

Abstract
The native kidney has a major role in lactate metabolism. The renal cortex appears to be the major lactate-consuming organ in the body after the liver. Under conditions of exogenous hyperlactatemia, the kidney is responsible for the removal of 25–30% of all infused lactate. Most of such removal is through lactate metabolism rather than excretion, although under conditions of marked hyperlactatemia such excretion can account for approximately 10–12% of renal lactate disposal. Indeed, nephrectomy results in an approximately 30% decrease in exogenous lactate removal. Importantly and differently from the liver, however, the kidney's ability to remove lactate is increased by acidosis. While acidosis inhibits hepatic lactate metabolism, it increases lactate uptake and utilization via gluconeogenesis by stimulating the activity of phospho-enolpyruvate carboxykinase. The kidney remains an effective lactate-removing organ even during endotoxemic shock. The artificial kidney also has a profound effect on lactate balance. If lactate-buffered fluids are used in patients who require continuous hemofiltration and who have pretreatment hyperlactatemia, the serum lactate levels can significantly increase. In some cases, this increase can result in an exacerbation of metabolic acidosis. If bicarbonate-buffered replacement fluids are used, a significant correction of the acidosis or acidemia can also be achieved. The clinician needs to be aware of these renal effects on lactate levels to understand the pathogenesis of hyperlactatemia in critically ill patients, and to avoid misinterpretations and unnecessary or inappropriate diagnostic or therapeutic activities.
Keywords: kidney, lactate
Introduction
Lactic acidosis is an important metabolic disorder associated with a poor outcome [1,2]. It is not surprising, therefore, that its pathogenesis has been, and continues to be, a great source of interest to critical care physicians [3]. All the information available from animal and human investigations indicates that lactate production and metabolism are two extraordinarily complex processes found in almost every organ, and that are perhaps as fundamental to intermediate metabolism as the generation and consumption of glucose. It is little surprise that the kidney should play a pivotal role in such processes, as it does in many other aspects of metabolism.
In the present review, the role of the native kidney as well as that of the artificial kidney in lactate production, lactate release, lactate uptake, lactate metabolism and lactate balance will be explored. There will be a strong focus on the clinical implications of such a role, with the aim of helping clinicians better understand the possible pathogenesis of the changes in blood lactate levels that they see in the intensive care unit every day.
The native kidney and lactate
There is strong evidence that, under normal physiological conditions, the native kidney is second only to the liver in removing lactate from the circulation and metabolizing it [1,4,5,6]. Such evidence is based on exogenous lactate infusion studies and nephrectomy studies in the rat, the dog and the sheep. These studies suggest that the native kidney's contribution to the removal of lactate is substantial, with the organ being responsible for the removal of approximately 20–30% of an exogenous load [1,5]. Such removal is mostly due to uptake and metabolism rather than urinary excretion. Indeed, even when the lactate level is artificially kept at approximately 10 mol/l to maximize urinary excretion, such excretion only accounts for 10–12% of the total removal of lactate achieved by the kidney [5]. When nephrectomy is performed, the half-life for lactate elimination is increased from 5.3 to 7.1 min and clearance is decreased from 45.3 to 32 ml/kg/min [5].
Various pathological conditions can be expected to influence the normal physiological role of the native kidney in lactate disposal. In studies of graded hemorrhage in the dog, for example, renal lactate uptake, which remains stable even with a blood loss close to 30% of the total volume, decreases sharply once blood loss reaches the 40% mark. Once such blood loss reaches 50% of the total blood volume (mean blood pressure of 38 mmHg with a 90% reduction in renal blood flow), renal lactate production occurs [7]. Reinfusion of shed blood does not restore renal lactate uptake to normal.
Acidosis also affects renal lactate uptake [1]. However, while acidosis significantly depresses hepatic uptake of lactate, acidosis enhances renal lactate metabolism [8,9,10]. Such adaptation takes place over 2–4 hours and occurs despite a reduction in renal blood flow. The renal contribution to lactate removal thus increases from 16% at a pH of 7.45 to 44% at a pH of 6.75 [4]. These changes will probably be important in human acidosis, and they compensate for approximately 50% of the hepatic loss of lactate metabolism.
