


Abstract
The Leishmania genome project reference strain, Leishmania major Friedlin, is trisomic for chromosome 1. The complete sequence of this chromosome has revealed that the genes are grouped into two large clusters of the polycistronic type, each borne by one DNA strand and located on each side of a 1.6-kb sequence often termed the switch region. Several hypotheses concerning the role of this switch region have been put forward (region of initiation of transcription for both gene clusters, origin of replication or centromeric sequence). In the present study, we have deleted this region on the three copies of chromosome 1 by sequential targeted replacements. The absence of the switch region did not alter the mitotic stability of the three deleted chromosomes. This region therefore does not appear necessary for chromosomal replication or segregation. However, during the third targeting round, which aimed at knocking out the last switch region, a fourth copy of chromosome 1 that retained this region appeared in all clones analysed. This suggests that the persistence of this switch region is necessary for parasite survival. We then showed that the presence/absence of the switch region did not act upon the expression of a resistance marker gene inserted beforehand into the left gene cluster of the same chromosomal molecule. This result suggests that the presence of this 1.6-kb sequence is not necessary for the expression of all genes on chromosome 1.
INTRODUCTION
Leishmania are protozoal parasites of the family of Trypanosomatids responsible for significant worldwide human morbidity and mortality. The clinical features vary from mild cutaneous lesions to fatal mucocutaneous or visceral involvement depending on the causative species. The genome structure of this organism has now been well explored; 36 heterologous chromosomes, sized from ∼0.3 to 3 Mb, are characterised for all Old World species, totalling ∼35 Mb (±2 Mb) for a haploid genome (1,2). Although Leishmania is considered to be essentially diploid, aneuploidy (mainly in the form of trisomic chromosomes) has been observed in natural isolates: thus, the Leishmania major Friedlin strain (LmF), the reference strain for the genome sequencing project, possesses three homologues of chromosome 1 (2,3). From the complete sequence of this ∼300-kb chromosome (GenBank accession no. AE001274), the three homologues appear to be almost identical in their coding regions; they are essentially distinguished by a size variation of ∼30 kb in the right non-coding subtelomeric end between chromosomes 1a and 1b/c (3,4).
The most remarkable finding of the Leishmania and Trypanosoma genome sequencing projects certainly is the gene organisation in these protozoa. Most of the genes are grouped into large (50–300 kb) clusters of the polycistronic type grouped on the same DNA strand. These clusters can be transcriptionally divergent or convergent depending on the chromosome region (5). The structure of LmF chromosome 1 is one of the most conspicuous examples of this organisation; two clusters containing 29 and 50 coding DNA sequences (CDSs), each borne by one DNA strand and oriented toward one telomere, are joined by an A/T-rich 1.6-kb sequence, hereafter termed the switch region (4). Obviously, the particular position of this region as well as its structural features (e.g. A/T richness) (6) have lead to several hypotheses concerning its potential role, whether it be the site of transcription initiation for both clusters (4,7), a replication origin (5,8) or even a centromere (4). Notably, a negative correlation between purine excess and cumulative GC skew with (here termed divergent) coding directions suggested that the switch region might be the replication origin on this chromosome (8). Here, we report the successive deletions of this region on chromosome 1 homologues and analyse the effects of these targeted replacements upon mitotic stability or transcription.
MATERIALS AND METHODS
Cell line cultures and transfection conditions
The L.major Friedlin (LmF) strain (MHOM/IL/81/Friedlin) has been described previously (2). Promastigote culture and transfection experiments were as described previously (9), except for the selection of transfectant clones, which was done on RPMI1640 low-melting agarose plates (0.6%). Depending on the resistance gene(s) inserted, transfectants were selected with 30 µg/ml hygromycin B (Gibco Life Technologies®), 10 µg/ml phleomycin (Euromedex®) and/or 100 µg/ml puromycin (Euromedex®). After 10 days, resistant cells were isolated and cultivated for 1 week, and their genomic DNA was analysed by pulsed field gel electrophoresis (PFGE).
