

Introduction
Dab1 is critical for Reelin-dependent neuron positioning events during vertebrate brain development [1,2]. Dab1 tyrosine phosphorylation increases in response to Reelin stimulation, and the tyrosine phosphorylation sites are required for Dab1 function in vivo [1]. In addition to tyrosine phosphorylation sites, the Dab1 protein contains an N-terminal phosphotyrosine-binding/protein interaction (PTB/PI) domain which can bind to phosphatidylinositol-4,5-bisphosphate and to the FXNPXY endocytosis signals found in various transmembrane proteins [3-6]. Both peptides and phosphoinositide head groups bind specifically and with micromolar affinity [3,7]. Consistent with this, most Dab1 fractionates with membranes from primary neurons [8], and immunofluorescence shows that Dab1 is in puncta in the neuron membrane [3].
Reelin induces Dab1 tyrosine phosphorylation by binding to its receptors, VLDLR and ApoER2, which also contain the FXNPXY signal that binds Dab1 [9,10]. Additional transmembrane proteins, including integrins and CNR [11-13], may also play a role, but clustering of VLDLR and ApoER2 is sufficient to induce Dab1 tyrosine phosphorylation in cultured neurons [14]. These results suggest that Reelin-induced clustering of VLDLR and ApoER2 may increase Dab1 tyrosine phosphorylation by clustering the FXNPXY Dab1 binding sequences or receptor-associated lipids on the cytoplasmic side of the membrane. This clustering might locally increase Dab1 density, either by recruiting more Dab1 from the cytosol or by clustering pre-bound Dab1 molecules [14]. Clustered Dab1 molecules may be more accessible to membrane-bound kinases or less accessible to phosphatases, or may undergo conformation changes that allow increased phosphorylation. However, the relative contributions of lipid binding and peptide binding to Dab1 function are not known. One possibility is that direct binding of Dab1 to the receptors relays the Reelin signal, while Dab1 binding to phosphoinositides determines the number of Dab1 molecules subject to both basal and Reelin-stimulated phosphorylation. Alternatively, or additionally, the phospholipid environment may be changed when the Reelin receptors are clustered, and this could cause an increase in the local density of Dab1. In this model, phosphoinositide binding but not receptor binding would be required for Reelin-stimulated Dab1 phosphorylation.
To further address the importance of receptor binding and phosphoinositide binding for Reelin-stimulated tyrosine phosphorylation of Dab1, we have made additional mutations in Dab1 and developed a transfection procedure for expressing Dab1 in primary neurons. Here we report that mutating a PTB domain residue that is critical for receptor binding [4,6,7] reduces Reelin-stimulated tyrosine phosphorylation of Dab1 in neurons. In contrast, mutating a residue that is important for phosphoinositide binding and membrane localization [4,6,7] reduces both basal and Reelin-stimulated Dab1 tyrosine phosphorylation in neurons. These results are consistent with a model in which the PTB domain has two functions: first, to recruit Dab1 to phosphoinositides in the membrane, increasing its basal phosphorylation, and, second, to bind membrane-associated Dab1 to Reelin receptors, permitting stimulation of phosphorylation by Reelin.
Materials and Methods
The Dab1 PTB domain (residues 1-178) or full length Dab1 was cloned into pCMX-EGFP [15](kind gift of J. Pines) cut with HindIII and KpnI to make C-terminal fusions. The reading frame was corrected by opening with Acc65I, blunting, and religating. A double PTB construct was made from pCMX PTB-EGFP by opening with SalI, filling in with Klenow, and inserting a blunt-ended HindIII fragment containing residues 23-178 of Dab1. Point mutations were made in pCMX PTB-EGFP using the QuickChange method (Stratagene). The mutant sequences were designed to introduce PvuII (K45Q) or StuI (R124Q) or destroy a BalI (K142Q) restriction site, respectively. Mutant colonies were identified by restriction digest and the complete insert was confirmed by sequencing. 293T cells were transfected with approximately 1 μg DNA using calcium phosphate, and examined by epifluorescence 16 hr after transfection. At 24 hr, the cells were lysed in 1% Nonidet P40 buffer, as described [16], and debris removed by centrifugation. Protein concentrations were equalized using the Bradford protein assay. Lysates were incubated with glutathione Sepharose beads loaded with GST or GST-VLDLR [17] for 1.5 hr at 4 C, then the beads were washed twice in the lysis buffer, eluted, and analyzed by SDS PAGE. Volumes of cell lysate equivalent to 15% of the volume incubated with the GST fusion proteins were analyzed on a parallel gel. Western blots were blocked with BSA and probed with antibodies to GFP (#1 814 460, Roche Diagnostics, Indianapolis, IN), essentially as described [18].