The effect of endotoxemia on renal lactate uptake has been studied in the dog. Bellomo et al. [11] have shown that, even during advanced endotoxemia and a reduction in renal blood flow of close to 30%, the kidney continues to removal lactate from the circulation (Fig. 1).
Figure 1.
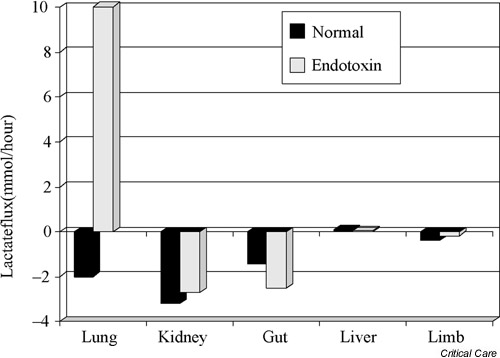
Histogram illustrating lactate fluxes across different regional beds in the endotoxemic dog. A negative value indicates removal/uptake, and a positive value indicates release. At baseline, there is lactate removal by the kidney. Lactate removal continues after the induction of endotoxemia. Reproduced from [11] with permission.
The fate of lactate within the kidney
It is clear from the evidence presented that the kidney is a major organ for lactate disposal and that such disposal only ceases under conditions of extreme (90%) decreases in renal perfusion. This information treats the kidney like a 'black box', however, and does not tell us whether there is uniformity within the kidney in terms of lactate metabolism and what the fate of lactate is within the organ. In this regard, it is important to appreciate that the fate of lactate within the kidney is complex, that it depends on a variety of hormonal and physiological stimuli, and that it differs from the medulla to the cortex.
The first observation concerning intrarenal lactate handling, as already highlighted, is that lactate is fully filtered by the glomerulus [12]. However, it is also almost completely re-absorbed in the proximal tubule [12]. Only a marked rise in blood lactate levels results in an increase in urinary excretion. Even then, lactate urinary losses are small in comparison with overall renal lactate metabolism, with only 2% of total lactate removal during exercise (plasma lactate >20 mmol/l) being achieved by urinary excretion [13].
Lactate uptake is the major mechanism of renal lactate removal and appears to be essentially confined to the cortex [5,14], as shown by radioisotopic methods in isolated, perfused rat kidney [14]. These studies also show that, in the absence of glucose and in the presence of starvation, the cortex produces negligible amounts of lactate. Once glucose is administered, the cortex continues to produce little, if any, lactate. The medulla, on the contrary, uses radiolabeled glucose and generates lactate from its glycolysis. The cortex simultaneously takes up the lactate released by the medulla and uses it for oxidation and gluconeogenesis. The cortex does not oxidize glucose directly.
These findings suggest the presence of a cortico-medullary glucose-lactate recycling system. The medulla consumes glucose (glycolysis) and generates lactate. The cortex takes up lactate to oxidize it for energy production and to generate glucose for release back to the medulla for medullary glycolysis and energy production. A similar recycling system may operate in the brain between neurons and astrocytes, and in the testis between Sertoli cells and spermatozoa.
To understand the pivotal role of lactate in intrarenal bioen-ergetics, it is important to note that lactate production from glucose correlates with the glomerular filtration rate (even though there is basal lactate production at zero glomerular filtration rate). The lactate production also correlates with the urine flow rate and sodium resorption. Lactate consumption, on the contrary, shows no correlation with any renal function [14].
When sodium reabsorption was inhibited with a loop diuretic [14], lactate production decreased by approximately 40%. When filtration was prevented, lactate production decreased by 50%. Prevention of filtration also inhibited consumption of lactate. These findings strongly suggest that glycolysis is needed for sodium reabsorption but that other renal transport functions exist that require lactate oxidation. These observations highlight the complexity of lactate metabolism within the kidney and challenge any naïve notion of lactate being a reliable marker of 'cell hypoxia' or 'anaerobic metabolism'. Indeed, the medulla has been shown by other investigators to produce lactate in spite of adequate substrate and oxygen supply [15].