For the study of chromosomal stability, two clones, termed cHBP1 and cHBP2 (cHBP for cΔ1.6::HYG/Δ1.6::BLE/Δ1.6::PAC/1.6), were cultivated without drug pressure, subcloning was carried out at different time intervals and the subclones cultivated in parallel in the presence or absence of drugs. For the study of transcription, clones cΔccp::HYG/Δ1.6::PAC 1, 2, 3 and 4 were selected with puromycin alone. These clones were then maintained in drug-free medium during 32 generations and then transferred cultivated in parallel in the presence or absence of drugs. Cell growth curves were established by daily cell counts on a Thoma haemocytometer after cultures were seeded with 2 × 105 cells ml–1 at day 0.
Construction of the transfection vector
The transfection vectors used for this study were derived from a home-constructed vector termed pVV (GenBank accession no. AF315645), the construction of which has been detailed elsewhere (9). In this study, the HYG (10), BLE (11) and PAC (12) genes (conferring resistance to hygromycin B, phleomycin and puromycin, respectively) were used as selection markers. The flanking DNAs were from the 5′ and 3′ untranslated regions of the L.major dihydrofolate reductase-thymidilate synthase (DHFRTS) gene (13). Two homologous recombining sequences were used for all knock-outs of the switch region, and two others for the insertion of the HYG gene into the CCP2 gene on the left part of chromosome 1: their positions on chromosome 1 (according to the complete sequence of the chromosome, GenBank accession no. AE001274) are 76471–77383 and 79049–79558, and 44147–44578 and 44837–45253, respectively. These recombining sequences were PCR amplified from LmF total DNA and introduced into both cloning sites of pVV vector. The resulting constructs were termed pVV-1.6/HYG, pVV-1.6/BLE, pVV-1.6/PAC and pVV-CCP2/HYG.
DNA preparation and electrophoretic techniques
Chromosome 1 was separated by PFGE as described by Wincker et al. (1), using a voltage of 7.5 V/cm and pulse times of 35 s for 72 h. Chromosomal DNA preparation (14) and enzymatic restriction analysis (15) were as described previously.
DNA probes
Chromosome 1-specific DNA probes P1.6, PCCP2, PORF20, P3 and P5 were PCR amplified from LmF total DNA, cloned into the pGEMT-Easy vector (Promega®) and sequenced. Their locations on chromosome 1 are indicated by the position of the primers used for their PCR amplification, referring to the complete sequence of chromosome 1 (GenBank accession no. AE001274): probe P1.6 (77384–79048) strictly encompasses the 1.6-kb switch region; probes PCCP (44837–45270), PORF20 (50717–51024), P3 (79559–80066) and P5 (171328– 172306) correspond to segments of chromosome 1 located 33 and 26 kb to the left side, and immediately and 93 kb to the right side, of the switch region, respectively. Homologues of chromosome 1 were distinguished by probe ST515, specific for the 81-bp minisatellite sequence LiSTIR1 (14) which is present in chromosome 1b/c and absent from the 1a homologue (3). The HYG, BLE and PAC probes were gel purified after electrophoresis of the corresponding enzyme-restricted pVV vector. The hybridisation conditions were as described by Ravel et al. (14).
Analysis of the recombination events
The replacements of the switch region on chromosome 1 by the drug resistance genes were analysed by double restriction with NheI and SpeI, followed by Southern analysis. NheI was selected for this analysis as its restriction sites encompass a ∼9-kb fragment comprising the switch region. The SpeI site had been introduced by the insertion of the linearised transfection vector (see Fig. 2). The integration on the same chromosomal molecule of the HYG and PAC genes into CCP2 and the switch region, respectively, was verified by an SpeI restriction.
Figure 2.

. Restriction analysis of the targeted replacements of the 1.6-kb switch region on chromosome 1 homologues. (A) Schematic representation of the NheI restriction fragment encompassing the switch region on the wild-type chromosome 1 (top line) and the mutated homologues (bottom line). In the first, the switch region (1.6) is comprised in an NheI fragment of ∼8.7 kb. The insertion of any of the three drug resistance genes (R) used in our study strictly replaces the switch region and introduces an SpeI site. The size of R is different for each of the three resistance genes. Therefore, an NheI–SpeI double restriction should reveal a different sized fragment (X) depending on the specific resistance gene inserted. 5′ and 3′ are the L.major DHFRTS 5′ and 3′ untranslated regions used in the vector construct. The scale of the schematic representation is not proportional. (B) An NheI–SpeI double digest of clone cHBP1 DNA was electrophoresed on an 0.7% agarose gel and subsequently Southern blotted and hybridised. Hybridisation with DNA probe P3 [shown in (A)] revealed four different sized fragments corresponding to four homologues of chromosome 1. Probe P1.6 (specific for the switch region) hybridised with an 8.7 kb fragment corresponding to the wild-type locus. The three remaining fragments (3.8, 4 and 4.4 kb in size) each hybridised with one of the drug resistance genes inserted (BLE, PAC and HYG, respectively); their respective size correlated with the expected size after replacement of the switch region by the resistance gene [X in (A)].