MAP2-EGFP under the control of the hybrid CAG enhancer/promoter was obtained from Dr. Stephanie Kaech, Oregon Health Sciences University [19]. Dab1EGFP was cut out of pCMX Dab1EGFP using HindIII and NheI and inserted in place of the HindIII to XbaI fragment containing MAP2EGFP. A HindIII to ClaI fragment containing part of the Dab1EGFP sequence was mutagenized as above to create S19A, K45Q and S114T mutations. After sequencing, the HindIII to ClaI fragment was replaced into non-mutagenized CAG-Dab1EGFP to minimize the risk of mutations elsewhere in the backbone. These constructs were sequenced again before use.
Neuron suspensions were prepared from E16.5 mouse embryo cerebral cortex essentially as described previously [20]. Following three washes in DMEM containing 10% horse serum, cell pellets containing approximately 107 cells were resuspended in electroporation solution (Neuron kit, Amaxa Inc., Gaithersberg, MD), mixed with 4 μg DNA, and nucleoporated (program O-06, Amaxa). After 10 min recovery, the neurons were plated in DMEM 10% horse serum onto two, 35-mm dishes coated with poly-L-lysine (Sigma) and E-C-L reagent (Upstate Biochemicals). The medium was changed after 4 hr to Neurobasal, 2% B27 supplement, 10 mM HEPES pH 7.4, containing antibiotics, glutamine and glutamic acid. Medium was changed every 2 days, omitting glutamic acid after day 4. On day 5 or 6, neurons were treated for 15 min with Neurobasal medium containing Reelin or control supernatant harvested from 293T cells stably transfected with Reelin or control expression plasmids [18]. Neurons were washed and lysed in NP40 buffer [18], and protein concentrations equalized. Immunoprecipitations were performed with mouse anti-GFP and protein G Sepharose.
Results
In order to study the subcellular localization of Dab1, it was tagged at either N or C terminus with enhanced green fluorescent protein (EGFP) and expressed in 293T cells. Both proteins were enriched at the cell surface, implying that the position of the GFP tag did not interfere with localization (data not shown). When the PTB domain of Dab1, tagged with EGFP, was expressed, it too was membrane associated (Fig. 1A, wildtype). Membrane association was increased when two Dab1 PTB domains were cloned in tandem (Fig. 1A, PTB2-EGFP), indicating that the membrane phosphoinositides are not limiting for Dab1 PTB domain binding in this system. We then attempted to inhibit Dab1 membrane localization by mutating basic residues. A molecular model [21] predicted a basic surface patch on the PTB domain composed of lysine residues 67, 69 or 100. However, mutating these residues to alanine or glutamine did not have noticeable effects on membrane association, and while a triple mutant was less conspicuously at the membrane it also showed reduced binding to the cytoplasmic tail of VLDLR in vitro, suggesting that it was not properly folded (data not shown). With the publication of three-dimensional structures of the Dab1 and Dab2 PTB domains, it became evident that residues 67, 69 and 100 do not contact phosphoinositides, and the binding site is actually composed of basic residues 45, 76, 81, 82, 124 and 142 [4,6]. We therefore targeted lysines 45 and 142 and arginine 124 for mutation to the uncharged but hydrophilic glutamine.
Figure 1.
