

Abstract
The cytosine C5 methyltransferase M.HaeIII recognises and methylates the central cytosine of its canonical site GGCC. Here we report that M.HaeIII can also, with lower efficiency, methylate cytosines located in a wide range of non-canonical sequences. Using bisulphite sequencing we mapped the methyl- cytosine residues in DNA methylated in vitro and in vivo by M.HaeIII. Methyl-cytosine residues were observed in multiple sequence contexts, most commonly, but not exclusively, at star sites (sites differing by a single base from the canonical sequence). The most frequently used star sites had changes at positions 1 and 4, but there is little or no methylation at star sites changed at position 2. The rate of methylation of non-canonical sites can be quite significant: a DNA substrate lacking a canonical site was methylated by M.HaeIII in vitro at a rate only an order of magnitude slower than an otherwise identical substrate containing the canonical site. In vivo methylation of non-canonical sites may therefore be significant and may have provided the starting point for the evolution of restriction–modification systems with novel sequence specificities.
INTRODUCTION
DNA cytosine C5 methyltransferases are a large group of enzymes whose structure is conserved in bacteria, archaea and eukaryotes. This class of enzymes has a common mechanism of catalysis and 10 highly conserved motifs making up the catalytic site and the cofactor binding site (1).
In eukaryotes, cytosine C5 methyltransferases are responsible for CpG methylation, which is involved in regulation of chromatin structure and gene expression (2). In bacteria, DNA methylation has roles in restriction–modification (R/M) systems, the control of DNA replication and post-replicative mismatch repair (3). Bacteria possessing a R/M system express a methylase and cognate restriction endonuclease which recognise the same target site. Methylation of the bacterial chromosome protects it from endonuclease digestion, but unmethylated DNA of infecting bacteriophage is digested by the endonuclease. Type II R/M systems are the most common, with over 200 DNA specificities (4).
The evolution of a R/M system with a novel specificity must necessarily involve the co-evolution of both a methylase and a restriction endonuclease; however, little is known about how this might occur (5–7). Both a methyltransferase and a restriction enzyme with novel sequence specificity must evolve, yet a simultaneous change in sequence specificity is not likely. Due to the lethal nature of a novel restriction enzyme without a cognate methyltransferase, the methyltransferase must change its specificity first. Several models for this co-evolution have been suggested, including duplication of the methyltransferase gene and its cognate restriction endonuclease gene (the two genes are frequently in close proximity) followed by changes in sequence specificity. This switch in specificity may start with the enhancement of activity towards a non-canonical site against which the enzyme already has low activity (5,6), as proposed for other enzymes (8,9). Star activity of restriction endonucleases, when the enzyme recognises and cuts at a sequence differing at only a single base pair from the canonical site, is a relatively well documented phenomenon (10,11). In contrast, DNA methylation is generally regarded as being highly sequence specific (3), with few published observations of non-canonical methylation. However, non-canonical, or aberrant, methylation has been observed with DNA methyltransferases at sites closely related to the normal target sequence (5,6,12–14).
Here we report an investigation into methylation of non-canonical sites by the cytosine C5 methyltransferase M.HaeIII which is part of a typical type II R/M system in the bacterium Haemophilus influenzae (biogroup aegyptius). M.HaeIII methylates the third base of the sequence GGCC in unmodified or hemimethylated duplex DNA. We have investigated the optimum conditions for methylation by M.HaeIII at canonical and non-canonical sites and examined the sequence preferences of M.HaeIII for non-canonical sites. We observe that M.HaeIII can methylate cytosines in a variety of sequence contexts both in vitro and in vivo.