RESULTS
Targeted replacement of the switch region by drug resistance genes in the three copies of LmF chromosome 1
Three homologues of chromosome 1 are present in LmF, sized 285 and 315 kb for chromosome 1a and chromosome 1b/c, respectively (both 1b and 1c therefore being indistinguishable on a PFGE karyotype). Three rounds of transfection were carried out in order to knock out the 1.6-kb switch region in these three homologues. Transfectant clones were studied by PFGE and Southern analysis to verify the successful integration of the resistance genes. After the first transfection round, the HYG gene integrated on chromosome 1b/c in clone cΔ1.6::HYG, termed cH (Fig. 1), in replacement of the switch region locus (see below). A second transfection was realised on this clone and the BLE gene was then found integrated on chromosome 1a, with the switch region absent from this chromosome (clone cΔ1.6::HYG/Δ1.6::BLE, termed cHB; Fig. 1). Lastly, clone cHB was transfected in order to replace the last copy of the switch region present on the third homologue of chromosome 1 (b/c) by the PAC gene. Twenty-four clones (termed cHBP) expressing the three drug resistances were then isolated and analysed as above. Three interesting points were then found. (i) The switch region was still present in all 24 clones, on chromosome 1b/c. (ii) In 23 clones, the PAC gene integrated at the level of chromosome 1a, but this did not knock out the BLE gene (see for example clone cHBP1 in Figure 1). (iii) In only one clone (cHBP2), PAC integrated at the level of chromosome 1b/c, but without knocking out HYG or the switch region in these homologues (Fig. 1). Two alternative hypotheses may explain these results: either two resistance genes had integrated the same chromosomal molecule (by a non-homologous recombination event) or the last transfection round had induced the formation of a fourth chromosome 1 homologue. We analysed the targeted replacements in more detail in order to settle this point.
Figure 1.
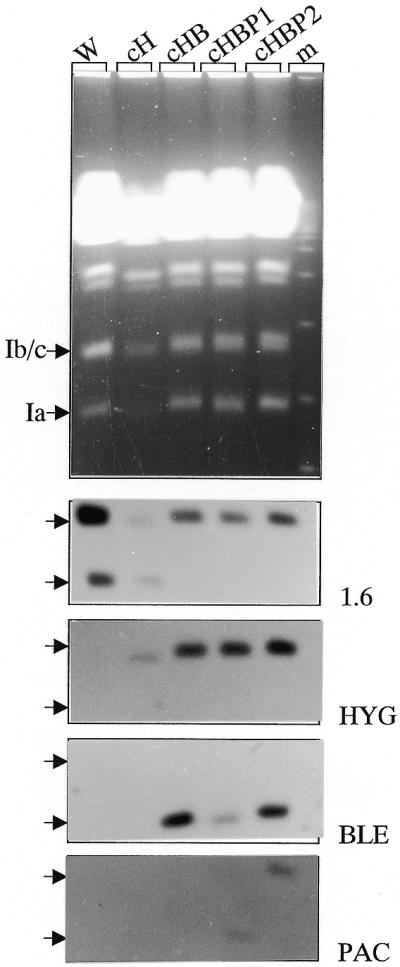
. Targeted replacement of the 1.6-kb switch region by drug resistance genes in the three homologues of chromosome 1 in L.major Friedlin. (Top) The small chromosomes of the wild-type strain (W) and of different transfectant clones (cH, cHB, cHBP1 and cHBP2; see text for details) were separated on an ethidium bromide-stained PFGE gel. (Bottom) The same gel was Southern blotted and sequentially hybridised with DNA probes P1.6, HYG, BLE and PAC. After transfection of clone cHB by the PAC-containing vector, 24 clones were isolated. The 1.6-kb switch region was present at the level of chromosome 1b/c in all clones. In 23 of them (e.g. cHBP1 here), PAC integrated at the level of chromosome 1a, where BLE remained present. In only one clone (cHBP2), PAC integrated at the level of chromosome 1b/c where the switch region and the HYG gene are also present. No hybridisation was observed with all these probes outside of the chromosome 1 size class, in particular in the compression zone (not shown). Moreover, in all clones, chromosome 1-specific DNA probes CCP2 and P5 hybridised at the same level as chromosome 1a and 1b/c in the wild-type strain (not shown), showing that no major rearrangement occurred in these chromosomes.