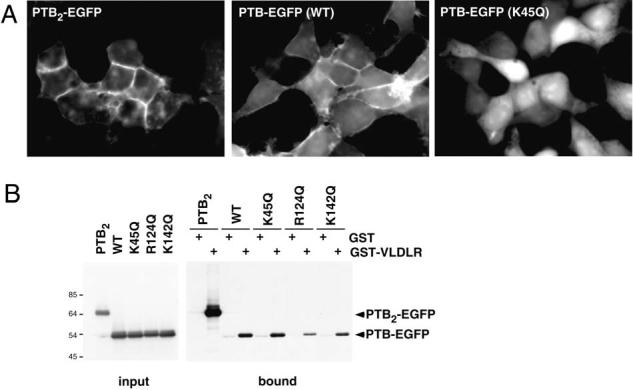
Membrane localization and VLDLR binding of the PTB/PI domain of Dab1. C-terminal EGFP fusions of wildtype or various mutant forms of the Dab1 PTB domain were expressed in 293T cells. (A) Epifluorescence images. Wildtype Dab1 PTB is enriched at cell edges and cell-cell junctions, indicating membrane association. K45Q mutant Dab1 PTB is not membrane associated, and a double PTB protein shows increased association. (B) Binding to the VLDLR C terminus, fused to GST, was assayed in vitro. The double PTB protein bound with higher avidity than the single PTB, and K45Q mutation had no effect on binding, in contrast to R124Q and K142Q.
While wildtype PTB-EGFP was clearly membrane localized, each of the three mutant forms K45Q, R124Q and K142Q were diffusely localized in the cytoplasm (Fig. 1A and data not shown). This suggests that mutating each of these basic residues reduces membrane association. We then tested whether the mutations affected binding of the PTB domain to the FXNPXY signal in the cytoplasmic tail of VLDLR. Cell lysates containing PTB-EGFP fusion proteins were mixed with immobilized GST fusion proteins containing the C terminus of the VLDLR.
Following washing, bound proteins were eluted and analyzed by Western blotting. As a positive control, PTB2-EGFP bound tightly to the VLDLR, as expected [22]. While wildtype and K45Q mutant PTB domains bound well to the VLDLR, the R124Q and K142Q mutants showed reduced binding, suggesting indirect effects on the peptide binding site, perhaps reflecting impaired protein folding (Fig. 1B). Thus the K45Q mutant was selected for further analysis.
In order to express Dab1 constructs in primary neurons, several transfection procedures were compared. We found that nucleoporation of CAG (cytomegalovirus enhancer, chicken beta actin promoter and first intron, rabbit beta globin 3′ flanking sequence [23]) constructs into freshly-dissociated embryonic cortex neurons gave reproducible expression levels (see Experimental Procedures). Transfected neurons were cultured for 4-6 days and stimulated for 15 min with Reelin-containing or control (Mock) supernatant. The expression of Dab1-EGFP relative to endogenous Dab1 could be visualized after SDS PAGE and Western blotting with antibodies to Dab1 (Fig. 2). Dab1-EGFP was expressed at 3-4-fold the level of endogenous Dab1. Following Reelin stimulation, endogenous Dab1 was phosphorylated on tyrosine to a similar level regardless of whether the cells were expressing Dab1-EGFP or a control protein, MAP2-EGFP (Fig. 2). The phosphorylation of Dab1-EGFP was difficult to determine from SDS PAGE of total neuron lysates, since it was obscured by other constitutively phosphorylated proteins of similar mobility. However, after immunoprecipitation with antibodies to GFP and SDS PAGE, Western blotting with antibodies to phosphotyrosine showed that Reelin stimulated the phosphorylation of Dab1-EGFP on tyrosine (Fig. 2). As expected, MAP2-EGFP was not tyrosine phosphorylated. These results show that ectopic Dab1 fusion proteins can interact with the Reelin-regulated phosphorylation machinery in neurons.
Figure 2.

Expression of proteins in primary neuron cultures. Mouse embryonic cortical neurons were electroporated with constructs expressing Dab1-EGFP or MAP2-EGFP. After 4-6 days, cells were stimulated for 15 min with control (mock, M) or Reelin-containing (R) medium, lysed, and analyzed. Western blots of total cell lysates and anti-GFP immunoprecipitates (IPs) are shown. Both Dab1-EGFP and MAP2-EGFP are expressed and detected with anti-GFP in total lysates or GFP IPs. Dab1-EGFP is expressed at higher level than endogenous Dab1 (lysates, anti-Dab1 blot; *, non-specific band). Reelin stimulated the tyrosine phosphorylation of both endogenous Dab1 (lysates, anti-pTyr blot) and Dab1-EGFP (GFP IPs, pTyr blot).
