

Summary
Considerable circumstantial evidence suggests that Aβ42 is the initiating molecule in Alzheimer's disease (AD) pathogenesis. However, the absolute requirement for Aβ42 for amyloid deposition has never been demonstrated in vivo. We have addressed this by developing transgenic models that express Aβ1-40 or Aβ1-42 in the absence of human amyloid β protein precursor (APP) overexpression. Mice expressing high levels of Aβ1-40 do not develop overt amyloid pathology. In contrast, mice expressing lower levels of Aβ1-42 accumulate insoluble Aβ1-42 and develop compact amyloid plaques, congophilic amyloid angiopathy (CAA), and diffuse Aβ deposits. When mice expressing Aβ1-42 are crossed with mutant APP (Tg2576) mice, there is also a massive increase in amyloid deposition. These data establish that Aβ1-42 is essential for amyloid deposition in the parenchyma and also in vessels.
Introduction
Overexpression of Alzheimer's disease (AD)-linked mutant human APP transgenes has been the most reliable means of promoting deposition of Aβ in the brains of transgenic mice. As they age, these mutant APP mice develop robust amyloid pathology and other AD-like features, including decreased synaptic density, reactive gliosis, and some cognitive deficits. However, these mutant APP mouse models show little evidence of overt neuronal loss and neurofibrillary tangle (NFT) pathology (Hardy and Selkoe, 2002; Price et al., 1998). One potential problem with most of the widely studied mutant APP mice is that the high level of overexpression of mutant human APP may confound the phenotype. Overexpression of APP results in overproduction of APP fragments, which may have neuroprotective or neurotoxic functions. For example, secreted APP generated by α-secretase (sAPPα) can be neuroprotective, whereas the carboxyl terminal fragment generated by β-secretase cleavage (CTFβ) and a caspase-cleaved fragment of APP (C31) can be neurotoxic (Lu et al., 2000; Mattson, 2004; Yankner et al., 1989). Moreover APP and fragments such as the APP intracellular domain have signaling functions that may also contribute to a phenotype (LaFerla, 2002). Consequently, mice that overexpress individual Aβ peptides in the absence of overexpression of APP allow testing of hypotheses regarding (1) the role of select Aβ species in the initiation and propagation of amyloid deposition in vivo and (2) the specific contribution of each Aβ peptide to the phenotype seen in AD mouse models.
Much of the data that support a pivotal role for Aβ42 in AD have come from the study of mutations in the APP and presenilin genes that cause early-onset familial forms of AD (Selkoe, 1998). The vast majority of these mutations selectively increase the relative levels of Aβ42. However, even in typical late-onset AD there is evidence that Aβ42, a minor Aβ species, usually representing less then 20% of the total Aβ secreted, is both the earliest form and the predominant species deposited in the brain parenchyma (Golde et al., 2000). In contrast, Aβ40, the major Aβ peptide secreted by cells, appears to be the predominant species deposited in the amyloid deposits in the cerebral vasculature (congophillic angiopathy, CAA) (Gravina et al., 1995; Iwatsubo et al., 1994). Transgenic mouse studies using mutant APP and PS transgenes have provided some insights into the effects that altering the ratio of Aβ40 and Aβ42 have on time to onset of deposition, type of deposit (e.g., diffuse versus compact), and extent of CAA (Borchelt et al., 1997; Herzig et al., 2004; Holcomb et al., 1998). However, such studies have not definitively identified which Aβ species are responsible for seeding amyloid deposition in either the parenchyma or vasculature.
To address this question, we have generated transgenic mice that express Aβ1-40 or Aβ1-42 without APP overexpression. For these studies we used cDNAs that express fusion proteins between the BRI protein, involved in amyloid deposition in Familial British (FBD) and Danish Dementia (FDD) and Aβ1-40 (BRI-Aβ40) or Aβ1-42 (BRI-Aβ42) (Lewis et al., 2001; Vidal et al., 1999, 2000)(Figure 1A). We have previously shown that transfection of BRI-Aβ cDNAs results in high-level expression and secretion of the encoded Aβ peptide through proteolytic cleavage of the fusion protein at a furin cleavage site immediately preceding Aβ (Lewis et al., 2001). Efficient secretion of Aβ from the BRI fusion protein distinguishes this approach from studies using Aβ minigene constructs that generate high levels of intra-cellular Aβ and minimal secreted Aβ (LaFerla et al., 1995). The BRI-Aβ transgenic mice we have generated provide substantial evidence that Aβ1-42 but not Aβ1-40 is sufficient to promote Aβ deposition in mice.
Figure 1.
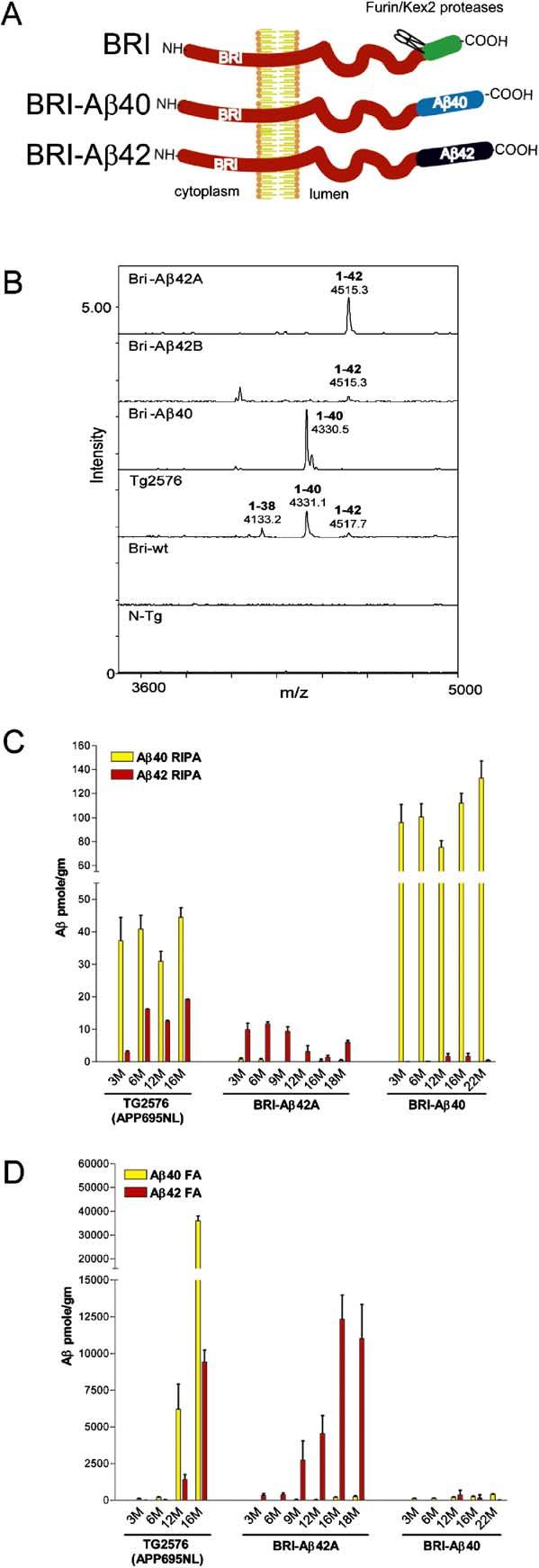