Restriction analysis of the switch region replacements
The replacement of the switch region by the different resistance genes should introduce an SpeI site immediately upstream of the resistance gene (Fig. 2A). The correct replacements were thus verified by NheI–SpeI double digestion of total genomic DNA (see Materials and Methods). In clone cHBP1 (Fig. 2B), three NheI–SpeI fragments of the expected sizes (3812, 4043 and 4442 bp) were recognised by DNA probes of the three resistance genes, BLE, PAC and HYG, respectively. This result rules out the hypothesis of non-homologous recombination described above, and confirms the insertion of the three markers on three distinct chromosome 1 homologues. In addition, hybridisation with the switch region probe revealed an 8.7 kb fragment corresponding to the NheI fragment on the wild-type chromosome (8676 bp from the complete sequence of chromosome 1) (see Fig. 2). These four polymorphic fragments hybridised with probe P3 (part of the CDS immediately to the right of the switch region). Therefore, they correspond on the one hand to the three mutated homologous loci that each integrated a resistance gene and on the other hand to the conserved wild-type locus. We concluded that four chromosome 1 homologues were present in clone cHBP1: two of the size of chromosome 1a (bearing either the BLE or the PAC gene) and two of the size of chromosome 1b/c (bearing either the HYG gene or the switch region). Similar restriction data were obtained in clone cHBP2, suggesting the presence of only one chromosome 1a and three homologues of the size of chromosome 1b/c (data not shown).
Mitotic stability of the switch region deletion mutant chromosomes
Clones cHBP1 and cHBP2 were cultivated without any drug pressure for 110 days (i.e. >130 generations in the conditions used here). Subcloning was carried out at different time intervals. The persistence of the resistance genes was directly tested by cultivating the subclones in parallel in the presence or absence of drugs. All the subclones tested conserved the three drug resistances. Moreover, the karyotypes of some of the subclones were verified by PFGE; all the subclones analysed exhibited a karyotype identical to that of the parental clone (not shown). Thus, the three mutant chromosomes bearing the switch region replacements proved mitotically stable in the absence of selective pressure. The presence of the switch region in cis therefore does not appear necessary to the mitotic stability of chromosome 1.
Role of the switch region in the transcription of chromosome 1
We then wished to further investigate the role of the switch region in the transcription of chromosome 1 gene clusters. In order to specifically identify the transcription of one of the homologues, in spite of the low nucleotide divergence between homologues in LmF (4), we inserted the HYG gene into the left gene cluster of chromosome 1a. The targeted sequence was the CCP2 gene, located at equal distance between the telomere and the switch region. HYG was inserted in the same orientation as CCP2. The selected clone (cΔccp::HYG) was then transfected with pVV-1.6/PAC, in order to replace the switch region by the PAC gene in a rightwards orientation (Fig. 3A). Four transfectants (cΔccp:: HYG/Δ1.6 ::PAC 1, 2, 3 and 4) that had integrated PAC in chromosome 1a were isolated. They were selected and cultivated without hygromycin. Southern analysis of the molecular karyotype of these clones (Fig. 3B) showed the insertion of both resistance genes and the loss of the switch region all on chromosome 1a. SpeI digestion followed by hybridisation with a probe specific for ORF20 (see Fig. 3A) revealed the expected 34 kb (34 453 bp) fragment (not shown), confirming that both resistance genes were located on the same chromosomal molecule in the four clones. The latter as well as cΔccp::HYG were then cultivated in parallel in the presence or absence of hygromycin. All growth curves proved identical (Fig. 3C), showing that the HYG gene was expressed in a similar manner whether the switch region was present or absent.