Next, we introduced wildtype, K45Q and S114T Dab1-EGFP constructs into primary neurons. S114T was chosen as a conservative mutation that was reported to decrease binding to an FXNPXY test peptide [4]. A less conservative S114Y substitution was recently reported to completely inhibit binding to an ApoER2 peptide, without compromising binding to phosphoinositide head groups [7]. In some experiments, a S19A mutant was also tested. S19 is homologous to S24 in the related protein, Dab2. Dab2 S24 is a site for protein kinase C modification [24]. After culturing for 4-6 days, the transfected neurons were stimulated as above and lysed and analyzed (Fig. 3). In a typical experiment, the expression levels of K45Q and S114T mutant Dab1-EGFP were greater than those of wildtype or S19A Dab1-EGFP (Fig. 3A). Because neuron cultures contain low levels of Reelin [25], and Dab1 turnover is increased following Reelin-stimulated tyrosine phosphorylation [26,27], increased levels of K45Q and S114T are consistent with decreased Reelin-dependent phosphorylation. Following acute Reelin stimulation, endogenous Dab1 in all cultures and wildtype, S19A and K45Q mutant Dab1-EGFP were tyrosine phosphorylated at increased levels (Fig. 3A). In contrast, only slight changes in tyrosine phosphorylation of S114T mutant Dab1-EGFP were detected following Reelin stimulation. This experiment was repeated three times with independently isolated neurons, and the results were quantified (Fig. 3B). While Reelin stimulated the tyrosine phosphorylation of wildtype and K45Q mutant Dab1-EGFP approximately 3.5-fold, the basal and stimulated stoichiometries of K45Q phosphorylation under mock and Reelin conditions were depressed to approximately one-third of wildtype. Such decreases are unlikely to occur by random chance (P<0.01 for mock treatment, P<0.05 for Reelin treatment). In addition, while S114T basal phosphorylation was not significantly different from wildtype, its phosphorylation after Reelin stimulation was significantly reduced relative to wildtype (P<0.01).
Figure 3.

Tyrosine phosphorylation of wildtype and mutant Dab1 in neurons. (A) Neuron cultures, electroporated with various constructs, were stimulated as in Fig. 2. Lysates and anti-GFPs IPs are shown. In all cultures, tyrosine phosphorylation of endogenous Dab1 was increased by Reelin (lysates, anti-pTyr blot). Dab1-EGFP levels were increased by the K45Q and S114T mutations, consistent with a lower stoichiometry of Reelin-induced tyrosine phosphorylation and reduced Reelin-dependent degradation (lysates and GFP IPs, anti-Dab1 blot). Dab1-EGFP tyrosine phosphorylation was obscured in total lysates by other phosphoproteins, but could be detected in anti-GFP IPs (GFP IPs, pTyr blot). All lanes in each panel were from the same blot, but the image was spliced to remove irrelevant lanes. (B). Quantification of results of three independent experiments. (B, left panel) Mean and standard error of the relative phosphorylation (R={pTyr in Dab1-EGFP}/{Dab1-EGFP protein}), normalized to mock-treated wildtype (R=1.0). (B, right panel) Mean and standard error of the stimulation by Reelin ({R, Reelin}/{R, mock}). P values (t test, single-tailed) are shown.
Discussion
We have found that K45Q mutation of Dab1 decreases membrane association without interfering with binding to the Reelin receptor, VLDLR. In neurons, the K45Q mutation reduces both unstimulated and Reelin-stimulated levels of tyrosine phosphorylation, but does not significantly impact the relative stimulation by Reelin. This result is consistent with a model in which phosphoinositide binding via K45 [4,6,7] is important for recruiting Dab1 to the vicinity of Reelin receptors, but does not provide a mechanism for changing Dab1 phosphorylation in response to Reelin. Thus, models in which reorganization of membrane phospholipids provides the sole mechanism for altering Dab1 tyrosine phosphorylation would not be supported by the results. On the other hand, the S114T mutation, which inhibits binding to FXNPXY sequences [4], probably without affecting phosphoinositide binding [7], does not affect basal phosphorylation but does inhibit Reelin-stimulated phosphorylation. This result is consistent with the Dab1-receptor interaction being dispensable for recruiting Dab1 to the membrane, but required for the Reelin signal to be transduced into an increase in Dab1 tyrosine phosphorylation.