Biochemical Analyses of Aβ Levels in BRI-Aβ Mice (A) Schematic of the BRI-Aβ fusion proteins. BRI-Aβ fusion constructs were designed to take advantage of the BRI protein that is cleaved by furin or a furin-like protease near the COOH-terminus to release a soluble 23 amino acid peptide in the wild-type protein. Aβ1-40 or Aβ1-42 was fused to the C terminus of the BRI protein at the furin cleavage site. This cleavage releases Aβ into the lumen or extracellular space, resulting in efficient secretion. (B) IP/MS analysis of Aβ isoforms in BRI-Aβ mice. The spectra from the BRI-Aβ42 lines had single peaks detected with a mass/charge ratio (m/z) of 4515 corresponding to human Aβ1-42. In BRI-40 mice, one peak was detected at m/z 4330 corresponding to human Aβ1-40. The shoulder of the 4330 peak corresponds to the Aβ40 sodium adduct ions. Peaks corresponding to human Aβ1-38, Aβ1-40, and Aβ1-42 were detected in extracts from Tg2576 and were absent in nontransgenic mice and BRI wild-type mice. (C) RIPA-soluble (RIPA) and (D) RIPA-insoluble formic acid extractable (FA) Aβ levels in hemibrains (n = 3-7 mice per age point per group) from BRI-Aβ mice were quantified by Aβ sandwich ELISAs. FA Aβ42 increased dramatically with age in the BRI-Aβ42 mice. There was no evidence for accumulation of insoluble Aβ40 in the BRI-Aβ40 mice with age.
Results
Generation of BRI-Aβ Transgenic Mice
The mouse prion promoter (MoPrp) (Borchelt et al., 1996) was used to drive BRI-Aβ40 and BRI-Aβ42 trans-gene expression. Linearized constructs were injected into fertilized oocytes from B6C3F1 mice. Multiple BRI-Aβ42 and BRI-Aβ40 founders were generated and maintained on a B6C3 background. Transgenic founders were screened by plasma-sandwich ELISAs. Six of the highest Aβ42-expressing founders, with plasma Aβ42 levels equivalent to or higher than Aβ42 levels in aged-matched Tg2576 mice (Hsiao et al., 1996), were selected for further breeding. Subsequently, the two lines that had the highest level of expression were chosen for further study. One BRI-Aβ1-40 founder had Aβ40 plasma levels higher than levels seen in age-matched Tg2576 mice (Kawarabayashi et al., 2001), and this line was also expanded.
Transgene expression and protein levels in the mice were determined by Northern blotting, in situ hybridization, and Western analysis (see Figure S1 in the Supplemental Data available online). Included in this analysis were BRI wild-type mice expressing the wild-type BRI protein under control of the MoPrP promoter. The BRI-Aβ40 line expressed the transgene at ∼2- to 3-fold higher levels than the BRI-Aβ42A mice. In all lines, BRI-Aβ mRNA was expressed in a pattern characteristic of the MoPrP promoter (Borchelt et al., 1996); with highest expression in the cerebellar granule cells and hippo-campus, followed by the cortex, pons, thalamus, and midbrain (Figures S1A and S1B). In the two highest expressing BRI-Aβ42 lines, BRI-Aβ42A and BRI-Aβ42B, the fusion transgene was expressed at levels approximating endogenous mouse APP expression (BRI-Aβ42A) or at approximately half this level (BRI-Aβ42B). Full-length BRI-Aβ proteins levels (∼37 kDa) were highest in BRI-Aβ40, followed by BRI-Aβ42A, with BRI-Aβ42B having the lowest protein levels (Figures S1D and S1E). Processed Aβ species (4 kDa) were detected in both the BRI-Aβ40 and BRI-Aβ42 transgenic mice by immunoprecipitation Western blotting (Figure S1F).
Full-length BRI-Aβ40 or BRI-Aβ42 did not show significant alterations with age (Figure S2). BRI-Aβ40 and BRI-Aβ42 mice have a normal lifespan with no obvious behavioral abnormalities.
Specific Expression of Aβ Peptide Isoforms in Transgenic Mice
Although 4 kDa Aβ species were detetected by Western immunoprecipitation, MALDI-TOF mass spectrometry (IP/MS) (Wang et al., 1996) was used to definitively identify the specific Aβ isoforms in PBS extracts from the brains of BRI-Aβ mice in comparison to Tg2576 (Figure 1B). The BRI-Aβ42 lines show single peaks corresponding to human Aβ1-42. The relative peak heights between the two lines were consistent with other data indicating that the BRI-Aβ42A mice had higher expression than line BRI-Aβ42B. In BRI-40 mice, a single peak was detected that corresponded to human Aβ1-40. Several peaks were detected in extracts from Tg2576 mice that corresponded to human Aβ1-38, Aβ1-40, and Aβ1-42. No peaks were detected in the wild-type BRI transgenic or nontransgenic mice.
BRI-Aβ42 but Not BRI-Aβ40 Mice Accumulate Detergent-Insoluble Aβ
Aβ peptides in the brains of BRI-Aβ42 and BRI-Aβ40 mice from 3 to 22 months of age were characterized by Aβ sandwich ELISAs (Figures 1C and 1D). Brains from Tg2576 mice were used as controls. RIPA-soluble Aβ1-40 levels in 3-month-old BRI-Aβ1-40 mice (80-120 pm/g) were ∼2- to 3-fold higher than levels of Aβ1-40 in 3-month-old Tg2576 mice (∼40 pm/gm). No appreciable change in the level of RIPA-soluble Aβ1-40 was observed up to 22 months of age, and there was no evidence for Aβ accumulation in the RIPA-insoluble formic acid extractable (FA) fraction. Levels of RIPA-soluble Aβ1-42 in 3-month-old BRI-Aβ42A and BRI-Aβ42B brains were ∼2- to 3- and ∼1- to 1.5-fold higher than the Aβ1-42 levels in 3- to 6-month-old Tg2576 brains. Both BRI-Aβ42 lines showed large increases in RIPA-insoluble Aβ1-42 levels with age. In the oldest BRI-Aβ42 mice, the levels of RIPA-insoluble Aβ1-42 exceed the Aβ1-42 levels seen in aged Tg2576 mice. Aβ42 and Aβ40 could be readily detected in plasma from the BRI-Aβ42 and BRI-Aβ40 mice, respectively (Figure S3). No Aβ40 can be detected in plasma from BRI-Aβ42 mice, and conversely, no Aβ42 was detected in plasma from BRI-Aβ40 mice.