MATERIALS AND METHODS
Oligonucleotides
Haesubplus: 5′-TTCGAGAAGCTGAGGCCGGCGTACC TGGAG-3′
Haesubminus: 5′-CTCCAGGTACGCCGGCCTCAGCTTC TCGAA-3′
Starsubplus: 5′-TTCGAGAAGCTGAAGCCGGCGTACC TGGAG-3′
Starsubminus: 5′-CTCCAGGTACGCCGGCTTCAGCTTC TCGAA-3′
GST1Fo: 5′-GAAATAGTTATGATTATGATTATGT TAAGT-3′
GST1Ba: 5′-TCTTTCTTATTATTCCACAACACCA TATAC-3′
GST2Fo: 5′-TAATTACCCTAAATTATTAAAACAT CCACA-3′
GST2Ba: 5′-GTTGTTTTGTTGTTTGGGGTAGTTG GGGAT-3′
Pet1Fo: 5′-GGGAGACCATGGTTTTGATTTTTTA TGTTGGATGTATTG-3′
Pet1Ba: 5′-CACAACGAGCTCAATAATCAACCCA CTAACACATTACAC-3′
Pet2Fo: 5′-GGGAGACCATGGTCTCAATCCTCTA CACCAAACACATC-3′
Pet2Ba: 5′-CACAACGAGCTCGATGTGTTGTGTG AGAAGATTGTGTATTGTTG-3′
cM1-Clon-F: 5′-ATCGATCGGTACCTTATTACTCTTT CTTGTTGTTCCACAGCGC-3′
cM1-Clon-B: 5′-TATCCTGAGCCATGGTGGTTACCCT GGGTTATTG-3′
Re-N-Flag-Bc: 5′-GCTGACAAGCTTAATAATTTTGTTT AACTTTAAGAAGGAGATATAGCCATG GACCTACAAGATGACGATGATAAAA ATTTAATTAGTCTTT-3′
In vitro methylation with S-adenosyl-l-[methyl-3H]methionine
Complementary oligonucleotides Haesubplus and Haesub minus were annealed by heating equal amounts (0.5 mM each in 10 mM Tris–HCl, pH 8.5) to 90°C for 5 min and then cooling slowly to 20°C to create a 30 bp template for methylation, termed Haesub (see Fig. 1A), containing a single canonical M.HaeIII methylation site (GGCC). Similarly, the DNA oligonucleotides Starsubplus and Starsubminus were annealed to create a second 30 bp template for methylation, termed Starsub (see Fig. 1A), which has no canonical HaeIII sites and differs from Haesub by only a single base pair.
Figure 1.

In vitro methylation rates of substrate DNA containing canonical and non-canonical sites by M.HaeIII. Two 30 bp substrates were used (A), differing by only a single base pair: Haesub contained a single canonical site (GGCC), shown in bold; and Starsub differed at one base pair, shown in bold, and lacked canonical sites. Initial rates of incorporation of methyl-3H into Haesub (continuous line, squares) and Starsub (dashed line, triangles) at various concentrations of glycerol (B), NaCl (C) and Tris–HCl (D). Measurements were taken in triplicate and error bars represent one standard error of the mean.
Incorporation of methyl-3H into the two DNA substrates by M.HaeIII was assayed. Reactions contained 10 nM M.HaeIII (New England Biolabs), 200 nM S-adenosyl-l-[methyl-3H]methionine (79.8 Ci/mmol; Perkin-Elmer) and 1.25 µM double-stranded DNA in 30 µl of a standard buffer (25 mM Tris–HCl pH 7.5, 25 mM NaCl, 10 mM dithioerythritol, 0.2 mg/ml BSA). In parallel reactions the concentration of NaCl was varied in the range 0–100 mM, Tris–HCl in the range 5–100 mM and glycerol in the range 0–50%, keeping the other two variables constant (see Fig. 1). Reactions were performed in triplicate, incubated at 37°C and stopped by the addition of 10 µl of 0.3 M sodium acetate and 320 µM unlabelled S-adenosyl-l-methionine. The rate of incorporation of methyl-3H into Haesub was linear for at least 20 min and the rate of incorporation into Starsub was linear for 2 h (data not shown). To measure the initial rate, reactions were quenched after 10 min (Haesub) or after 40 min (Starsub). Thirty-five microlitres of the quenched reaction was spotted onto a Whatman DE81 filter. Filters were washed four times with 2 ml of 0.2 M ammonium hydrogen carbonate, twice with 1 ml of ethanol, air dried and counted using a Beckmann LS6000SC scintillation counter in 3 ml of scintillant (National Diagnostics).
