

Abstract
This review discusses the possible involvement of a variety of genetic polymorphisms on the course of meningococcal disease. It has been shown that several common genetic polymorphisms can either influence the susceptibility to meningococcal disease or can account for a higher mortality rate in patients. Gene polymorphisms concerning antibody receptors, lipopolysaccharide (LPS) binding receptors or proteins, innate complement proteins as well as cytokines and hemostatic proteins are described. The study of genetic polymorphisms might provide important insights in the pathogenesis of meningococcal disease and could make it possible to identify individuals who are at risk of either contrcting or dying from meningococcal disease.
Keywords: genetic polymorphisms, meningococcal sepsis, review
Introduction
Neisseria meningitidis is an intracellular Gram-negative diplococcus that can cause serious illnesses in children as well as in adults. The spectrum of disease varies from a common cold to life-threatening disorders including meningitis and/or a fulminant septic shock. Meningococcal sepsis is characterised by a sudden onset of fever and a petechial or purpuric rash, which can be followed by hypotension and multiple organ failure. Mortality rates can be as high as 40% [1]. Meningococcal sepsis is characterised by an exceptionally high level of lipopolysaccharide (LPS) found in blood or cere-brospinal fluid. Additional features of meningococal sepsis include a severe capillary leak syndrome caused by endothelial damage due to circulating mediators, neutrophils and platelets and disseminated intravascular coagulation leading to microthrombi. Late sequelae include skin necrosis and occasionally the amputation of limbs.
Rates of carriage are estimated to be 10% in the general population when cultured by nasopharyngeal swabbing, and up to 45% when cultured from tonsillar tissue [2]. In contrast, typical annual incidences of meningococcal disease are 5.9 per 100,000 in Ireland, 3.5 per 100,000 in The Netherlands and 0.9–1.5 per 100,000 population in the United States [1,3,4]. Thus, despite a high carriage rate, progression to invasive disease occurs rarely. The clinical picture of patients with meningococcal infections admitted to the hospital varies from mild bacteraemia or chronic meningococcaemia to a lethal septic shock syndrome. This raises two questions: Why do some patients die within hours despite intensive treatment? Why is meningococcal bacteraemia in other patients a self-limiting disorder? We propose that this can be explained by variability in host genetic factors. Convincing evidence for the importance of the genetic background in relation to susceptibility to infectious disease was provided by Sørensen et al. [5]. They showed that adult adoptees, of whom a biological parent had died from an infection before the age of 50, had a fivefold higher risk of dying from an infectious cause than control subjects. In general, host genetic factors include gene mutations, resulting in an absent or deficient protein, which predisposes for disease. Gene mutations are very rare and are estimated to be involved in less than 1% of meningococcal disease cases. However, genetic polymorphisms are stable gene variants that occur rather frequently in the general population and do not follow simple patterns of heritability. Usually they have minor effects on the regulation or function of proteins, but these subtle changes might very well have important consequences for susceptibility to disease. Furthermore, it has been shown that some genetic polymorphisms influence the severity of the course of a disease and therefore can account for higher mortality rates [6]. Thus, studying these polymorphisms might answer some of the questions about meningococcal disease. This review attempts to describe the current knowledge of the role of genetic influences in severe meningococcal infections (Fig. 1).
Figure 1.
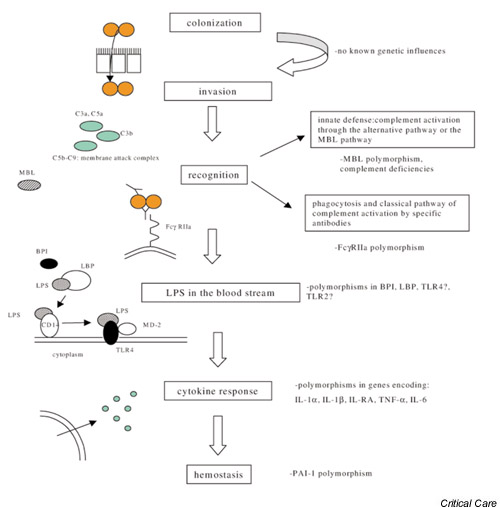
The possible involvement of genetic polymorphisms on the course of meningococcal disease. Partially adapted from [32].
