776-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rose J, Armoundas AA, Tian Y, DiSilvestre D, Burysek M, Halperin V, O’Rourke B, Kass DA, Marban E, Tomaselli GF. The molecular basis of altered expression of potassium currents in failing rabbit myocardium. Am J Physiol Heart Circ Physiol. 2005;288:H2077–H2087. doi: 10.1152/ajpheart.00526.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akar FG, Wu RC, Deschenes I, Armoundas AA, Piacentino V, III, Houser SR, Tomaselli GF. Phenotypic differences in transient outward K+ current of human and canine ventricular myocytes: insights into molecular composition of ventricular Ito. Am J Physiol Heart Circ Physiol. 2004;286:H602–H609. doi: 10.1152/ajpheart.00673.2003. [DOI] [PubMed] [Google Scholar]
- 34.Bueno OF, De Windt LJ, Tymitz KM, Witt SA, Kimball TR, Klevitsky R, Hewett TE, Jones SP, Lefer DJ, Peng CF, Kitsis RN, Molkentin JD. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000;19:6341–6350. doi: 10.1093/emboj/19.23.6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang Q, Molkentin JD. Redefining the roles of p38 and JNK signaling in cardiac hypertrophy: dichotomy between cultured myocytes and animal models. J Mol Cell Cardiol. 2003;35:1385–1394. doi: 10.1016/j.yjmcc.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Petrich BG, Wang Y. Stress-activated MAP kinases in cardiac remodeling and heart failure: new insights from transgenic studies. Trends Cardiovasc Med. 2004;14:50–55. doi: 10.1016/j.tcm.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 37.Kobayashi T, Yamada Y, Nagashima M, Seki S, Tsutsuura M, Ito Y, Sakuma I, Hamada H, Abe T, Tohse N. Contribution of KChIP2 to the developmental increase in transient outward current of rat cardiomyocytes. J Mol Cell Cardiol. 2003;35:1073–1082. doi: 10.1016/s0022-2828(03)00199-8. [DOI] [PubMed] [Google Scholar]
- 38.Xu H, Dixon JE, Barry DM, Trimmer JS, Merlie JP, McKinnon D, Nerbonne JM. Developmental analysis reveals mismatches in the expression of K+ channel alpha subunits and voltage-gated K+ channel currents in rat ventricular myocytes. J Gen Physiol. 1996;108:405–419. doi: 10.1085/jgp.108.5.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosati B, Pan Z, Lypen S, Wang HS, Cohen I, Dixon JE, McKinnon D. Regulation of KChIP2 potassium channel beta subunit gene expression underlies the gradient of transient outward current in canine and human ventricle. J Physiol. 2001;533:119–125. doi: 10.1111/j.1469-7793.2001.0119b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dixon JE, McKinnon D. Quantitative analysis of potassium channel mRNA expression in atrial and ventricular muscle of rats. Circ Res. 1994;75:252–260. doi: 10.1161/01.res.75.2.252. [DOI] [PubMed] [Google Scholar]
- 41.Rosati B, Grau F, Rodriguez S, Li H, Nerbonne JM, McKinnon D. Concordant expression of KChIP2 mRNA, protein and transient outward current throughout the canine ventricle. J Physiol. 2003;548:815–822. doi: 10.1113/jphysiol.2002.033704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.LaMorte VJ, Thorburn J, Absher D, Spiegel A, Brown JH, Chien KR, Feramisco JR, Knowlton KU. Gq- and ras-dependent pathways mediate hypertrophy of neonatal rat ventricular myocytes following alpha 1-adrenergic stimulation. J Biol Chem. 1994;269:13490–13496. [PubMed] [Google Scholar]
- 43.Wu D, Katz A, Lee CH, Simon MI. Activation of phospholipase C by alpha 1-adrenergic receptors is mediated by the alpha subunits of Gq family. J Biol Chem. 1992;267:25798–25802. [PubMed] [Google Scholar]
- 44.Husain S, Young D, Wingard CJ. Role of PKCalpha and PKCiota in phenylephrine-induced contraction of rat corpora cavernosa. Int J Impot Res. 2004;16:325–333. doi: 10.1038/sj.ijir.3901164. [DOI] [PubMed] [Google Scholar]
- 45.Rybin VO, Buttrick PM, Steinberg SF. PKC-lambda is the atypical protein kinase C isoform expressed by immature ventricle. Am J Physiol. 1997;272:H1636–H1642. doi: 10.1152/ajpheart.1997.272.4.H1636. [DOI] [PubMed] [Google Scholar]
- 46.Baines CP, Molkentin JD. STRESS signaling pathways that modulate cardiac myocyte apoptosis. J Mol Cell Cardiol. 2005;38:47–62. doi: 10.1016/j.yjmcc.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 47.Bueno OF, Molkentin JD. Involvement of extracellular signal-regulated kinases 1/2 in cardiac hypertrophy and cell death. Circ Res. 2002;91:776–781. doi: 10.1161/01.res.0000038488.38975.1a. [DOI] [PubMed] [Google Scholar]
- 48.Selvetella G, Hirsch E, Notte A, Tarone G, Lembo G. Adaptive and maladaptive hypertrophic pathways: points of convergence and divergence. Cardiovasc Res. 2004;63:373–380. doi: 10.1016/j.cardiores.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 49.Kaab S, Dixon J, Duc J, Ashen D, Nabauer M, Beuckelmann DJ, Steinbeck G, McKinnon D, Tomaselli GF. Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. J Gen Physiol. 1998;98:1383–1393. doi: 10.1161/01.cir.98.14.1383. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.