The concentration of S-adenosylmethionine in the assays (200 nM) was lower than the Km of M.HaeIII for S-adenosylmethionine (336 nM; H.M.Cohen, unpublished results); however, the use of low concentrations of cofactor with high specific radioactivity was necessary to allow sensitive detection of methyl group incorporation.
Determining the methylation pattern of DNA in vitro
The chicken glutathione-S-transferase M1-1 gene (15) was amplified from chicken liver cDNA (Clontech) using primers cM1-Clon-F and cM1-Clon-B, digested with NcoI and KpnI and ligated into pGEM4z (Promega). PCR amplification of this plasmid, using the standard –21M13 forward and M13 reverse sequencing primers, yielded an 832 bp template for methylation. The methylation reaction contained 100 nM M.HaeIII (New England Biolabs), 73 nM PCR fragment (purified using the Wizard PCR purification kit; Promega), and 80 µM S-adenosyl methionine in 50 µl of buffer (25 mM NaCl, 25 mM Tris–HCl pH 8.5, 5 mM DTT, 40% glycerol). The reaction was incubated for 8 h at 37°C, with an additional 80 µM S-adenosyl methionine added after 4 h. The DNA was purified (Wizard PCR purification kit), treated with bisulphite and sequenced as described below. An unmethylated sample of the PCR fragment was treated with bisulphite in a parallel reaction.
Determination of the methylation pattern of DNA in vivo
The M.HaeIII gene was amplified by PCR from H.influenzae (biogroup aegyptius) (ATCC 11116) using oligonucleotides HaeIII-FoNC and HaeIII-Bc (16), digested with KpnI and HindIII and ligated into pGEM4Z (Promega). The gene was amplified from this plasmid by PCR using oligonucleotides Re-N-Flag-Bc and LMB2-Nest (16), digested with NcoI and SacI, ligated into pET30a (Invitrogen) creating pET-MHaeIII, and transformed into Escherichia coli C41(DE3) (17). The expressed protein has N-terminal His6, S and Flag tags. Cells were grown in 2× YT (18) at 37°C to OD 0.6, induced by the addition of 0.3 mM isopropyl-β-d-thiogalactoside and transferred to 30°C for 4 h. M.HaeIII was purified from the cells using NiNTA agarose (Qiagen) according to the manufacturer’s instructions. pET-MHaeIII plasmid was also prepared from the induced cells using the Qiagen Plasmid Midi-prep kit. Fifty-six nanograms of plasmid was linearised by digestion with SacI, purified (Qiaquick PCR purification kit; Qiagen), treated with bisulphite and sequenced as described below. pET30a (containing no methyltransferase gene) was prepared from E.coli C41(DE3) after induction, as above, for use in a parallel bisulphite reaction.
Bisulphite sequencing
Purified methylated DNA was treated with bisulphite as described (19). Strand 1 of the bisulphite-converted GST gene was amplified using primers GST1Fo and GST1Rev, strand 2 was amplified with GST2Fo and GST2Rev. PCR products were cloned using the TOPO TA Cloning kit (Invitrogen) and sequenced. Strand 1 of the bisulphite-converted pET plasmid was amplified using Pet1Fo and Pet1Rev and strand 2 was amplified using Pet2Fo and Pet2Rev. PCR products were digested with NcoI and SacI, cloned in pIVEX2.2 (Roche) and sequenced.
RESULTS
The relative rates of methylation of two substrates, Haesub, which contains a single canonical M.HaeIII methylation site (GGCC), and Starsub, which has no canonical HaeIII sites and differs from Haesub by only a single base pair (Fig. 1A), were determined under a variety of different conditions in vitro (Fig. 1). It is well known that the star activity of restriction enzymes is greatly enhanced under conditions of low ionic strength or high glycerol concentrations; therefore, we investigated whether the star activity of M.HaeIII could be enhanced under similar conditions.