Colonisation and invasion
Colonisation and invasion of the nasopharyngeal mucosa are the first steps in the pathogenesis of meningococcal disease. In short, the filamentous pili proteins on the bacterial surface bind to the CD46 receptor, after which Opa and Opc proteins can bind to their respective receptors: CD66 and a heparan sulphate proteoglycan receptor [7,8,9,10]. Invasion of the mucosal cells by meningococci then occurs by endocyto-sis [11]. Active or passive smoking is a well-known risk factor for colonisation, but no genetic risk factors in the host for colonisation or invasion have yet been established [12].
Recognition, phagocytosis and complement
Fc-γ receptor polymorphisms
Antibodies against Neisseria meningitidis are important for complement-mediated killing through the classical pathway as well as for phagocytosis of the bacteria. Polymorphonuclear leucocytes possess three major classes of receptor for the constant regions of IgG (FcγR): FcγRI (CD64), FcγRIIa (CD 32) and FcγRIIIb (CD16). FcγRIIa is the only receptor capable of binding IgG1, IgG2 as well as IgG3 [13]. Two allotypic forms of this receptor are known, namely FcγRIIa-H131 and FcγRIIa-R131, based on a single amino acid difference; these are respectively a histidine (H) or an arginine (R) residue at position 131 [13]. Homozygous FcγRIIa-R/R131 are much less effective in binding IgG2 and are therefore less effective in the phagocytosis of Neisseria meningitidis, Haemophilus influenzae type b and Staphylococcus aureus [14,15]. This allotype was present in 44% of children surviving a meningococcal septic shock, compared with 23% in a healthy population [14]. Another study showed an association between the FcγRIIa-R/R131 allotype and susceptibility to meningococcal disease in individuals older than 5 years [16]. However, this accounts for less than 30% of all patients, because most pediatric patients suffering from meningococcal disease are younger than 4 years. In all age groups, a correlation was found between FcγRIIa allotype and severity of meningococcal disease, indicated by a longer duration of hospitalisation and a higher percentage of complications for patients with the FcγRIIa-R/R131 allotype. Neu-trophils with the heterozygous allotype FcγRIIa-R/H131 showed intermediate levels of phagocytosis, resulting in intermediate associations with susceptibility to and severity of meningococcal disease [16]. The role of FcγR polymorphisms in complement-deficient individuals has been studied extensively because these people depend heavily on phagocytosis in their defence against meningococci. The distribution of the different allotypes was no different in individuals with late-complement-component deficiency (LCCD) from that in the general population, but again the severity of disease was positively correlated with the FcγRIIa-R/R131 allotype [17]. Thus, LCCD patients with this allotype are not more susceptible to meningococcal disease, but owing to ineffective phagocytosis their course of disease is significantly more severe. However, the combination of FcγRIIa-R/R131 and FcγRIIIb-NA2/NA2, which is another genotype associated with a decreased capacity for phagocytosis by neutrophils in vitro, was associated with a higher susceptibility to meningococcal disease of LCCD patients [18].
Complement deficiency
Activation of the complement cascade eventually lead to the formation of a membrane attack complex, which results in the lysis and cell death of the bacterium. In particular, deficiencies of the alternative pathway and terminal component deficiencies seem to have a large effect on susceptibility to, as well as severity of, meningococcal disease, emphasising the importance of the complement-dependent defence against this particular pathogen. Individuals with LCCD, for example, have a 7000–10,000-fold higher risk of symptomatic meningococcal infections. However, mortality rates of meningococcal disease in these patients are much lower: 1.5% compared with 19% in the general population. Furthermore, the first episode of meningococcal disease occurs at a median age of 17 years, whereas in the general population the median age of occurrence is 3 years [19]. Other complement-component deficiencies associated with an increased susceptibility to meningococcal disease are deficiencies in properdin and factor D, both components of the alternative pathway. Because no genetic polymorphisms are known in these complement components, detailed discussions about these diseases will be omitted from this review.
Mannose-binding lectin polymorphisms
Specific antibodies, as well as proteins of the innate immune system, can activate the complement system. One of these innate pathways is activated by mannose-binding lectin (MBL). This protein binds to sugars on microbial cell walls and activates two mannose-binding-lectin-associated serine proteases: MASP-1 and MASP-2 [20]. Defects in MBL can result in impaired complement activation through this pathway. The MBL gene determines the amount of MBL present in plasma. Three variant alleles of exon 1 of this gene have been identified [21,22,23]. Individuals who are homozygous for one of these variants or carry two different variant alleles have plasma concentrations of less than 1% of wild-type concentrations, whereas heterozygotes have plasma concentrations of about 10% [24]. A large study in children with meningococcal disease revealed that the rates of homozygous as well as heterozygous MBL variant genotypes were much higher in patients than in controls (7.7% compared with 1.5%, and 29.9% compared with 22.1% respectively). Strikingly, the results of the same study suggested a milder course of disease in individuals with variant genotypes, as is also seen in individuals with LCCD [25]. However, MBL variant genotypes are much more common than LCCD in the general population (especially in African and Oriental populations) and could therefore account for a larger number of meningococcal disease cases [21].