Extensive Vascular and Parenchymal Amyloid Pathology in BRI-Aβ42 but Not BRI-Aβ40 Mice
The biochemical analysis indicated that BRI-Aβ42 mice selectively accumulated detergent-insoluble Aβ as they aged and that the BRI-Aβ40 mice did not. Immuno-staining with anti-Aβ antibodies and thioflavin S staining showed that both lines of BRI-Aβ42 mice, but not BRI-Aβ40 mice, developed compact amyloid deposits and diffuse Aβ deposits with age (Figure 2). In contrast to other mutant APP transgenic mice (Hsiao et al., 1996; Price et al., 1998), cerebellar Aβ accumulation was the earliest and most consistent feature in both lines of BRI-Aβ42 mice. In the BRI-Aβ42A mice, cored plaques were observed as early as 3 months of age in the molecular layer, with diffuse Aβ deposition becoming more prominent with age. Forebrain pathology occurred later and was more variable; occasional extracellular plaques were observed in the entorhinal/piriform cortices and the hippocampus at 6 months of age, but were not consistently present until at least 12 months in BRI-Aβ42A mice. The oldest mice had more widespread pathology, developing thioflavin S-positive-cored and diffuse plaques in the cerebellum, cortex, hippocampus, and olfactory bulb, in a similar manner to pathology described for Tg2576 (Figure S4). Onset of amyloid pathology in the BRI-Aβ42B mice was more variable than in the BRI-Aβ42A mice; by 20 months of age, these mice had widespread cerebellar and forebrain pathology with a similar plaque distribution to that seen in the BRI-Aβ42A mice. The greatest amlyoid plaque burden was detected with N-terminal antibodies such as Bam-10 and 82E1 (Figures 2 and 4). Immunostaining with Aβ40 or Aβ42 end-specific antibodies was consistent with the notion that the deposited Aβ was almost entirely composed of Aβ42 (Figure 2E). We could not rule out that trace amounts of Aβ40, derived from endogenous mouse APP, or limited proteolysis, were present in the core of some large compact plaques. Despite similar levels of BRI expression at the mRNA level, there was no pathology apparent in the brains of BRI wild-type mice at any age (up to 24 months; Figure 2L).
Figure 2.
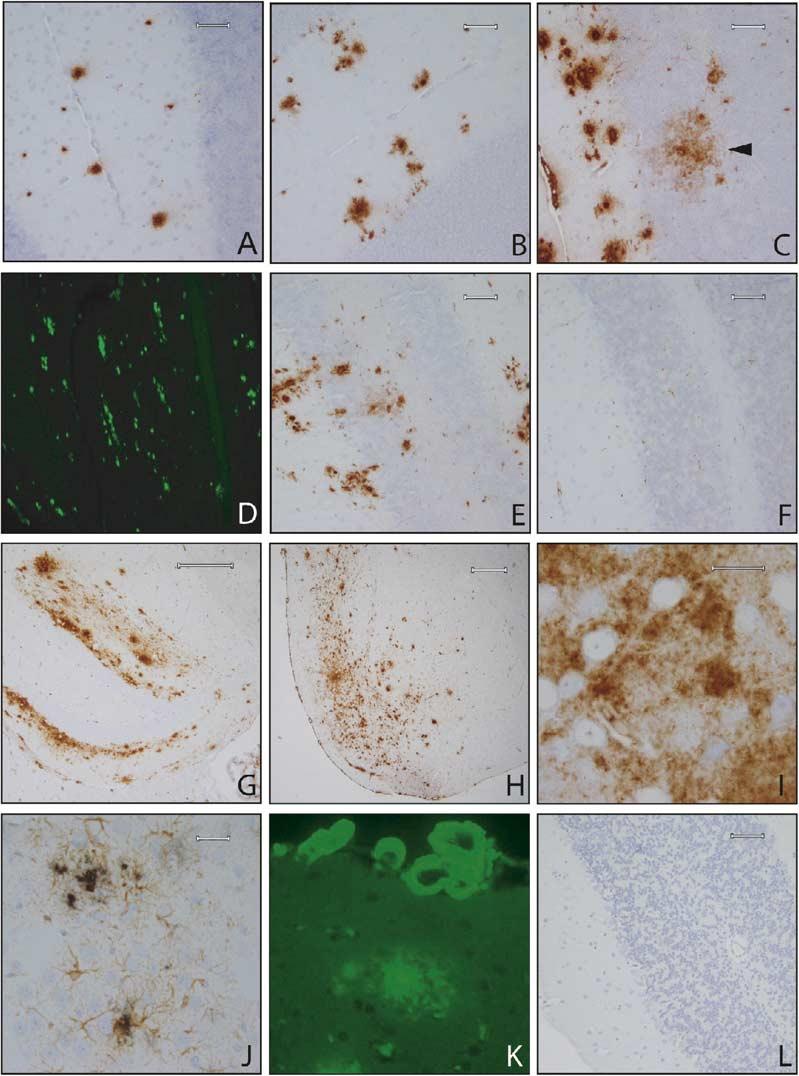
Amyloid Pathology in BRI-Aβ Mice Progression of amyloid deposition (total Aβ immunostaining with 33.1.1 raised against Aβ1-16) in the cerebellum of BRI-Aβ42A mice at 3 months (A), 6 months (B), and 16.5 months (C). BRI-Aβ42 mice develop both diffuse (arrow in [C]) and compact thioflavin S-positive deposits (D). Plaques were immunoreactive with Aβ42 end-specific antibodies (E). No pathology was evident even in 16.5-month-old BRI-Aβ40 mice, immuno-stained with a panel of Aβ antibodies or thioflavin S (F). Old (19 month) BRI-Aβ42 mice show widespread forebrain Aβ deposits, particularly in the molecular layer of the hippocampus (G) and the entorhinal/piriform cortex (H). The entorhinal Aβ deposits were largely diffuse, and neurons were often observed within these diffuse deposits (I). Increased gliosis was associated with plaque deposition (J). Sections were immunostained with anti-GFAP (brown reaction product) followed by immunostaining for Aβ using the antibody BAM-10 (black reaction product). CAA was progressive and severe in old mice (K); vessels were stained with thioflavin S. A dense cored plaque is also visible. The vascular deposits were fibrillar, as demonstrated by electron microscopy (see Figure 3). (L) wild-type BRI mice had no amyloid pathology (total Aβ immunohistochemistry) at ages up to 24 months of age. Scale bars: 60 μm (A—F), 200 μm (G), 300 μm (H), 20 μm (I), 30 μm (J).