Under all conditions tested, the rate of methylation of the non-canonical substrate, Starsub, was significant, ranging between 2.8 and 50.8% of the rate with the canonical substrate, Haesub, under the same conditions (Fig. 1). Both the absolute rates and relative rates of methylation of the two substrates were dependent on the reaction conditions. The fastest rate of methylation of Haesub is achieved in 25 mM NaCl and 25–50 mM Tris–HCl. The reaction is inhibited by glycerol, with an almost 4-fold reduction in rate caused by the addition of glycerol to 50% (Fig. 1B). Unlike Haesub the rate of methylation of Starsub is increased by the addition of glycerol (Fig. 1B), and is higher in 100 mM NaCl than at lower salt concentrations (Fig. 1C). The dependence on Tris–HCl concentration mirrors that of HaeSub (Fig. 1D) and the optimum pH for methylation of both substrates is between 7.0 and 8.0 (data not shown).
Under the optimal conditions for methylation of Haesub the substrate lacking a canonical site, Starsub, is methylated 35.7-fold more slowly. However, the highest rate of non-canonical methylation occurred in 25 mM Tris–HCl, 25 mM NaCl and 50% glycerol, when Haesub is methylated only 1.97-fold faster than Starsub.
The pattern of DNA methylation by M.HaeIII was determined both in vitro and in vivo by bisulphite modification and sequencing of methylated DNA (19). This chemical treatment results in deamination of all the unmethylated cytosines to uracil, which is copied as thymine in the subsequent PCR. C5-methylated cytosines react more slowly and remain unconverted (Fig. 2).
Figure 2.
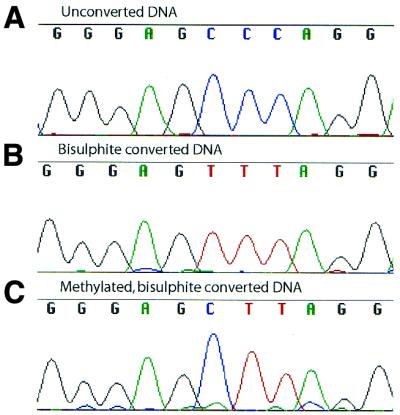
Typical sequences of the methylation template before bisulphite treatment (A), after bisulphite conversion and cloning (B) and after in vitro methylation by M.HaeIII, bisulphite treatment and cloning (C).
To assay methylation in vitro, an 832 bp DNA fragment encoding the chicken glutathione-S-transferase M1-1 gene was incubated with purified M.HaeIII in a buffer chosen to maximise the rate of non-canonical DNA methylation (see Fig. 1). For methylation of pET-MHaeIII in vivo, the rate of methylation was also enhanced by overexpressing M.HaeIII in E.coli. A total of 3.1 mg of M.HaeIII was purified from 7.8 g of bacteria (data not shown) which corresponds to an approximate intracellular concentration of 9.15 µM enzyme (assuming a density of 1 g/ml and an Mr of the tagged protein of 43 412 Da).
After methylation, the DNA fragment encoding glutathione-S-transferase was purified from the in vitro reactions and pET-MHaeIII was purified from the induced E.coli. This DNA was then treated with bisulphite (19). Sequencing of M.HaeIII-methylated, bisulphite-treated DNA before cloning only revealed methyl cytosine at canonical sites (data not shown), as this method displays an average of all the strands. Cloning the bisulphite-treated DNA allowed the analysis of individual strands (Table 1 and Fig. 3). 22 870 bases of in vitro-methylated DNA and 12 730 bases of in vivo-methylated DNA were sequenced. The percentage of canonical HaeIII sites methylated in vitro was 90.3% and the percentage of canonical sites in pET-MHaeIII methylated in vivo was 100%. In addition to these sites, a further 0.9% of all other cytosines in the PCR fragment and 0.6% in pET-MHaeIII remained unconverted. More than half of these unconverted cytosines were M.HaeIII star sites, with unconverted cytosines observed at seven of the nine possible star sites. Additionally, 28 unconverted cytosines were observed in sequence contexts differing by two or three bases from the canonical site; 19 in vitro and nine in vivo.