The role of LPS and signal transduction
Meningococcal LPS, which is released in enormous amounts during meningococcal sepsis, consists of a polysaccharide part and lipid A, which is responsible for the toxicity of the molecule. LPS can bind to LPS-binding protein (LBP) which is produced by hepatocytes and forms a complex [26]. This complex is recognised by the LPS receptor CD14 on polymorphic nuclear neutrophils and macrophages [27]. The CD14 receptor itself does not have a transmembrane part and therefore cannot directly transduce the LPS signal [28]. It has been shown more recently that the Toll-like receptor 4 (TLR4) and its cofactor MD2 are essential in the process of LPS signal transduction [29,30,31]. TLR4/MD-2 initiates the LPS-induced cell response by signalling through the cell membrane, by way of several signal pathways including activation of the nuclear factor NF-κβ, leading to gene replication and the production of pro-inflammatory mediators in the cell nucleus [32]. The response of TLR4/MD-2 on LPS is enhanced by the presence of CD14 [28], but much is still unknown about this process.
Theoretically, polymorphisms of the LBP gene could affect the structure of LBP and therefore influence the host defence against LPS. Two LBP genotypes have been studied, in both of which a single nucleotide is exchanged for another, resulting in a different amino acid: T292G (Cys98 → Gly) and C1306T (Pro436 → Leu). No significant differences in the distribution of these genotypes were found between a group of patients with sepsis and healthy controls. However, there was a gender-related relationship between the Cys98 → Gly polymorphism and sepsis: a significantly higher proportion of male patients had at least one Gly98allele than did the control group. It was suggested that this unfavourable association might account for the worse prognosis for male patients with sepsis than that for female patients [33]. There is some evidence that common mutations in the tlr4 gene are associated with LPS hyporesponsiveness in humans [34]. In addition, an excess of rare amino acid polymorphisms in the TLR4 have been shown to have a deleterious effect in humans and are present at significantly higher frequencies in patients with meningococcal sepsis than in healthy individuals [35] (B. Beutler, personal communication). Furthermore, it has been shown that an LPS-deficient mutant of Neisseria meningitidis is capable of inducing the release of pro-inflammatory cytokines by macrophages as a result of signalling via TLR2 [36]. Therefore, genetic alterations in the tlr2 gene might also prove to be important in influencing the response to LPS in humans.
Another protein with a high affinity for LPS is bactericidal/permeability-increasing protein (BPI), which is stored within neu-trophilic granules and has been studied as an adjunctive treatment drug for severe meningococcal sepsis [37]. Three different polymorphisms of the BPI gene were studied as well, but no significant difference in genotype distribution was found between patients and controls [33]. It should be noted that this study was undertaken in a general sepsis population and the results might therefore not be representative of meningococcal sepsis.
Cytokine response
Tumour necrosis factor-α
Mononuclear phagocytes release TNF-α upon stimulation with LPS. The level of TNF-α varies greatly in patients with meningococcal disease. The genetic influence on TNF-α release has been shown clearly by several studies. For example, Westendorp et al. [38] found that 60% of the variation in TNF-α production seems to be genetically determined. It has been assumed that a high level of circulating TNF-α is associated with a severe course of disease [39]. However, those authors also found that family members of non-survivors showed a low TNF and high production of interleukin-10 (IL-10) when whole blood was stimulated with LPS [38]. This suggests that an anti-inflammatory cytokine genotype is unfavourable for the outcome of meningococcal disease, which is in contrast with the association of high TNF levels and the adverse outcome shown by other studies [39,40]. A biallelic gene polymorphism in the restriction site of NcpI within the gene encoding TNF-β is associated with a high level of circulating TNF-α, but the influence of this polymorphism on the outcome in meningococcal sepsis is unclear [41]. Furthermore, a genetic polymorphism has been found in the promotor region of TNF-α, consisting of a single base replacement at position -308 (guanosine versus adenosine), but there is a lack of consensus about the relevance of this polymorphism [42,43,44]. In summary, it is clear that TNF-α production during meningococcal sepsis is greatly influenced by genetic factors, but the impact of these factors is still under discussion.