The strong effect of the S114T mutation in this assay differs from the mild effect of an F158V mutation introduced into the dab1 gene in vivo [28]. F158V mutation inhibits binding of Dab1 to FXNPXY sequences [3,4,7], but supported normal brain development in the mouse; a mild phenotype was revealed only when the mutant allele was hemizygous. Moreover, F158V Dab1 tyrosine phosphorylation stoichiometry seemed normal in vivo [28]. One possible explanation for the discrepancy is that mutating F158 is less inhibitory for Reelin receptor binding than mutation of S114 [7].
While our experiments were being completed, Stolt et al. [29] used lentivirus to express wildtype and mutant forms of myc epitope-tagged Dab1 in primary neuron cultures. They found that K45E mutant Dab1 did not associate with neuron membranes and was not tyrosine phosphorylated in response to Reelin. A S114T F158V double mutant Dab1 did associate with membranes but did not undergo Reelin-stimulated phosphorylation. Basal tyrosine phosphorylation was not detected in their experiments. Our finding that a K45Q mutant still responds to Reelin, albeit with reduced basal and stimulated phosphorylation levels, may be due to the different amino acid substitution used, the epitope tag used, or the relative expression level. It is possible that the K45Q mutant, expressed at lower levels, would not have sufficient affinity for membranes to be phosphorylated at all.
In another very recent development, Huang et al. [30] showed that K45A mutant Dab1 is not tyrosine phosphorylated when expressed artificially in 293T cells. It also fails to activate endogenous Src when over-expressed. They also made use of neomycin to disrupt phosphoinositide-dependent interactions. In both 293T cells and neurons, neomycin inhibited Dab1 tyrosine phosphorylation. These observations are consistent with our finding that basal phosphorylation of Dab1 requires phosphoinositide interactions.
In conclusion, the Dab1 PTB domain contains two important binding activities, for phospholipids and Reelin receptors. Some other adaptor proteins, like IRS1, contain two domains, that cooperate for phospholipid binding and receptor interaction [31]. In Dab1 these two functions are subsumed into a single domain, and both functions are required for an efficient Reelin response. Our findings suggest a model in which phosphoinositides recruit Dab1 to the membrane, where it is activated directly by Reelin receptors.
Acknowledgements
We thank Jonathon Pines, Joachim Herz and Stephanie Kaech for plasmids, Lakhu Keshvara and John O'Bryan for interesting discussions, and Nate Allen and Susan Veals for their comments on the manuscript. This work was supported by grant CA41072 from the National Cancer Institute. Reelin-stimulated tyrosine phosphorylation of the Dab1 adaptor protein is required during brain development for Reelin-dependent neuron positioning in the cerebral cortex and various other laminated regions. Dab1 contains an amino-terminal PTB/PI domain through which it can bind to Reelin receptors and membrane phosphoinositides. The relative contributions of these binding activities were unknown. Here, we identify a mutation in the PTB domain of Dab1 that inhibits membrane localization without inhibiting receptor binding. In neurons, this mutation reduces both basal and Reelin-stimulated Dab1 tyrosine phosphorylation. In contrast, a mutation that inhibits receptor binding reduces Reelin-stimulated but not basal tyrosine phosphorylation. These results support a model in which phospholipids recruit Dab1 to membranes but do not play a direct role in relaying the Reelin signal, while direct Dab1-receptor interaction is responsible for relaying the Reelin signal but not for membrane recruitment.