Figure 3.


. Analysis of clone cΔccp::HYG/Δ1.6::PAC 1 bearing a double mutation on chromosome 1a. (A) Schematic map of the left part of chromosome 1a, including the two targeted sequences and the transfection vector sequences inserted; the arrows indicate the transcription sense orientation of the marker resistance genes. The wild-type chromosome 1 has only one SpeI site located at the right end of the chromosome (position 267 268 bp). The double insertion of HYG and PAC introduced two novel SpeI sites. The collinearity of both resistance genes on the chromosome was verified by the hybridisation of probe PORF20 on a ∼34-kb SpeI fragment. (B) PFGE separation of the small chromosomes of strain LmF (a), clone cΔccp::HYG (b) and clone cΔccp::HYG/Δ1.6::PAC 1 (c). The gel was Southern blotted and hybridised sequentially with the P1.6, HYG and PAC probes. (C) Comparison of the growth kinetics of clones cΔccp::HYG (a) and cΔccp::HYG/Δ1.6::PAC 1 (b) and 2 (c) in hygromycin-containing medium over 7 days (J0–J7). Identical curves were obtained in drug-free medium (not shown).
DISCUSSION
In the present study, we have focused on the as yet unclear role of the 1.6-kb sequence located at the junction between the two large divergent gene clusters of L.major chromosome 1. We performed targeted deletions of this sequence on the three chromosome 1 homologues present in this strain. Nevertheless, we were unable to create null mutants for this switch region, as a fourth wild-type copy of chromosome 1 persisted after the last transfection round, leading to tetrasomy for this chromosome. Other authors have experienced similar difficulties in disrupting essential genes in the Leishmania genome, due to intrachromosomal amplification, creation of extrachromosomal elements (16), genomic rearrangements of the duplication–transposition type (17), or duplication of whole chromosomes (18). We therefore conclude that the persistence of this 1.6-kb sequence is essential for parasite survival. In a second part of the study, we examined possible changes in the expression of the HYG gene inserted into the left gene cluster of chromosome 1a after we deleted the switch region on the same chromosome. The potential role of this region, as well as a number of mechanisms in which it is supposedly involved (replication, transcription, segregation), are discussed below.
The first hypothesis tested is whether this sequence is necessary for chromosomal stability, i.e. is a single replication origin (8) or a centromeric sequence (4). Our results show that its presence in cis is not necessary for chromosomal stability. Indeed, chromosomes which did not bear this sequence were mitotically stable, hence they were able to replicate and segregate normally. Regarding centromeric function, this finding is in agreement with recent studies suggesting that the sequences involved in chromosomal segregation may be located in the subtelomeric part of chromosome 1 (19). Regarding chromosome replication, although our results do not support the presence of an origin of replication in the switch region, they do not formally rule out this hypothesis. Indeed, in Saccharomyces cerevisiae, the knock-out of a functional autonomous replicating sequence (ARS) can be compensated by the activation of a neighbouring ARS (20,21). Moreover, a trans-complementation by an ARS present on the other homologue might be responsible for the maintenance of a chromosome fragment without origin (22). Here, the difficulty encountered in completely knocking out the switch region from the genome might be explained by the second hypothesis. In this case, the only remaining 1.6-kb region (on the fourth homologue) might complement the absence of origin on the three others. More elements will obviously be necessary to settle this point.
The second hypothesis that has been proposed about the role of the switch region is that it is a key element in the initiation/regulation of transcription of the two gene clusters (4). The difficulty in knocking it out of the genome might be due to the fact that all essential genes present on chromosome 1 would remain untranscribed on the mutated homologues. However, our results do not fit with the latter hypothesis. First, the three resistance genes replacing the switch region in the knockouts were expressed. Secondly, when we deleted the switch region on a homologue of chromosome 1 tagged with the HYG gene integrated in the left gene cluster, the growth kinetics of the double mutants showed that the absence of the switch region did not act upon the expression of this collinear gene.
The persistence of the switch region might then have another more trivial explanation: its knockout may have disrupted the splice site of the first gene in each of the opposing polycistronic transcription units [similar to exopolyphosphatase and poly(A) export protein genes, respectively]. If either of these genes were essential, one would never generate a switch region null mutant. Finally, one cannot rule out another more speculative hypothesis: that the switch region, which does not appear to contain any open reading frames, might contain essential non-coding RNAs (e.g. the recently identified miRNAs) (23).