IL-1
The family of genes encoding IL-1 has several members: IL1A and IL1B encode the pro-inflammatory mediators Il-1α and IL-1β respectively, whereas IL1RN encodes the anti-inflammatory IL-1 receptor antagonist. Single-nucleotide polymorphisms occur in all these genes, resulting in biallelic genotypes; these have been studied by Read et al. [43]. The allelic distribution within IL1A had no significant consequences, but individuals homozygous at position -511 within IL1B were more likely to die from meningococcal disease. Furthermore, individuals carrying the rare allele (either homozygous or heterozygous) of IL1RN were also more likely to die. A combination of these two genotypes leads to an even higher mortality rate of up to 42% [43]. Another polymorphism within IL1RN, studied in a general sepsis population, is located within intron 2, which contains a variable number of base-pair repeats. Five different alleles can be identified on the basis of the size of this repeat. The allele frequency of IL-1raA2 was increased in patients with severe sepsis compared with healthy individuals [45]. Thus, this gene polymorphism might influence susceptibility to meningococcal sepsis.
IL-6
IL-6 is a pro-inflammatory cytokine that is usually present in high concentrations in patients with meningococcal sepsis. A well-known polymorphism in the IL-6 promotor region is a G → C transition at position -174. The G/G genotype is associated with higher levels of IL-6 and this is correlated with high mortality in general in patients with sepsis or septic shock [46,47,48]. As with meningococcal disease, patients with a G/G genotype had a threefold higher risk of death than patients with all other genotypes [49]. This corresponds well to the observation that higher levels of IL-6 are associated with a fatal outcome of meningococcal disease [50]. No differences in allele frequencies were found between patients and healthy controls [49].
Haemostasis
Plasminogen-activator-inhibitor-1
An important fibrinolysis gene polymorphism associated with meningococcal disease concerns plasminogen-activator-inhibitor-1 (PAI-1), a protease secreted by endothelial cells, thrombocytes and hepatocytes. It forms a complex with plasminogen activator, which is a proteolytic enzyme involved in fibrinolysis. Meningococcal endotoxin induces extremely high levels of PAI-1 in plasma, which can eventually lead to massive coagulation. An intensively studied polymorphism within the promotor region of the PAI-1 gene is a biallelic single base-pair insertion/deletion polymorphism, resulting in either four or five guanosine bases at position -675. The homozygous 4G/4G genotype predisposes to higher PAI-1 concentrations on stimulation with IL-1β, and it has been shown that the level of plasma PAI-1 is significantly higher in non-survivors of meningococcal septic shock [51,52]. A study by Hermans et al. [53] showed that a 4G/4G genotype in patients was associated with a twofold greater risk of death from meningococcal disease compared with those with the 4G/5G or 5G/5G genotype. Susceptibility to disease was not affected by the genotype [54]. The same association with poor prognosis was found in severely injured patients after trauma [55]. It seems that 4G/4G patients respond to various stimuli with high PAI-1 levels, resulting in impaired fibrinolysis and microcirculation; they therefore have a higher risk for mortality. Many other polymorphisms concerning coagulation and fibrinolysis are described in other reviews [56].
Conclusions and future perspectives
It is clear that host genetic factors can be important in the various stages of meningococcal infections, but much is still unknown about the genetic background of contracting meningococcal disease. Genetic polymorphisms are commonly prevalent, and newly found polymorphisms will have to be tested for their clinical relevance to susceptibility to and severity of meningococcal disease. Possibly, gene polymorphisms also function at the important level of colonisation and invasion of meningococci, which would make possible the identification of individuals at high risk. In addition, it is most likely that individuals with certain combinations of several polymorphisms within the above-described genes have the highest overall risk of dying from meningococcal disease. It is therefore of great importance that molecular-genetic techniques that can be easily and rapidly used in clinical practice are implemented in the study of the interaction of multiple polymorphisms in severe meningococcal infection.
Competing interests
None declared.
Abbreviations
IL = interleukin; LBP = lipopolysaccharide-binding protein; LCCD = late-complement-component deficiency; LPS = lipopolysaccharide; MBL = mannose-binding lectin; PAI-1 = plasminogen-activator-inhibitor-1; TLR = Toll-like receptor; TNF = tumour necrosis factor.