Figure 4.

Mouse Aβ and Modified Aβ Peptides Are Present in the Plaques of BRI-Aβ42 Mice (A) Senile plaques in the temporal gyrus from an AD brain, Braak stage VI, immunostained with anti-Aβ1-16 (33.1.1). (B) A serial section of the AD tissue in (A) immunostained with anti-rodent Aβ that does not detect human Aβ deposited in AD. (C) Total Aβ immunostaining using antibody 33.1.1, in the cerebellum of a 17-month-old BRI-Aβ mouse shows multiple cored plaques, a subset of these plaque cores are immunopositive for rodent Aβ in a serial section (D). Arrows in (C) and (D) indicate the same plaques immunostained with 33.1.1 and rodent Aβ, respectively. (E) Diffuse and dense cored plaques in the cerebellum were immunopositive with an anti-Aβ1-16 (82E1, IBL), a subset of these plaques were stained by an antibody that recognizes a truncated N-terminal modified pyrogluta-mate form of Aβ (N3pE Aβ) (F). Arrows in (E) and (F) indicate the same plaques immunostained with anti-Aβ1-16 and anti-N3pE Aβ, respectively. Scale bars: 200 μm (A—D) and 60 μm (E and F).
Congophilic amyloid angiopathy (CAA) was absent in the BRI-Aβ40 mice, but was a prominent feature in the BRI-Aβ42 mice (Figure 2K). EM analysis demonstrated that the CAA was fibrillar and closely associated with the basal lamina (Figures 3A and 3B), similar to CAA observed in AD. EM analysis of extracellular amyloid plaques showed dense amyloid cores with radiating fibrils (Figure 3C). Many bundles of dystrophic neurites were also present at the periphery of the dense cored extracellular plaques. Compact plaques were small relative to those seen in Tg2576 mice (mean plaque diameter 38 ± 5 μm at 3 months to 53 ± 3 μm at 16-18 months of age). Reactive astrogliosis was associated with the compact plaques (Figure 2J). There was little evidence of tau pathology, with only rare CP13-positive dystrophic neurites seen in association with the dense cored plaques in the oldest mice.
Figure 3.
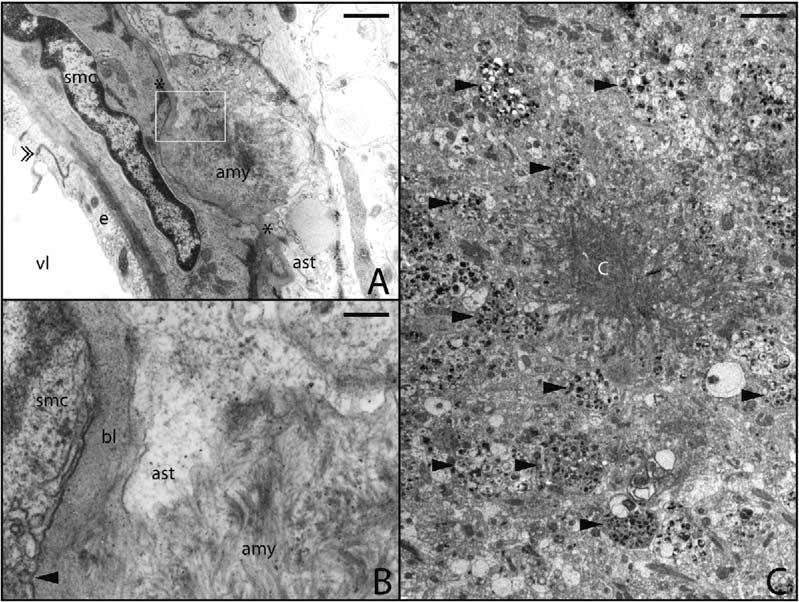
Electron Microscopy of Congo-philic Amyloid Angiopathy and Aβ Plaques in BRI-Aβ42 Mice CAA was pronounced in BRI-Aβ42 mice. (A) Amyloid deposit (amy) at the outer basal lamina (marked with *) of an arteriole found at the cortical surface. An interendothelial junction is indicated by the double arrowhead. The boxed area in (A) is enlarged in (B). (B) Note the disruption of basal lamina (bl) between the astrocyte (ast) and smooth muscle cells (smc) by bundles of amyloid fibrils (amy). Arrowhead points to surface vesicles of SMC. Many of the extracellular amyloid plaques in the BRI-Aβ mice had dense amlyoid aggregates with radiating fibrils (C). There were many dystrophic neurites (arrow-heads) present at the periphery of these plaques. vl, lumen; e, endothelium; SMC, smooth muscle cell; Ast, astrocytes. Scale bars: 1 μm (A), 30 nm (B), and 5 μm (C).
To determine if endogenous mouse Aβ was present in the deposits, a monoclonal antibody raised against aggregated mouse Aβ42 (32.4.1) that specifically recognizes an epitope in mouse Aβ1-16 and does not cross-react with human Aβ by ELISA (data not shown) was used to stain brain sections from human AD and BRI-Aβ42 mice. This antibody did not detect plaques from human AD tissue (Figures 4A and 4B). In BRI-Aβ42 mice, the antibody labeled a subset of the Aβ plaque cores, particularly in the cerebellum, and some diffuse plaques in older mice (Figures 4C and 4D). These data indicate that endogenous mouse Aβ is present in the plaques. We also immunostained sections from the BRI-Aβ42 mice with an antibody specific for the N-terminal truncated and modified pyroglutamate Aβ species Aβ[N3pe]. A proportion of the cored plaques and, to a lesser extent, the diffuse amyloid were immuno-positive for Aβ[N3pe], showing that this N-terminal modified pyroglutamate Aβ species was present in the plaques in the BRI-Aβ mice (Figures 4E and 4F).