On the whole, this work provides direct functional evidence that the main hypotheses about the role of the switch region (whether single origin of replication or region of initiation of the transcription of both chromosome 1 gene clusters) (4–8) are not validated by experimental results. The bioinformatic analysis of five switch regions located on different chromosomes did not allow identification of structural elements strictly common to all, with the exception of a high intrinsic DNA curvature (6). Although their peculiar position between two convergent or divergent gene clusters made them candidates as major regulating/functional sequences, it is not impossible that their role is restricted to the transcription of the sole adjacent genes. The publication of the complete sequence of the LmF genome, predicted for late 2002 (24), should allow a more systematic structural and functional analysis of these regions, and thus help to define their possible role in chromosome function.
Acknowledgments
ACKNOWLEDGEMENTS
We gratefully acknowledge the expert technical help of Michèle Lefebvre. We also thank Etienne Schwob for helpful advice and discussion. Finally, we are grateful to the reviewers for their pertinent questioning, which led us to more precise and extended conclusions. This study received financial support from the Centre National de la Recherche Scientifique (CNRS), the GDR 1077 of the CNRS/DGA-DSP and the CNRS programme ‘Puces à ADN’ (no. 00N65/0004).
REFERENCES
- 1.Wincker P., Ravel,C., Blaineau,C., Pagès,M., Jauffret,Y., Dedet,J.P. and Bastien,P. (1996) The Leishmania genome comprises 36 chromosomes conserved across widely divergent human pathogenic species. Nucleic Acids Res., 24, 1688–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ravel C., Dubessay,P., Blackwell,J.M., Ivens,A.C., Bastien,P. and the Leishmania Genome Network (1998) The complete chromosomal organization of the reference strain of the Leishmania Genome Project, L.major ‘Friedlin’. Parasitol. Today, 14, 301–303. [DOI] [PubMed] [Google Scholar]
- 3.Sunkin S.M., Kiser,P., Myler,P.J. and Stuart,K. (2000) The size difference between Leishmania major Friedlin chromosome one homologues is localized to sub-telomeric repeats at one chromosomal end. Mol. Biochem. Parasitol., 109, 1–15. [DOI] [PubMed] [Google Scholar]
- 4.Myler P.J., Audelman,L., De Vos,T., Hixson,G., Kiser,P., Lemley,G., Magness,C., Rickel,E., Sisk,E., Sunkin,S., Swartzell,S., Westlake,T., Bastien,P., Fu,G., Ivens,A. and Stuart,K. (1999) Leishmania major Friedlin chromosome 1 has an unusual distribution of protein-coding genes. Proc. Natl Acad. Sci. USA, 96, 2902–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Myler P.J., Sisk,E., McDonagh,P.D., Martinez-Calvillo,S., Schnaufer,A., Sunkin,S.M., Yan,S., Madhubala,R., Ivens,A. and Stuart,K. (2000) Genomic organisation and gene function in Leishmania. Biochem. Soc. Trans., 28, 527–531. [DOI] [PubMed] [Google Scholar]
- 6.Tosato V., Ciarloni,L., Ivens,A.C., Rajandream,M.A., Barrell,B.G. and Bruschi,C.V. (2001) Secondary DNA structure analysis of the coding strand switch regions of five Leishmania major Friedlin chromosomes. Curr. Genet., 40, 186–194. [DOI] [PubMed] [Google Scholar]
- 7.Myler P.J., Beverley,S.M., Cruz,A.K., Dobson,D.E., Ivens,A.C., McDonagh,P.D., Madhubala,R., Martinez-Calvillo,S., Ruiz,J.C., Saxena,S., Sisk,E., Sunkin,S.M., Worthey,E., Yan,S. and Stuart,K.D. (2001) The Leishmania genome project: new insights into gene organization and function. Med. Microbiol. Immunol. (Berl.), 190, 9–12. [DOI] [PubMed] [Google Scholar]