References
- Rosenstein NE, Perkins BA, Stephens DS, Popovic T, Hughes JM. Meningococcal disease. N Engl J Med. 2001;344:1378–1388. doi: 10.1056/NEJM200105033441807. [DOI] [PubMed] [Google Scholar]
- Sim RJ, Harrison MM, Moxon ER, Tang CM. Underestimation of meningococci in tonsillar tissue by nasopharyngeal swabbing. Lancet. 2000;356:1653–1654. doi: 10.1016/s0140-6736(00)03162-7. [DOI] [PubMed] [Google Scholar]
- Fogarty I, Cafferkey MT, Moloney AC. Meningococcal disease in the Republic of Ireland: 1995. Commun Dis Rep CDR Rev. 1997;7:R9–R13. [PubMed] [Google Scholar]
- Anonymous Bacterial Meningitis in the Netherlands. 2001. Annual Report 1999. Amsterdam: Netherlands Reference Laboratory for Bacterial Meningitis.
- Sorensen TI, Nielsen GG, Andersen PK, Teasdale TW. Genetic and environmental influences on premature death in adult adoptees. N Engl JMed. 1988;318:727–732. doi: 10.1056/NEJM198803243181202. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski D. Genetic dissection of the molecular pathogenesis of severe infection. Intensive Care Med. 2000;26(Suppl 1):S89–S97. doi: 10.1007/s001340051124. [DOI] [PubMed] [Google Scholar]
- Kallstrom H, Liszewski MK, Atkinson JP, Jonsson AB. Membrane cofactor protein (MCP or CD46) is a cellular pilus receptor for pathogenic Neisseria. Mol Microbiol. 1997;25:639–647. doi: 10.1046/j.1365-2958.1997.4841857.x. [DOI] [PubMed] [Google Scholar]
- Virji M, Alexandrescu C, Ferguson DJ, Saunders JR, Moxon ER. Variations in the expression of pili: the effect on adherence of Neisseria meningitidis to human epithelial and endothelial cells. Mol Microbiol. 1992;6:1271–1279. doi: 10.1111/j.1365-2958.1992.tb00848.x. [DOI] [PubMed] [Google Scholar]
- Virji M, Makepeace K, Ferguson DJ, Watt SM. Carcinoembryonic antigens (CD66) on epithelial cells and neutrophils are receptors for Opa proteins of pathogenic neisseriae. Mol Microbiol. 1996;22:941–950. doi: 10.1046/j.1365-2958.1996.01551.x. [DOI] [PubMed] [Google Scholar]
- de Vries FP, Cole R, Dankert J, Frosch M, van Putten JP. Neisseria meningitidis producing the Opc adhesin binds epithelial cell proteoglycan receptors. Mol Microbiol. 1998;27:1203–1212. doi: 10.1046/j.1365-2958.1998.00763.x. [DOI] [PubMed] [Google Scholar]
- Stephens DS, Hoffman LH, McGee ZA. Interaction of Neisseria meningitidis with human nasopharyngeal mucosa: attachment and entry into columnar epithelial cells. J Infect Dis. 1983;148:369–376. doi: 10.1093/infdis/148.3.369. [DOI] [PubMed] [Google Scholar]
- Stuart JM, Cartwright KA, Robinson PM, Noah ND. Effect of smoking on meningococcal carriage. Lancet. 1989;2:723–725. doi: 10.1016/s0140-6736(89)90781-2. [DOI] [PubMed] [Google Scholar]
- Warmerdam PA, van de Winkel JG, Vlug A, Westerdaal NA, Capel PJ. A single amino acid in the second Ig-like domain of the human Fc gamma receptor II is critical for human IgG2 binding. J Immunol. 1991;147:1338–1343. [PubMed] [Google Scholar]
- Bredius RG, Derkx BH, Fijen CA, de Wit TP, de Haas M, Weening RS, van de Winkel JG, Out TA. Fc gamma receptor IIa (CD32) polymorphism in fulminant meningococcal septic shock in children. J Infect Dis. 1994;170:848–853. doi: 10.1093/infdis/170.4.848. [DOI] [PubMed] [Google Scholar]
- Bredius RG, de Vries CE, Troelstra A, van Alphen L, Weening RS, van de Winkel JG, Out TA. Phagocytosis of Staphylococcus aureus and Haemophilus influenzae type B opsonized with polyclonal human IgG1 and IgG2 antibodies. Functional hFc gamma RIIa polymorphism to IgG2. J Immunol. 1993;151:1463–1472. [PubMed] [Google Scholar]