Enhanced Pathology in BRI-Aβ42 × Tg2576 Bigenic Mice
Hemizygous BRI-Aβ42A mice were crossed with hemizygous APPsweTg2576 mice, and the amount of Aβ deposition in the bigenic BRI-Aβ42/Tg2576 offspring, age-matched singly transgenic BRI-Aβ42A, and Tg2576 and nontransgenic littermates was analyzed by biochemical and immunohistochemical methods. Amyloid deposition initially develops in the cerebellum of BRI-Aβ42 mice and is extensive by 12 months of age, whereas, forebrain pathology only consistently develops after 12 months of age. In contrast, amyloid deposition in Tg2576 mice only occurs in the forebrain after 6 months of age, and no cerebellar deposits are routinely observed. For this reason, we analyzed the hindbrain (including cerebellum) and forebrain Aβ levels independently at 6, 10, and 14.5 months (Figure 5). Anti-Aβ immunohistochemsitry showed marked augmentation of Aβ deposition throughout the cortex, hippocampus, and cerebellum. Representative sections of the entorhinal/piriform cortical areas from 14.5-month-old BRI-Aβ42A, Tg2576, and bigenic BRI-Aβ42/Tg2576 litter-mate mice are shown in Figures 5A-5C. RIPA-soluble and RIPA-insoluble FA Aβ levels in both the forebrain and hindbrain were analyzed by Aβ ELISA. At 6 months of age, the levels of RIPA-soluble Aβ in the forebrain (Figure S5A), hindbrain (data not shown), and plasma (Figure S5B) of the crossed mice were consistent with an additive effect of the two transgenes with respect to Aβ production, providing assurance that alteration in transgene expression would not confound these studies. A marked increase in both RIPA-insoluble FA extracted Aβ40 and Aβ42 was seen in the forebrain of bigenic mice compared to single transgenic BRI-Aβ42 or Tg2576 littermates (Figure 5D). The increase in FA Aβ levels was greater than that expected for a simple additive effect of Aβ levels in BRI-Aβ42 and Tg2576 (∼30-to 40-fold increase at 10 months of age in the fore-brain). Additionally, although Aβ deposition does not occur in the cerebellum of Tg2576, Aβ40 and Aβ42 levels were substantially elevated in the hindbrain of the bigenic mice compared to the single transgenic littermates (Figure 5E). Taken together these data suggest that Aβ42, by itself, accelerates amyloid deposition in the Tg2576 mice.
Figure 5.

Enhanced Pathology in BRI-Aβ42 × Tg2576 Bigenic Mice Transgenic mice expressing both BRI-Aβ42 and mutant APPswe(Tg2576) have enhanced senile plaque pathology in the forebrain compared with their singly transgenic littermates and significantly elevated insoluble Aβ levels in the forebrain and hindbrain. Entorhinal/piriform cortex of 14.5-month-old BRI-Aβ42A mouse (A), 14.5-month-old Tg2576 (B), 14.5-month-old BRI-Aβ42 × Tg2576 bigenic mouse (C) immunostained with anti-total Aβ (33.1.1). All mice were littermates. FA Aβ levels in the forebrain (D) and hindbrain (E) of an aging series of bigenic BRI-Aβ/Tg2576 compared with their single transgenic littermates. Scale bars: 300 μm (A—C).
Discussion
We have developed transgenic mice expressing BRI-Aβ40 or BRI-Aβ42 fusion proteins. Biochemical analyses of these mice demonstrate that these transgenes selectively express the encoded Aβ peptide in vivo. Since there is no pathological phenotype in mice over-expressing the BRI wild-type protein under control of the MoPrP, any resultant phenotype is likely to be attributed to the specific Aβ peptide expressed. BRI-Aβ40 mice express 2- to 3-fold higher levels of Aβ1-40 than Tg2576 mice, but do not develop Aβ deposits by 24 months of age. In contrast, the BRI-Aβ42 lines, which have ∼5- to 10-fold lower transgene expression than BRI-Aβ40 mice, develop robust parenchymal and cerberovascular Aβ deposits as early as 3 months of age. Reactive gliosis was observed in the BRI-Aβ42 mice, but the mice had neither obvious neuronal cell loss nor prominent neurofibrillary pathology. The lack of a more robust AD-like phenotype in the BRI-Aβ42 mice suggests that the absence of a more prominent AD phenotype in mutant APP mice cannot be attributed to protection conferred by overexpression of other APP derivatives (e.g., sAPPα) (Mattson, 2004), although this can only be definitely tested by crossing the BRI-Aβ42 mice into an APP knockout background. Bigenic BRI-Aβ42/Tg2576 mice show a marked increase in the extent of Aβ deposition and validate the physiologic relevance of selectively increasing Aβ42 via the BRI fusion proteins. Such data are entirely consistent with the increases in deposition produced by APP and PS mutant transgenes that shift γ-secretase cleavage to augment Aβ42 production and accelerate Aβ deposition (Borchelt et al., 1997; Holcomb et al., 1998).
The pattern of amyloid deposition in BRI-Aβ42 mice generally followed the expression pattern of the PrP transgene (Borchelt et al., 1996) and was similar to Tg2576 pathology except for the presence of a large number of plaques in the cerebellum of BRI-Aβ42 mice. The extensive Aβ42 deposition in the cerebellum and severe CAA in the BRI-Aβ42 mice mimics the pattern of amyloid deposition seen in FBD (Ghiso et al., 2000; Holton et al., 2001; Vidal et al., 1999). Though high levels of transgene expression are seen in the cerebellum of both Tg2576 and BRI-Aβ42 mice, we do find that the level of RIPA-soluble Aβ in the nondepositing Tg2576 forebrain is ∼50% higher than that found in the hind-brain and that BRI-Aβ42 mice generate significantly higher Aβ levels in their hindbrain compared to Tg2576 mice. This may indicate that the level of enzymatic processing of BRI-Aβ42 and APP varies in different brain regions and might in turn partially account for the difference in the distribution of amlyoid deposition in Tg2576 and BRI-Aβ42 mice and between AD and FBD. However, the data from the BRI mice are potentially confounded by the fact that cerebellar Aβ deposits are present by 3 months of age, preventing us from definitively establishing steady-state Aβ levels prior to deposition.