Table 1. The sequence context of C5 methyl cytosines detected in DNA methylated by M.HaeIII in vitro and in vivo.
| Site | Sites analysed | Unconverted cytosines | % Unconverted | |
|---|---|---|---|---|
| Cytosines unconverted in vitro | ||||
| All cytosines | All cytosines | 6861 | 226 | 3.3 |
| GGCC | 187 | 169 | 90.3 | |
| Star sites | 1261 | 38 | 3.0 | |
| Non-GGCC | 6674 | 57 | 0.9 | |
| Star sites position 1 | AGCC | 112 | 15 | 13.4 |
| CGCC | 150 | 2 | 1.3 | |
| TGCC | 75 | 0 | 0 | |
| A/C/TGCC | 337 | 17 | 5.0 | |
| Star sites position 2 | GACC | 38 | 1 | 2.6 |
| GCCC | 171 | 0 | 0.0 | |
| GTCC | 228 | 0 | 0.0 | |
| GA/C/TCC | 437 | 1 | 0.2 | |
| Star sites position 4 | GGCA | 131 | 5 | 3.8 |
| GGCG | 150 | 9 | 6.0 | |
| GGCT | 206 | 6 | 2.9 | |
| GGCA/G/T | 487 | 20 | 4.1 | |
| Non-star sitesa | AGCA | 152 | 4 | 2.6 |
| CTCC | 360 | 3 | 0.8 | |
| CTCT | 57 | 1 | 1.8 | |
| GCCA | 152 | 1 | 0.7 | |
| GCCG | 132 | 1 | 0.8 | |
| TCCA | 380 | 3 | 0.8 | |
| All non-star | 5413 | 19 | 0.4 | |
| Cytosines unconverted in vivo | ||||
| All cytosines | All cytosines | 3437 | 124 | 3.6 |
| GGCC | 104 | 104 | 100 | |
| Star sites | 843 | 11 | 1.3 | |
| Non-GGCC | 3333 | 20 | 0.6 | |
| Star sites position 1 | AGCC | 42 | 5 | 11.9 |
| CGCC | 154 | 3 | 1.9 | |
| TGCC | 55 | 1 | 1.8 | |
| A/C/TGCC | 251 | 9 | 3.6 | |
| Star sites position 2 | GACC | 19 | 0 | 0 |
| GCCC | 171 | 0 | 0 | |
| GTCC | 57 | 0 | 0 | |
| GA/C/TCC | 247 | 0 | 0 | |
| Star sites position 4 | GGCA | 70 | 0 | 0 |
| GGCG | 218 | 2 | 0.9 | |
| GGCT | 57 | 0 | 0 | |
| GGCA/G/T | 345 | 2 | 0.6 | |
| Non-star sitesa | CACG | 88 | 2 | 2.3 |
| CTCT | 77 | 1 | 1.3 | |
| GCCA | 174 | 2 | 1.1 | |
| GCCG | 161 | 1 | 0.6 | |
| All sites | 2490 | 9 | 0.4 | |
Individual clones of methylated, bisulphite-treated DNA were sequenced. In total, the occurrence of methyl cytosines in 22 870 bases of in vitro-methylated DNA and 12 730 bases of in vivo-methylated DNA was analysed. In star sites, bases different from the wild-type sequence are indicated in bold type.
aOnly those non-star site cytosines that were observed more than once in vitro and in vivo are listed.
Figure 3.

Frequency of occurrence of C5 methyl-cytosine groups in a glutathione-S-transferase gene methylated in vitro by M.HaeIII determined by bisulphite treatment and sequencing of individual clones. The sequences of both strands of the gene are shown, with canonical HaeIII sites shown in red. Bars above the sequence indicate unconverted cytosines in the top strand and bars below the unconverted cytosines in the bottom strand. The height of the bars represent the percentage of unconverted cytosines at each position with a scale of 0–50%. All canonical HaeIII sites were unconverted in >50% of the clones analysed. PCR amplification of the top strand yields a 583 bp product and the bottom strand gives a 480 bp product.
However, unconverted cytosines, in addition to arising from DNA methylation, may also be due to incomplete chemical reaction of the DNA with bisulphite or mutagenesis in the subsequent PCR causing a T to C transition. To test the background level of unconverted cytosines, the unmethylated PCR fragment and unmethylated pET30a were subjected to bisulphite sequencing. A total of 7640 bases of the bisulphite-treated, cloned glutathione-S-transferase gene and 3350 bases of the bisulphite-treated, cloned, pET30a were sequenced. The percentage of cytosines remaining unconverted in these samples was 0.7 and 0.1%, respectively. No bias in the sequence context of unconverted cytosines was observed: of the 64 permutations of NNCN, 51 were never observed unconverted, 12 were observed unconverted only once, and only one site (CGCC) was observed unconverted twice. Methylated DNA, in contrast, has a strongly biased pattern of non-canonical unconverted cytosines that cannot be explained simply by incomplete reaction with bisulphite (Fig. 3).