- Platonov AE, Shipulin GA, Vershinina IV, Dankert J, van de Winkel JG, Kuijper EJ. Association of human Fc gamma RIIa (CD32) polymorphism with susceptibility to and severity of meningococcal disease. Clin Infect Dis. 1998;27:746–750. doi: 10.1086/514935. [DOI] [PubMed] [Google Scholar]
- Platonov AE, Kuijper EJ, Vershinina IV, Shipulin GA, Westerdaal N, Fijen CA, van de Winkel JG. Meningococcal disease and polymorphism of FcgammaRIIa (CD32) in late complement component-deficient individuals. Clin Exp Immunol. 1998;111:97–101. doi: 10.1046/j.1365-2249.1998.00484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijen CA, Bredius RG, Kuijper EJ, Out TA, de Haas M, De Wit AP, Daha MR, De Winkel JG. The role of Fcgamma receptor polymorphisms and C3 in the immune defence against Neisseria meningitidis in complement-deficient individuals. Clin Exp Immunol. 2000;120:338–345. doi: 10.1046/j.1365-2249.2000.01208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa JE, Densen P. Infectious diseases associated with complement deficiencies. Clin Microbiol Rev. 1991;4:359–395. doi: 10.1128/cmr.4.3.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel S, Vorup-Jensen T, Stover CM, Schwaeble W, Laursen SB, Poulsen K, Willis AC, Eggleton P, Hansen S, Holmskov U, Reid KB, Jensenius JC. A second serine protease associated with mannan-binding lectin that activates complement. Nature. 1997;386:506–510. doi: 10.1038/386506a0. [DOI] [PubMed] [Google Scholar]
- Lipscombe RJ, Sumiya M, Hill AV, Lau YL, Levinsky RJ, Summer-field JA, Turner MW. High frequencies in African and non-African populations of independent mutations in the mannose binding protein gene. Hum Mol Genet. 1992;1:709–715. doi: 10.1093/hmg/1.9.709. [DOI] [PubMed] [Google Scholar]
- Madsen HO, Garred P, Kurtzhals JA, Lamm LU, Ryder LP, Thiel S, Svejgaard A. A new frequent allele is the missing link in the structural polymorphism of the human mannan-binding protein. Immunogenetics. 1994;40:37–44. doi: 10.1007/BF00163962. [DOI] [PubMed] [Google Scholar]
- Sumiya M, Super M, Tabona P, Levinsky RJ, Arai T, Turner MW, Summerfield JA. Molecular basis of opsonic defect in immun-odeficient children. Lancet. 1991;337:1569–1570. doi: 10.1016/0140-6736(91)93263-9. [DOI] [PubMed] [Google Scholar]
- Lipscombe RJ, Sumiya M, Summerfield JA, Turner MW. Distinct physicochemical characteristics of human mannose binding protein expressed by individuals of differing genotype. Immunology. 1995;85:660–667. [PMC free article] [PubMed] [Google Scholar]
- Hibberd ML, Sumiya M, Summerfield JA, Booy R, Levin M. Association of variants of the gene for mannose-binding lectin with susceptibility to meningococcal disease. Meningococcal Research Group. Lancet. 1999;353:1049–1053. doi: 10.1016/s0140-6736(98)08350-0. [DOI] [PubMed] [Google Scholar]
- Schumann RR, Leong SR, Flaggs GW, Gray PW, Wright SD, Mathison JC, Tobias PS, Ulevitch RJ. Structure and function of lipopolysaccharide binding protein. Science. 1990;249:1429–1431. doi: 10.1126/science.2402637. [DOI] [PubMed] [Google Scholar]
- Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249:1431–1433. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- Delude RL, Savedra R, Jr, Zhao H, Thieringer R, Yamamoto S, Fenton MJ, Golenbock DT. CD14 enhances cellular responses to endotoxin without imparting ligand-specific recognition. Proc Natl Acad Sci USA. 1995;92:9288–9292. doi: 10.1073/pnas.92.20.9288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltorak A, Smirnova I, He X, Liu MY, Van Huffel C, McNally O, Birdwell D, Alejos E, Silva M, Du X, Thompson P, Chan EK, Ledesma J, Roe B, Clifton S, Vogel SN, Beutler B. Genetic and physical mapping of the Lps locus: identification of the toll-4 receptor as a candidate gene in the critical region. Blood Cells Mol Dis. 1998;24:340–355. doi: 10.1006/bcmd.1998.0201. [DOI] [PubMed] [Google Scholar]