As has been reported from biochemical data in one APP transgenic line (Pype et al., 2003), we find immunohistochemical evidence for codeposition of mouse Aβ within a subset of plaques. Such data are not surprising given that in vitro data demonstrate that human and mouse Aβ can coassemble into aggregates (Fung et al., 2004). We also find evidence for truncation and modification of the deposited Aβ; a subset of plaques are labeled by an antibody recognizing Aβ[N3pe]. The most prominent staining with this antibody was seen in the compact cerebellar plaques of old (18 month) BRI-Aβ42 mice. Since the cerebellum is the initial site of deposition in these mice, such data would be consistent with postdeposition modification of the deposited Aβ.
A comparison of the various transgenic models of Aβ deposition reveals that there is a great deal of variability with respect to the age at which deposition begins, the nature and composition of the Aβ deposits (compact versus diffuse; proportion of Aβ40 versus Aβ42), and the extent of CAA (Borchelt et al., 1997; Chishti et al., 2001; Games et al., 1995; Herzig et al., 2004; Holcomb et al., 1998; Hsiao et al., 1996; Mucke et al., 2000; Sturchler-Pierrat et al., 1997). One factor that clearly regulates Aβ deposition is the relative levels of Aβ42, which can be manipulated by either expressing APP transgenes with FAD-linked mutations (e.g., APP V717I/F mutations) or with coexpression of mutant presenilin transgenes that shift γ-secretase cleavage to favor increased production of Aβ42. Increasing relative Aβ42 levels lowers the age when deposition first occurs, increases the amount of diffuse Aβ, and promotes parenchymal versus cerebrovascular deposition. In contrast, a high ratio of Aβ40 to Aβ42 was associated with severe CAA in the absence of parenchymal amyloid (Herzig et al., 2004). However, because these mutations have other physiological effects, it was possible that other alterations in function contributed to the phenotype. Our data critically extend these observations and provide unequivocal in vivo evidence that, in the absence of mutations within the Aβ sequence, Aβ42 expression appears to be necessary for both parenchymal and cerebrovascular amyloid deposition. Thus, although the Aβ40/Aβ42 ratio influences the extent of vascular versus parenchymal deposition, Aβ42 appears to be required to “seed” both types of deposits.
In vitro biophysical studies of synthetic Aβ demonstrate that Aβ42 aggregates more readily than Aβ40, presumably because Aβ42 nucleates more efficiently. Following nucleation, there seems to be little kinetic or thermodynamic differences between elongation of fibrils composed of either Aβ40 or Aβ42 (Hasegawa et al., 1999; Jarrett et al., 1993; Jarrett and Lansbury, 1993). Although numerous trans-acting factors such as ApoE (Bales et al., 1999; Holtzman et al., 2000) and Aβ degrading enzymes can modulate Aβ deposition (Farris et al., 2003; Ohno et al., 2004), alterations in these factors do not result in complete reversal of the deposition phenotype. Thus, the simplest explanation for the complete lack of pathology in the BRI-Aβ40 mice would be the relative inability of Aβ40 to initiate nucleation events capable of promoting amyloid deposition. Similar in vivo results have been obtained in a Drosophila model using a different fusion protein strategy (Iijima et al., 2004). Expression of Aβ1-42, but not Aβ1-40, promoted peptide accumulation of diffuse intracellular and extracellular deposits in the transgenic flies. Significantly, such Aβ42 accumulation was associated with neurodegeneration.
These findings raise an important question regarding the possible role of intracellular Aβ in Alzheimer's disease. Previous studies with transgenic mice expressing Aβ minigenes that are not efficiently secreted suggest that intracellular Aβ can be neurotoxic (LaFerla et al., 1995), as do studies of direct injection of aggregated Aβ into cultured primary neurons (Zhang et al., 2002). However, none of these studies prove or disprove a role of intracellular Aβ in AD pathogenesis. The BRI-Aβ mice that we have developed are clearly distinct from these other “minigene” models in that they efficiently secrete Aβ. To date we have little evidence for accumulation of intracellular Aβ in these mice; thus, they provide an excellent tool to study the effects of secreted Aβ independently of APP transgenes.
Collectively our data provide further support for the hypothesis that Aβ42 is the initiating molecule in the pathogenesis of AD (Hardy and Selkoe, 2002; Younkin, 1998). These data provide definitive in vivo confirmation of predictions inferred from (1) in vitro studies of Aβ aggregation, (2) effects of PS and certain APP mutations on Aβ production and deposition, and (3) analyses of Aβ peptides in the human AD brain (Gravina et al., 1995; Iwatsubo et al., 1994). Although our studies show that Aβ42 is necessary to initiate aggregation of Aβ, they do not demonstrate that aggregation of Aβ42, by itself, is sufficient to induce all the hallmark pathologies of AD in mice, including neurofibrillary tangles and cell loss. This may reflect a simple limitation of the mouse models that we and others employ, or could point to some more complex aggregation phenomena that may require interaction of Aβ42 with Aβ40 or other Aβ species to drive AD pathogenesis. In any case, the BRI-Aβ transgenic mice that we have developed provide models for further studies of the potential physiologic and pathologic roles of Aβ40 and Aβ42 in vivo.
Experimental Procedures
Generation of Transgenic Mice
The BRI-Aβ40, BRI-Aβ42 fusion constructs (Lewis et al., 2001), or BRI wild-type constructs were subcloned into the MoPrP/bluescript vector (Borchelt et al., 1996) at the XhoI restriction site. The transgene was linearized by NotI restriction digest, gel purified, and digested with β-agarase according to the manufacturer's conditions (New England Biolabs). The linearized construct was injected into the pronuclei of single-cell embryos from C3B6F1 × B6 mice and implanted into pseudopregnant females. Founder pups were identified by PCR for the fusion construct or wild-type construct against an internal PS2 control PCR. Founder mice were back-crossed to C3B6F1 mice, and the transgenic lines were maintained on a C3B6 hybrid strain. All animal procedures were conducted with Mayo Clinic's animal care and use committee approval.
Bigenic BRI-Aβ42 × Tg2576 Mice
Hemizygous BRI-Aβ42A mice were mated with Tg2576 mice expressing mutant APPsweunder control of the hamster prion promoter (Hsiao et al., 1996), generating bigenic BRI-Aβ42/Tg2576, BRI-Aβ42, Tg2576, and nontransgenic offspring. Tg2576 mice were maintained on a B6/SJL background. For all studies, age- and strain-matched littermates were used.