DISCUSSION
Traditionally, DNA methyltransferases are viewed as highly sequence specific (3). However, recent observations, including this study, suggest that methylation of non-canonical sites may be a common feature of DNA methyltransferases.
Non-canonical methylation was first reported for the adenine N6 methyltransferase M.EcoRI (14) and has since been reported for two other adenine N6 methyltransferases, M.FokI and M.EcoRV. Twenty-seven oligonucleotides were tested as substrates for methylation by the M.FokI DNA methyltransferase (5). The enzyme has two domains, each methylating one strand of its asymmetric recognition site, GGATG. Whilst the N-terminal domain only methylated one star site, the C-terminal domain methylated most of the star sites tested and two sites differing by two base pairs from the canonical site. M.EcoRV (canonical site GATATC) is also capable of methylating five different degenerate EcoRV recognition sites, including one site with three altered bases (GATC) (12,20).
Methylation of non-canonical sites has also been reported for three cytosine C5 methyltransferases (13). Overexpression of M.HpaII (canonical site CCGG), Dcm and EcoRII (CCAGG) methyltransferases in E.coli causes an increase in the C–T mutation rate at the canonical sites for methylation, but also at some non-canonical sites (CAGG and CCGGG). Restriction digestion of the plasmid isolated from E.coli overexpressing M.EcoRII showed that the DNA was partially protected from digestion at the site CCCGGG. Methylation of this star site by M.EcoRII was confirmed using a synthetic oligonucleotide in vitro.
In these studies, methylation of a target cytosine or adenine was examined in a limited number of sequence contexts. Here we attempted to explore the full range of sequence contexts for methylation of cytosine residues by M.HaeIII.
The pattern of methylation was investigated both in vitro, using purified enzyme, and in vivo in E.coli overexpressing methyltransferase. The pattern of non-canonical DNA methylation was similar in vitro and in vivo. Of the 63 permutations of NNCN (excepting GGCC), 25 were unconverted by bisulphite modification at least once in the clones analysed. Methylation of non-canonical sites was not uniform, either in vitro or in vivo, M.HaeIII methylates star sites more frequently than other sites. Only at two of the nine star sites was methylation not detected either in vitro or in vivo. The enzyme shows less discrimination at the first and fourth base of its recognition sequence, with the central cytosine of AGCC being methylated more frequently than all other non-canonical cytosines, in vitro and in vivo. The star sites GTCC and GCCC were never observed unconverted, and in only one case was the third base of GACC unconverted, indicating a greater stringency for recognition of the second base of the sequence.
In addition to star site methylation, DNA methylated in vitro and in vivo contained unconverted cytosines at sites differing from the canonical site by two or three base pairs. However, it is difficult to be sure whether cytosines observed unconverted only once are due to methylation or to the incomplete conversion of cytosines by bisulphite. Although conversion was very efficient (over 99.3% of cytosines converted), some of the cytosines observed in the methylated DNA were almost certainly due to incomplete conversion. However, some sites, notably AGCA, were observed multiple times in methylated DNA but never in unmethylated DNA, and are likely to be the result of methylation by M.HaeIII. This site combines the base alterations of two star sites that are methylated at an appreciable level by M.HaeIII in vitro (AGCC and GGCA).
In E.coli overexpressing M.HaeIII, DNA is hypermethylated to the extent that the central cytosine of the sequence AGCC is methylated in 11.9% of cases and methylation is also seen at other star sites (Table 1). These high levels of methylation suggests that there might still be significant levels of non-canonical DNA methylation in Haemophilus aegyptius, despite the lower concentration of M.HaeIII.