- Beutler B. Endotoxin, toll-like receptor 4, and the afferent limb of innate immunity. Curr Opin Microbiol. 2000;3:23–28. doi: 10.1016/s1369-5274(99)00046-6. [DOI] [PubMed] [Google Scholar]
- Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal. 2001;13:85–94. doi: 10.1016/s0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- Hubacek JA, Stuber F, Frohlich D, Book M, Wetegrove S, Ritter M, Rothe G, Schmitz G. Gene variants of the bactericidal/permeability increasing protein and lipopolysaccharide binding protein in sepsis patients: gender-specific genetic predisposition to sepsis. Crit Care Med. 2001;29:557–561. doi: 10.1097/00003246-200103000-00015. [DOI] [PubMed] [Google Scholar]
- Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K, Watt JL, Schwartz DA. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25:187–191. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- Smirnova I, Hamblin M, McBride C, Di Rienzo A. Excess of rare amino acid polymorphisms in the Toll-like receptor 4 in humans. Genetics. 2001;158:1657–1664. doi: 10.1093/genetics/158.4.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pridmore AC, Wyllie DH, Abdillahi F, Steeghs L, van der Ley P, Dower SK, Read RC. A lipopolysaccharide-deficient mutant of Neisseria meningitidis elicits attenuated cytokine release by human macrophages and signals via toll-like receptor (TLR) 2 but not via TLR4/MD2. J Infect Dis. 2001;183:89–96. doi: 10.1086/317647. [DOI] [PubMed] [Google Scholar]
- Levin M, Quint PA, Goldstein B, Barton P, Bradley JS, Shemie SD, Yeh T, Kim SS, Cafaro DP, Scannon PJ, Giroir BP. Recombinant bactericidal/permeability-increasing protein (rBPI21) as adjunctive treatment for children with severe meningococcal sepsis: a randomised trial. rBPI21 Meningococcal Sepsis Study Group. Lancet. 2000;356:961–967. doi: 10.1016/s0140-6736(00)02712-4. [DOI] [PubMed] [Google Scholar]
- Westendorp RG, Langermans JA, Huizinga TW, Elouali AH, Verweij CL, Boomsma DI, Vandenbroucke JP, Vandenbrouke JP. Genetic influence on cytokine production and fatal meningo-coccal disease. Lancet. 1997;349:170–173. doi: 10.1016/s0140-6736(96)06413-6. [DOI] [PubMed] [Google Scholar]
- Waage A, Halstensen A, Espevik T. Association between tumour necrosis factor in serum and fatal outcome in patients with meningococcal disease. Lancet. 1987;i:355–357. doi: 10.1016/s0140-6736(87)91728-4. [DOI] [PubMed] [Google Scholar]
- Westendorp RG, Langermans JA, de Bel CE, Meinders AE, Vandenbroucke JP, van Furth R, van Dissel JT. Release of tumor necrosis factor: an innate host characteristic that may contribute to the outcome of meningococcal disease. J Infect Dis. 1995;171:1057–1060. doi: 10.1093/infdis/171.4.1057. [DOI] [PubMed] [Google Scholar]
- Stuber F, Petersen M, Bokelmann F, Schade U. A genomic polymorphism within the tumor necrosis factor locus influences plasma tumor necrosis factor-alpha concentrations and outcome of patients with severe sepsis. Crit Care Med. 1996;24:381–384. doi: 10.1097/00003246-199603000-00004. [DOI] [PubMed] [Google Scholar]
- Nadel S, Newport MJ, Booy R, Levin M. Variation in the tumor necrosis factor-alpha gene promoter region may be associated with death from meningococcal disease. J Infect Dis. 1996;174:878–880. doi: 10.1093/infdis/174.4.878. [DOI] [PubMed] [Google Scholar]
- Read RC, Camp NJ, di Giovine FS, Borrow R, Kaczmarski EB, Chaudhary AG, Fox AJ, Duff GW. An interleukin-1 genotype is associated with fatal outcome of meningococcal disease. J Infect Dis. 2000;182:1557–1560. doi: 10.1086/315889. [DOI] [PubMed] [Google Scholar]