In Situ Hybridization
Sagital cryostat sections (15 μm) were fixed in 4% paraformaldehyde, dehydrated, and hybridized with a transgene-specific oligomer (5'-ACACAGTCGATAAATAAAGAAACCCAGGTGATCAATG-3') 3'-end labeled with α35S dATP. Sections were hybridized at 37°C overnight in buffer containing 4× SSC, 1× Denhardt's solution, 50% w/v deionized formamide, 10% w/v dextran sulfate, 200 mg/μl herring sperm DNA, and 0.03% β-mercaptoethanol. Control sections were hybridized in the presence of a 50- to 100-fold molar excess of unlabeled oligonucleotide. After hybridization, the sections were stringently washed (1× SSC at 50°C), dehydrated, and exposed to β max hyperfilm (Amersham) for 7-10 days.
Northern Blotting
Total RNA was extracted from the brains of male and female BRI-Aβ mice using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Nontransgenic and mutant BRI transgenic mice were used as controls. Total RNA (15 μg) was run on a MOPS-formaldehyde denaturing gel and transferred to nitrocellulose membranes by capillary action. The blot was probed with a 32Pradiolabeled oligonucleotide probe specific for both mouse and human BRI, stripped, and reprobed with a 32P-radiolabeled APP cDNA probe and subsequently with a 32P-radiolabeled histone probe. Band densities were quantified using phosphorimage screens (Kodak) and Imagequant software (Molecular Dynamics).
Western Analysis
Proteins were extracted from brain tissues by sonication in a 2% SDS/1% protease inhibitor cocktail solution. The resulting homogenate was centrifuged at 100,000 × g for 1 hr at 4°C. The supernatant containing the SDS-soluble fraction was aliquoted and stored at -80°C. Total protein concentrations were determined using the “BCA Protein Assay kit” (Pierce). Protein extracts from BRI-Aβ mice were run on a SDS denaturing gel and transferred to Immobilon P membranes. Tg2576 and wild-type BRI transgenic lines were included as controls. Blots were probed with monoclonal antibodies to human Aβ (6E10 and BAM10) followed by anti-mouse HRP or anti-rabbit HRP conjugated secondary antibody, respectively. Blots were stripped and reprobed with anti-β-actin or α-tubulin as a loading control.
Immunoprecipitation Western Analysis
Brain samples were homogenized in 2% SDS and diluted in RIPA buffer. Protein G was added and the sample mixed at 4°C for 1 hr prior to centrifugation for 60 s at 13000 rpm. The rabbit polycolonal Aβ antibody, 3160, with Protein G (Pierce) was used to immuno-precipitate the Aβ peptide at 4°C overnight. Then samples were centrifuged and the supernatants run on SDS denaturing gels (Biorad) and transferred to 0.2 μm nitrocellose membranes. Blots were boiled for 10 min in 0.1 M PBS and probed with the monoclonal anti-Aβ, 6E10, followed by anti-mouse HRP. Tg2576 and nontransgenic samples and Aβ peptides were run as controls.
Mass Spectrometry
Six-month-old brains from the BRI-Aβ42, BRI-Aβ40, Tg2567, wild-type BRI lines, and nontransgenic littermates were homogenized in PBS plus protease inhibitor solution to extract soluble proteins. Human Aβ peptides were isolated by immunoprecipitation using two monoclonal anti-Aβ antibodies (6E10 and 4G8) and analyzed by a MALDI-TOF mass spectrometry (Wang et al., 1996).
Aβ Sandwich ELISAs
Mouse hemibrains (excluding the olfactory bulb) for BRI-Aβ40 and BRI-Aβ42 studies or forebrain and hindbrain for BRI-Aβ42 × Tg2576 studies were homogenized in RIPA buffer and ultracentrifuged to separate RIPA-soluble and -insoluble fractions. The insoluble proteins were extracted using 70% formic acid. Formic acid samples were neutralized with Tris base buffer, and all samples were appropriately diluted (from 10- to 5000-fold). For plasma analysis of Aβ, blood samples were centrifuged at 3000 rpm for 10 min at 4°C and the plasma decanted and diluted appropriately. Aβ levels were determined by end-specific sandwich ELISAs (Kawarabayashi et al., 2001) using Ab9 (anti Aβ1-16 of Aβ) as the capture antibody and 21.3.1 (anti Aβ 35-42) or 13.1.1 (anti Aβ 35-40) as the detection antibodies for Aβ42 and Aβ40, respectively (n = 3-7 mice per age point per group). Nontransgenic mice (n = 13) and Tg2576 mice at 3, 6, 12 and 18 months (n = 3 at each time point) were included on each ELISA plate as controls.
Immunohistochemistry
Paraffin-embedded tissue sections (5 μm) from BRI-Aβ40, BRI-Aβ42, and wild-type BRI mice (3-22 months of age, minimum of n = 3 per age group) were immunostained with Aβ antibodies, including anti-total Aβ (Bam 10, 1:50,000, Sigma; 6E10, 1:10,000, Signet; 33.1.1, 1:1,000; T. Golde; 82E1, 1:1000, IBL), anti-Aβ40 (13.1.1., 1:1,000, T. Golde), anti-Aβ42 (21.3.1., 1:1,000, T. Golde), 4G8 (Aβ17-24; 1:5,000, Signet), anti-Aβ[n3pE] (8E1, 3rd N-terminal residue converted to pyroglutamate, 1:1000, IBL), and anti-rodent Aβ (32.4.1. 1:1000, C. Eckman/T. Golde) on a DAKO autostainer using standard avidin or streptavidin biotin peroxidase methods. Additional sections were immunostained with thioflavin S, anti-tau antibodies (CP13, 1:100, P. Davies), and glial markers (GFAP, 1:100, Biogenex).
Electron Microscopy
BRI-Aβ mice were perfused with saline followed by 2.5% glutaraldehyde/2% paraformaldehyde/0.1 M cacodylate buffer. The entorhinal cortex was removed, cut into 2 mm3pieces, postfixed in 1% osmium tetroxide and 1% uranyl acetate, dehydrated in a series of alcohol and propylene oxide, infiltrated, and embedded in EPON 812. Thin sections were stained with uranyl acetate and lead citrate prior to examination with a Philips 208S electron microscope.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. D. Borchelt for the MoPrP vector; F. Conkle, E. Kirkham, T. Hammond, and the veterinary medicine staff for animal maintenance; and L. Rousseau and V. Phillips for tissue processing and histology. NIA RO1 AG022595-01 to E.M., NCI CA88325 to R.W. and the Mayo Clinic Alzheimer's Disease Research Center supported this study. Additional resources from the Mayo Foundation provided by a gift from Robert and Clarice Smith were used to support the Tg2576 mouse colony.
Footnotes
Supplemental Data: The Supplemental Data for this article can be found online at http://www.neuron.org/cgi/content/full/47/2/191/DC1/.