Classification terms: Development and regeneration > Cell differentiation and migration
References
- 1.Jossin Y. Neuronal migration and the role of reelin during early development of the cerebral cortex. Mol. Neurobiol. 2004;30:225–251. doi: 10.1385/MN:30:3:225. [DOI] [PubMed] [Google Scholar]
- 2.Rice DS, Curran T. Role of the reelin signaling pathway in central nervous system development. Annu. Rev. Neurosci. 2001;24:1005–1039. doi: 10.1146/annurev.neuro.24.1.1005. [DOI] [PubMed] [Google Scholar]
- 3.Howell BW, Lanier LM, Frank R, Gertler FB, Cooper JA. The disabled 1 phosphotyrosine-binding domain binds to the internalization signals of transmembrane glycoproteins and to phospholipids. Mol. Cell. Biol. 1999;19:5179–5188. doi: 10.1128/mcb.19.7.5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yun M, Keshvara L, Park CG, Zhang YM, Dickerson JB, Zheng J, Rock CO, Curran T, Park HW. Crystal structures of the dab homology domains of mouse disabled 1 and 2. J. Biol. Chem. 2003;278:36572–36581. doi: 10.1074/jbc.M304384200. [DOI] [PubMed] [Google Scholar]
- 5.Homayouni R, Rice DS, Sheldon M, Curran T. Disabled-1 binds to the cytoplasmic domain of amyloid precursor-like protein 1. J. Neurosci. 1999;19:7507–7515. doi: 10.1523/JNEUROSCI.19-17-07507.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stolt PC, Jeon H, Song HK, Herz J, Eck MJ, Blacklow SC. Origins of peptide selectivity and phosphoinositide binding revealed by structures of Disabled-1 PTB domain complexes. Structure (Camb) 2003;11:569–579. doi: 10.1016/s0969-2126(03)00068-6. [DOI] [PubMed] [Google Scholar]
- 7.Stolt PC, Vardar D, Blacklow SC. The dual-function disabled-1 PTB domain exhibits site independence in binding phosphoinositide and peptide ligands. Biochemistry. 2004;43:10979–10987. doi: 10.1021/bi049092l. [DOI] [PubMed] [Google Scholar]
- 8.Bock HH, Jossin Y, Liu P, Forster E, May P, Goffinet AM, Herz J. Phosphatidylinositol 3-kinase interacts with the adaptor protein Dab1 in response to Reelin signaling and is required for normal cortical lamination. J. Biol. Chem. 2003;278:38772–38779. doi: 10.1074/jbc.M306416200. [DOI] [PubMed] [Google Scholar]
- 9.Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, Herz J. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999;24:481–489. doi: 10.1016/s0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- 10.D'Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T. Reelin is a ligand for lipoprotein receptors. Neuron. 1999;24:471–479. doi: 10.1016/s0896-6273(00)80860-0. [DOI] [PubMed] [Google Scholar]
- 11.Schmid RS, Jo R, Shelton S, Kreidberg JA, Anton ES. Reelin, Integrin and Dab1 Interactions during Embryonic Cerebral Cortical Development. Cereb. Cortex. 2005 doi: 10.1093/cercor/bhi041. [DOI] [PubMed] [Google Scholar]
- 12.Dulabon L, Olson EC, Taglienti MG, Eisenhuth S, McGrath B, Walsh CA, Kreidberg JA, Anton ES. Reelin binds alpha3beta1 integrin and inhibits neuronal migration. Neuron. 2000;27:33–44. doi: 10.1016/s0896-6273(00)00007-6. [DOI] [PubMed] [Google Scholar]
- 13.Senzaki K, Ogawa M, Yagi T. Proteins of the CNR family are multiple receptors for Reelin. Cell. 1999;99:635–647. doi: 10.1016/s0092-8674(00)81552-4. [DOI] [PubMed] [Google Scholar]
- 14.Strasser V, Fasching D, Hauser C, Mayer H, Bock HH, Hiesberger T, Herz J, Weeber EJ, Sweatt JD, Pramatarova A, Howell B, Schneider WJ, Nimpf J. Receptor clustering is involved in Reelin signaling. Mol. Cell. Biol. 2004;24:1378–1386. doi: 10.1128/MCB.24.3.1378-1386.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Umesono K, Murakami KK, Thompson CC, Evans RM. Direct repeats as selective response elements for the thyroid hormone, retinoic acid, and vitamin D3 receptors. Cell. 1991;65:1255–1266. doi: 10.1016/0092-8674(91)90020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arnaud L, Ballif BA, Forster E, Cooper JA. Fyn tyrosine kinase is a critical regulator of Disabled-1 during brain development. Curr. Biol. 2003;13:9–17. doi: 10.1016/s0960-9822(02)01397-0. [DOI] [PubMed] [Google Scholar]