The in vitro methylation rates (Fig. 1) provide further support for this suggestion as the enzyme concentration (10 nM) and DNA concentration (1.25 µM sites) were very close to those predicted in bacteria (50–500 nM methylase and ∼1–10 µM sites) (3). Even under the conditions where the enzyme showed highest specificity for its canonical site, the rate of methylation of non-canonical substrate was still only 36.7-fold lower than the rate with the canonical substrate. This corresponds to a 303-fold lower rate of methylation per cytosine, based on two canonical target cytosines in Haesub and 17 non-canonical cytosines in Starsub.
The existence of a large number of R/M systems with a wide range of sequence specificities raises the question of how methylases and restriction enzymes were able to co-evolve to recognise new target sites. It has been proposed that many enzymes can catalyse the conversion of molecules which are not their principle substrate with low efficiency, and that new enzymes may evolve by improvements in the enzyme’s ability to catalyse the conversion of one of these poor substrates (8,9,21). Promiscuous activities as low as described above (and indeed lower) can provide a selective advantage and thereby a starting point for the evolution of a new activity (22). In particular, it has been proposed that protein–DNA interactions evolve by relaxing an existing specificity (namely, increasing promiscuity) and then restricting it to the new target; this principle has recently been used to alter the site specificity of Cre recombinase (23). High levels of star activity by restriction endonucleases would be lethal unless the cognate methylase also had the ability to methylate and protect these star sites. The problems associated with non-canonical methylation are probably less severe. These include the energetic cost and the 10-fold higher mutation rate of C5-methylated cytosines compared with unmethylated cytosines (24,25), and the different response of prokaryotic DNA-interacting proteins, for example, methyl-directed restriction enzymes, to methylated and unmethylated DNA. In fact the selective disadvantage attributed to a methyltransferase with two distinct recognition sites (the canonical site and a newly arisen specificity) is comparable to that caused by expressing two different methyltransferases, whereas natural populations of bacteria may contain in excess of 10 different R/M systems (26). Hence, it is likely that a DNA methyltransferase with a new specificity arises before the cognate restriction endonuclease.
If DNA methylases possess a significant level of activity at non-canonical sites then this could have served as a starting point for evolutionary divergence towards a new R/M site (5,6). Type II R/M systems recognise symmetrical DNA targets 2–8 bp in length so the evolution of a new specificity would usually involve a change of at least two base pairs in the target site. The ability of M.HaeIII to methylate cytosines in a variety of non-canonical sequence contexts indicates that such changes in specificity may not represent a large evolutionary barrier.
REFERENCES
- 1.Kumar S., Cheng,X., Klimasauskas,S., Mi,S., Posfai,J., Roberts,R.J. and Wilson,G.G. (1994) The DNA (cytosine-5) methyltransferases. Nucleic Acids Res., 22, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vertino P.M. (1999) Eukaryotic DNA methyltransferases. In Cheng,X. and Blumenthal,R.M. (eds), S-Adenosylmethionine-Dependent Methyltransferases. World Scientific, Singapore, pp. 341–372.
- 3.Dryden D.T.F. (1999) Bacterial DNA methyltransferases. In Cheng,X. and Blumenthal,R.M. (eds), S-Adenosylmethionine-Dependent Methyltransferases. World Scientific, Singapore, pp. 283–340.