References
- Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, Fishman CE, DeLong CA, Piccardo P, Petegnief V, et al. Proc. Natl. Acad. Sci. USA. Vol. 96. 1999. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer's disease; pp. 15233–15238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet. Anal. 1996;13:159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 2001;276:21562–21570. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intra-cellular domain in vivo. Proc. Natl. Acad. Sci. USA. 2003;100:4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung J, Frost D, Chakrabartty A, McLaurin J. Interaction of human and mouse Abeta peptides. J. Neurochem. 2004;91:1398–1403. doi: 10.1111/j.1471-4159.2004.02828.x. [DOI] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, et al. Alzheimer-type neuropathology in transgenic mice over-expressing V717F β-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Ghiso J, Vidal R, Rostagno A, Miravalle L, Holton JL, Mead S, Revesz T, Plant G, Frangione B. Amyloidogenesis in familial British dementia is associated with a genetic defect on chromosome 13. Ann. N Y Acad. Sci. 2000;920:84–92. doi: 10.1111/j.1749-6632.2000.tb06908.x. [DOI] [PubMed] [Google Scholar]
- Golde TE, Eckman CB, Younkin SG. Biochemical detection of Abeta isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer's disease. Biochim. Biophys. Acta. 2000;1502:172–187. doi: 10.1016/s0925-4439(00)00043-0. [DOI] [PubMed] [Google Scholar]
- Gravina SA, Ho L, Eckman CB, Long KE, Otvos L, Jr., Younkin LH, Suzuki N, Younkin SG. Amyloid beta protein (A beta) in Alzheimer's disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at Abeta 40 or A beta 42(43) J. Biol. Chem. 1995;270:7013–7016. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hasegawa K, Yamaguchi I, Omata S, Gejyo F, Naiki H. Interaction between Abeta(1-42) and Abeta(1-40) in Alzheimer's beta-amyloid fibril formation in vitro. Biochemistry. 1999;38:15514–15521. doi: 10.1021/bi991161m. [DOI] [PubMed] [Google Scholar]
- Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, Danner S, Abramowski D, Sturchler-Pierrat C, Burki K, et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat. Neurosci. 2004;7:954–960. doi: 10.1038/nn1302. [DOI] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- Holton JL, Ghiso J, Lashley T, Rostagno A, Guerin CJ, Gibb G, Houlden H, Ayling H, Martinian L, Anderton BH, et al. Regional distribution of amyloid-Bri deposition and its association with neurofibrillary degeneration in familial British dementia. Am. J. Pathol. 2001;158:515–526. doi: 10.1016/S0002-9440(10)63993-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. USA. 2000;97:2892–2897. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Iijima K, Liu HP, Chiang AS, Hearn SA, Konsolaki M, Zhong Y. Dissecting the pathological effects of human Abeta40 and Abeta42 in Drosophila: a potential model for Alzheimer's disease. Proc. Natl. Acad. Sci. USA. 2004;101:6623–6628. doi: 10.1073/pnas.0400895101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of A beta 42(43) and Abeta 40 in senile plaques with and-specific Abeta monoclonals: evidence that an initially deposited species is A beta 42(43) Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- Jarrett JT, Lansbury PT., Jr. Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- Jarrett JT, Berger EP, Lansbury PT., Jr. The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J. Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat. Rev. Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Tinkle BT, Bieberich CJ, Haudenschild CC, Jay G. The Alzheimer's Abeta peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nat. Genet. 1995;9:21–30. doi: 10.1038/ng0195-21. [DOI] [PubMed] [Google Scholar]
- Lewis PA, Piper S, Baker M, Onstead L, Murphy MP, Hardy J, Wang R, McGowan E, Golde TE. Expression of BRI-amyloid beta peptide fusion proteins: a novel method for specific high-level expression of amyloid beta peptides. Biochim. Biophys. Acta. 2001;1537:58–62. doi: 10.1016/s0925-4439(01)00054-0. [DOI] [PubMed] [Google Scholar]
- Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, Bredesen DE. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat. Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF. BACE1 Deficiency Rescues Memory Deficits and Cholinergic Dysfunction in a Mouse Model of Alzheimer's Disease. Neuron. 2004;41:27–33. doi: 10.1016/s0896-6273(03)00810-9. [DOI] [PubMed] [Google Scholar]
- Price DL, Tanzi RE, Borchelt DR, Sisodia SS. Alzheimer's disease: genetic studies and transgenic models. Annu. Rev. Genet. 1998;32:461–493. doi: 10.1146/annurev.genet.32.1.461. [DOI] [PubMed] [Google Scholar]
- Pype S, Moechars D, Dillen L, Mercken M. Characterization of amyloid beta peptides from brain extracts of transgenic mice overexpressing the London mutant of human amyloid precursor protein. J. Neurochem. 2003;84:602–609. doi: 10.1046/j.1471-4159.2003.01556.x. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol. 1998;8:447–453. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc. Natl. Acad. Sci. USA. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal R, Frangione B, Rostagno A, Mead S, Revesz T, Plant G, Ghiso J. A stop-codon mutation in the BRI gene associated with familial British dementia. Nature. 1999;399:776–781. doi: 10.1038/21637. [DOI] [PubMed] [Google Scholar]
- Vidal R, Revesz T, Rostagno A, Kim E, Holton JL, Bek T, Bojsen-Moller M, Braendgaard H, Plant G, Ghiso J, Fran-gione B. A decamer duplication in the 3' region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred. Proc. Natl. Acad. Sci. USA. 2000;97:4920–4925. doi: 10.1073/pnas.080076097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Sweeney D, Gandy SE, Sisodia SS. The profile of soluble amyloid beta protein in cultured cell media. Detection and quantification of amyloid beta protein and variants by immunoprecipitation-mass spectrometry. J. Biol. Chem. 1996;271:31894–31902. doi: 10.1074/jbc.271.50.31894. [DOI] [PubMed] [Google Scholar]
- Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science. 1989;245:417–420. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]
- Younkin SG. The role of Abeta 42 in Alzheimer's disease. J. Physiol. (Paris) 1998;92:289–292. doi: 10.1016/s0928-4257(98)80035-1. [DOI] [PubMed] [Google Scholar]
- Zhang Y, McLaughlin R, Goodyer C, LeBlanc A. Selective cytotoxicity of intracellular amyloid beta peptide 1-42 through p53 and Bax in cultured primary human neurons. J. Cell Biol. 2002;156:519–529. doi: 10.1083/jcb.200110119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.