- 17.Stockinger W, Brandes C, Fasching D, Hermann M, Gotthardt M, Herz J, Schneider WJ, Nimpf J. The reelin receptor ApoER2 recruits JNK-interacting proteins-1 and -2. J. Biol. Chem. 2000;275:25625–25632. doi: 10.1074/jbc.M004119200. [DOI] [PubMed] [Google Scholar]
- 18.Ballif BA, Arnaud L, Cooper JA. Tyrosine phosphorylation of disabled-1 is essential for Reelin-stimulated activation of Akt and Src family kinases. Brain Res. Mol. Brain Res. 2003;117:152–159. doi: 10.1016/s0169-328x(03)00295-x. [DOI] [PubMed] [Google Scholar]
- 19.Kaech S, Parmar H, Roelandse M, Bornmann C, Matus A. Cytoskeletal microdifferentiation: a mechanism for organizing morphological plasticity in dendrites. Proc. Natl. Acad. Sci. USA. 2001;98:7086–7092. doi: 10.1073/pnas.111146798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herrick TM, Cooper JA. A hypomorphic allele of dab1 reveals regional differences in reelin-Dab1 signaling during brain development. Development. 2002;129:787–796. doi: 10.1242/dev.129.3.787. [DOI] [PubMed] [Google Scholar]
- 21.O'Bryan JP. The PTB domain: A modular domain with multiple functions. Curr. Opin. Drug Disc. Devel. 1999;2:505–518. [PubMed] [Google Scholar]
- 22.Mishra SK, Keyel PA, Hawryluk MJ, Agostinelli NR, Watkins SC, Traub LM. Disabled-2 exhibits the properties of a cargo-selective endocytic clathrin adaptor. EMBO J. 2002;21:4915–4926. doi: 10.1093/emboj/cdf487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 24.Tseng CP, Ely BD, Pong RC, Wang Z, Zhou J, Hsieh JT. The role of DOC-2/DAB2 protein phosphorylation in the inhibition of AP-1 activity. An underlying mechanism of its tumor-suppressive function in prostate cancer. J. Biol. Chem. 1999;274:31981–31986. doi: 10.1074/jbc.274.45.31981. [DOI] [PubMed] [Google Scholar]
- 25.Koch S, Strasser V, Hauser C, Fasching D, Brandes C, Bajari TM, Schneider WJ, Nimpf J. A secreted soluble form of ApoE receptor 2 acts as a dominant-negative receptor and inhibits Reelin signaling. EMBO J. 2002;21:5996–6004. doi: 10.1093/emboj/cdf599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arnaud L, Ballif BA, Cooper JA. Regulation of protein tyrosine kinase signaling by substrate degradation during brain development. Mol. Cell. Biol. 2003;23:9293–9302. doi: 10.1128/MCB.23.24.9293-9302.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bock HH, Jossin Y, May P, Bergner O, Herz J. Apolipoprotein E receptors are required for reelin-induced proteasomal degradation of the neuronal adaptor protein Disabled-1. J. Biol. Chem. 2004;279:33471–33479. doi: 10.1074/jbc.M401770200. [DOI] [PubMed] [Google Scholar]
- 28.Herrick TM, Cooper JA. High affinity binding of Dab1 to Reelin receptors promotes normal positioning of upper layer cortical plate neurons. Brain Res. Mol. Brain Res. 2004;126:121–128. doi: 10.1016/j.molbrainres.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 29.Stolt PC, Chen Y, Liu P, Bock HH, Blacklow SC, Herz J. Phosphoinositide binding by the disabled-1 PTB domain is necessary for membrane localization and Reelin signal transduction. J. Biol. Chem. 2005;280:9671–9677. doi: 10.1074/jbc.M413356200. [DOI] [PubMed] [Google Scholar]
- 30.Huang Y, Shah V, Liu T, Keshvara L. Signaling through Disabled 1 requires phosphoinositide binding. Biochem. Biophys. Res. Commun. 2005;331:1460–1468. doi: 10.1016/j.bbrc.2005.04.064. [DOI] [PubMed] [Google Scholar]
- 31.Yenush L, White MF. The IRS-signalling system during insulin and cytokine action. BioEssays. 1997;19:491–500. doi: 10.1002/bies.950190608. [DOI] [PubMed] [Google Scholar]