- 4.Roberts R.J. and Macelis,D. (2001) REBASE—restriction enzymes and methylases. Nucleic Acids Res., 29, 268–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedrich T., Fatemi,M., Gowhar,H., Leismann,O. and Jeltsch,A. (2000) Specificity of DNA binding and methylation by the M.FokI DNA methyltransferase. Biochim. Biophys. Acta, 1480, 145–159. [DOI] [PubMed] [Google Scholar]
- 6.Beck C., Cranz,S., Solmaz,M., Roth,M. and Jeltsch,A. (2001) How does a DNA interacting enzyme change its specificity during molecular evolution? A site-directed mutagenesis study at the DNA binding site of the DNA-(adenine-N6)-methyltransferase EcoRV. Biochemistry, 40, 10956–10965. [DOI] [PubMed] [Google Scholar]
- 7.Kobayashi I. (2001) Behaviour of restriction–modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res., 29, 3742–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Brien P.J. and Herschlag,D. (1999) Catalytic promiscuity and the evolution of new enzymatic activities. Chem. Biol., 6, R91–R105. [DOI] [PubMed] [Google Scholar]
- 9.Matsumura I. and Ellington,A.D. (2001) In vitro evolution of beta-glucuronidase into a beta-galactosidase proceeds through non-specific intermediates. J. Mol. Biol., 305, 331–339. [DOI] [PubMed] [Google Scholar]
- 10.Robinson C.R. and Sligar,S.G. (1995) Heterogeneity in molecular recognition by restriction endonucleases: osmotic and hydrostatic pressure effects on BamHI, PvuII and EcoRV specificity. Proc. Natl Acad. Sci. USA, 92, 3444–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao W., Mayer,A.N. and Barany,F. (1995) Stringent and relaxed specificities of TaqI endonuclease: interactions with metal cofactors and DNA sequences. Biochemistry, 34, 2276–2283. [DOI] [PubMed] [Google Scholar]
- 12.Taylor J.D., Goodall,A.J., Vermote,C.L. and Halford,S.E. (1990) Fidelity of DNA recognition by the EcoRV restriction/modification system in vivo. Biochemistry, 29, 10727–10733. [DOI] [PubMed] [Google Scholar]
- 13.Bandaru B., Gopal,J. and Bhagwat,A.S. (1996) Overproduction of DNA cytosine methyltransferases causes methylation and C→T mutations at non-canonical sites. J. Biol. Chem., 271, 7851–7859. [DOI] [PubMed] [Google Scholar]
- 14.Woodbury C.P.J., Downey,R.L. and von Hippel,P.H. (1980) DNA site recognition and overmethylation by the EcoRI methylase. J. Biol. Chem., 255, 11526. [PubMed] [Google Scholar]
- 15.Liu L.F. and Tam,M.F. (1991) Nucleotide sequence of a class mu glutathione S-transferase from chicken liver. Biochim. Biophys. Acta, 1090, 343–344. [DOI] [PubMed] [Google Scholar]
- 16.Tawfik D.S. and Griffiths,A.D. (1998) Man-made cell-like compartments for molecular evolution. Nat. Biotechnol., 16, 652–656. [DOI] [PubMed] [Google Scholar]
- 17.Miroux B. and Walker,J.E. (1996) Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol., 260, 289–298. [DOI] [PubMed] [Google Scholar]
- 18.Russell D.W. and Sambrook,J. (2001) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, New York.
- 19.Paulin R., Grigg,G.W., Davey,M.W. and Piper,A.A. (1998) Urea improves efficiency of bisulphite-mediated sequencing of methylcytosine in genomic DNA. Nucleic Acids Res., 26, 5009–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeltsch A., Christ,F., Fatemi,M. and Roth,M. (1999) On the substrate specificity of DNA methyltransferases. Adenine-N6 DNA methyltransferases also modify cytosine residues at position N4. J. Biol. Chem., 274, 19538–19544. [DOI] [PubMed] [Google Scholar]
- 21.Jensen R.A. (1976) Enzyme recruitment in evolution of new function. Annu. Rev. Microbiol., 30, 409–425. [DOI] [PubMed] [Google Scholar]
- 22.James L.C. and Tawfik,D.S. (2001) Catalytic and binding poly-reactivities shared by two unrelated proteins: the potential role of promiscuity in enzyme evolution. Protein Sci., 10, 2600–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buchholz F. and Stewart,A.F. (2001) Alteration of Cre recombinase site specificity by substrate-linked protein evolution. Nat. Biotechnol., 19, 1047–1052. [DOI] [PubMed] [Google Scholar]
- 24.Lieb M. (1991) Spontaneous mutation at a 5-methylcytosine hotspot is prevented by very short patch (VSP) mismatch repair. Genetics, 128, 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coulondre C., Miller,J.H., Farabaugh,P.J. and Gilbert,W. (1978) Molecular basis of base substitution hotspots in Escherichia coli. Nature, 274, 775–780. [DOI] [PubMed] [Google Scholar]
- 26.Takata T., Aras,R., Tavakoli,D., Ando,T., Olivares,A.Z. and Blaser,M.J. (2002) Phenotypic and genotypic variation in methylases involved in type II restriction–modification systems in Helicobacter pylori. Nucleic Acids Res., 30, 2444–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]