- Brinkman BM, Zuijdeest D, Kaijzel EL, Breedveld FC, Verweij CL. Relevance of the tumor necrosis factor alpha (TNF alpha)-308 promoter polymorphism in TNF alpha gene regulation. J Inflamm. 1996;46:32–41. [PubMed] [Google Scholar]
- Fang XM, Schroder S, Hoeft A, Stuber F. Comparison of two polymorphisms of the interleukin-1 gene family: interleukin-1 receptor antagonist polymorphism contributes to susceptibility to severe sepsis. Crit Care Med. 1999;27(7):1330–1334. doi: 10.1097/00003246-199907000-00024. [DOI] [PubMed] [Google Scholar]
- Hack CE, De Groot ER, Felt-Bersma RJ, Nuijens JH, Strack Van Schijndel RJ, Eerenberg-Belmer AJ, Thijs LG, Aarden LA. Increased plasma levels of interleukin-6 in sepsis. Blood. 1989;74:1704–1710. [PubMed] [Google Scholar]
- Takala A, Jousela I, Jansson SE, Olkkola KT, Takkunen O, Orpana A, Karonen SL, Repo H. Markers of systemic inflammation predicting organ failure in community-acquired septic shock. Clin Sci (Colch) 1999;97:529–538. [PubMed] [Google Scholar]
- Harris MC, Costarino AT, Jr, Sullivan JS, Dulkerian S, McCawley L, Corcoran L, Butler S, Kilpatrick L. Cytokine elevations in critically ill infants with sepsis and necrotizing enterocolitis. J Pediatr. 1994;124:105–111. doi: 10.1016/s0022-3476(94)70264-0. [DOI] [PubMed] [Google Scholar]
- Balding J, Livingstone WJ, Healy M, White B, Mynett-Johnson L, Cafferkey M, Smith OP. G to C transition in the promotor region of the IL6 gene is associated with disease outcome in meningococcal sepsis [abstract P1065]. In XVIII International Society on Thrombosis and Haemostasis. 2001. http://www.cartesian-secure.com/isth2001/iAbstract/html/absP1065.html
- Waage A, Brandtzaeg P, Halstensen A, Kierulf P, Espevik T. The complex pattern of cytokines in serum from patients with meningococcal septic shock. Association between interleukin 6, interleukin 1, and fatal outcome. J Exp Med. 1989;169:333–338. doi: 10.1084/jem.169.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson SJ, Wiman B, Hamsten A, Green F, Humphries S, Henney AM. The two allele sequences of a common polymorphism in the promoter of the plasminogen activator inhibitor-1 (PAI-1) gene respond differently to interleukin-1 in HepG2 cells. J Biol Chem. 1993;268:10739–10745. [PubMed] [Google Scholar]
- Kornelisse RF, Hazelzet JA, Savelkoul HF, Hop WC, Suur MH, Borsboom AN, Risseeuw-Appel IM, van der Voort E, de Groot R. The relationship between plasminogen activator inhibitor-1 and proinflammatory and counterinflammatory mediators in children with meningococcal septic shock. J Infect Dis. 1996;173:1148–1156. doi: 10.1093/infdis/173.5.1148. [DOI] [PubMed] [Google Scholar]
- Hermans PW, Hibberd ML, Booy R, Daramola O, Hazelzet JA, de Groot R, Levin M. 4G/5G promoter polymorphism in the plasminogen-activator-inhibitor-1 gene and outcome of meningo-coccal disease. Meningococcal Research Group. Lancet. 1999;354:556–560. doi: 10.1016/s0140-6736(99)02220-5. [DOI] [PubMed] [Google Scholar]
- Westendorp RG, Hottenga JJ, Slagboom PE. Variation in plasminogen-activator-inhibitor-1 gene and risk of meningococcal septic shock. Lancet. 1999;354:561–563. doi: 10.1016/S0140-6736(98)09376-3. [DOI] [PubMed] [Google Scholar]
- Menges T, Hermans PW, Little SG, Langefeld T, Boning O, Engel J, Sluijter M, de Groot R, Hempelmann G. Plasminogen- activator-inhibitor-1 4G/5G promoter polymorphism and prognosis of severely injured patients. Lancet. 2001;357:1096–1097. doi: 10.1016/S0140-6736(00)04311-7. [DOI] [PubMed] [Google Scholar]
- Hazelzet JA, Hermans PW. Hemostatic gene polymorphisms and meningococcal sepsis. Sepsis. 2001;4:233–237. doi: 10.1023/A:1012965007673. [DOI] [Google Scholar]