- 8.McDonagh P.D., Myler,P.J. and Stuart,K. (2000) The unusual gene organization of Leishmania major chromosome 1 may reflect novel transcription processes. Nucleic Acids Res., 28, 2800–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dubessay P., Ravel,C., Bastien,P., Lignon,M.F., Ullman,B., Pagès,M. and Blaineau,C. (2001) Effect of large targeted deletions on the mitotic stability of an extra chromosome mediating drug-resistance in Leishmania. Nucleic Acids Res., 29, 3231–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cruz A., Titus,R. and Beverley,S.M. (1993) Plasticity in chromosome number and testing of essential genes in Leishmania by targeting. Proc. Natl Acad. Sci. USA, 90, 1599–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jefferies D., Tebabi,P., Le Ray,D. and Pays,E. (1993) The ble resistance gene as a new selectable marker for Trypanosoma brucei: fly transmission of stable procyclic transformants to produce antibiotic resistant bloodstream forms. Nucleic Acids Res., 21, 191–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Freedman D.J. and Beverley,S.M. (1993) Two more independent selectable markers for stable transfection of Leishmania. Mol. Biochem. Parasitol., 62, 37–44. [DOI] [PubMed] [Google Scholar]
- 13.Ryan K.A., Dasgupta,S. and Beverley,S.M. (1993) Shuttle cosmid vectors for the trypanosomatid parasite Leishmania. Gene, 131, 145–150. [DOI] [PubMed] [Google Scholar]
- 14.Ravel C., Wincker,P., Bastien,P., Blaineau,C. and Pagès,M. (1995) A polymorphic minisatellite sequence in the subtelomeric regions of chromosome I and V in Leishmania infantum. Mol. Biochem. Parasitol., 74, 31–34. [DOI] [PubMed] [Google Scholar]
- 15.Ravel C., Wincker,P., Blaineau,C., Britto,C., Bastien,P. and Pagès,M. (1996) Medium range restriction maps of five chromosomes of Leishmania infantum and localization of size-variable regions. Genomics, 35, 509–516. [DOI] [PubMed] [Google Scholar]
- 16.Curotto de Lafaille M.A. and Wirth,D.F. (1992) Creation of null/+ mutants of the α-tubulin gene in Leishmania enrietti by gene cluster deletion. J. Biol. Chem., 267, 23839–23846. [PubMed] [Google Scholar]
- 17.Dumas C., Ouellette,M., Tovar,J., Cunningham,M.L., Fairlamb,A.H., Tamar,S., Olivier,M. and Papadopoulou,B. (1997) Disruption of the trypanothione reductase gene of Leishmania decreases its ability to survive oxidative stress in macrophages. EMBO J., 16, 2590–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cruz A.K., Titus,R. and Beverley,S.M. (1993) Plasticity in chromosome number and testing of essential genes in Leishmania by targeting. Proc. Natl Acad. Sci. USA, 90, 1599–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dubessay P., Ravel,C., Bastien,P., Stuart,K., Dedet,J.P., Blaineau,C. and Pagès,M. (2002) Mitotic stability of a CDS-free version of Leishmania major chromosome 1 generated by targeted chromosome fragmentation. Gene, 289, 151–159. [DOI] [PubMed] [Google Scholar]
- 20.Dershowitz A. and Newlon,C.S. (1993) The effect on chromosome stability of deleting replication origins. Mol. Cell. Biol., 13, 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vujcic M., Miller,C.A. and Kowalski,D. (1999) Activation of silent replication origins at autonomously replicating sequence elements near HML locus in budding yeast. Mol. Cell. Biol., 19, 6098–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tabrizifard S. and Newlon,C.S. (2001) Maintenance of an originless fragment in Saccharomyces cerevisiae. In Eukaryotic DNA Replication. Cold Spring Harbor Laboratory Meeting, Sept. 5–9th, Cold Spring Harbor, NY, p. 274.
- 23.Storz G. (2002) An expanding universe of noncoding RNAs. Science, 296, 1260–1263. [DOI] [PubMed] [Google Scholar]
- 24.Almeida R., Norrish,A., Levick,M., Vetrie,D., Freeman,T., Vilo,J., Ivens,A., Lange,U., Stober,C., McCann,S. and Blackwell,J.M. (2002) From genomes to vaccines: Leishmania as a model. Philos. Trans. R. Soc. Lond. B Biol. Sci., 357, 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]