If the medulla produces lactate through glycolysis, such a metabolic pathway appears a straightforward and 'natural' way to provide energy for the medulla's tasks. If the cortex does not produce anything but minute quantities of lactate and rather takes up this substrate, however, what is then the fate of lactate within the cortex? The answer to this question is predictably complex, and depends on the pathophysiological state of the organism, on the hormonal milieu, on the demands imposed on the organ and on the nutrients available.
For example, some investigators [5] have found that 22.4% of total renal CO2 production in chronic acidosis is derived from lactate oxidation, while 47.4% is so derived in alkalosis. At the same time, conversion of lactate to glucose (gluconeogenesis in the cortex) during acidosis accounted for the addition of 6.7 mol/min glucose to the renal vein, while it only accounted for the addition of 2 mol/min glucose in alkalosis [5]. When stoichiometric calculations are performed, it can be shown that these two pathways of lactate metabolism (oxidation and gluconeogenesis) account for 100% of radiola-beled lactate utilization [5]. These findings are supported by other studies [16].
The importance of renal gluconeogenesis to the overall balance of glucose and to the maintenance of glucose homeostasis has been studied in detail under normal physiological circumstances and during insulin-induced hypoglycemia in the awake dog, and indeed in humans by cannulation of the renal vein [17,18]. The findings of these investigations indicate that renal lactate uptake could account for approximately 40% of postabsorptive renal glucose production and for 60% of renal glucose production during hypoglycemia. Such glucose production results in a fivefold to 10-fold increase in glucose release into the renal vein after insulin-induced hypo-glycemia, which adds a further 4 g glucose to the systemic circulation every hour. Lactate may thus be the major gluconeogenetic precursor in the kidney under some conditions, and contributes significantly to the glycemic impact of other renal-specific precursors of gluconeogenesis such as glycerol [17], alanine and glutamine [19].
The artificial kidney and lactate
The use of the artificial kidney has a clinically significant impact on lactate balance and on plasma lactate concentrations. This impact may derive from lactate removal as well as from lactate administration. Lactate clearance during intermittent hemodialysis or intermittent hemofiltration has not been formally studied, but is probably similar to that of other small molecules given the molecular weight of lactate. Assuming a small molecular clearance of 200 ml/min, lactate clearance during dialysis would reach approximately 20% of endogenous clearance. The impact of such clearance on lactate levels, however, has not been studied. Lactate has not been traditionally used as a buffer for intermittent hemodialysis. There is therefore little specific information on the use of lactate-buffered dialysate on lactate levels and on acid–base balance in dialysis patients [20].
When intermittent hemofiltration is used and lactate-based replacement fluid is administered at high rates (approximately 200 mmol/hour), however, a significant increase in plasma lactate levels can be easily demonstrated [21] (Fig. 2). Although the clinical significance of such increases in lactate levels is unknown, this iatrogenic phenomenon needs to be appreciated to avoid misdiagnosis. The magnitude of this phenomenon (average peak increase of 3 mmol/l at 3 hours) also needs to be understood to separate it from other factors, which may simultaneously be operative in determining the patient's lactate levels.
Figure 2.
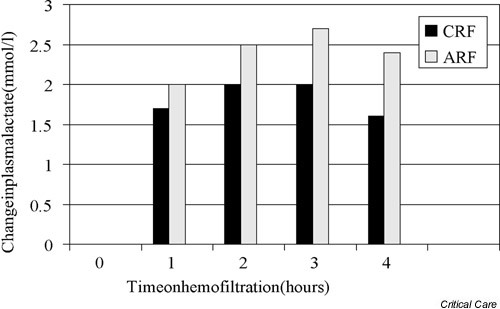
Histogram illustrating the mean increment in plasma lactate concentration induced by intermittent machine hemofiltration with the exogenous administration of approximately 200 mmol/hour lactate in patients with acute renal failure (ARF) or chronic renal failure (CRF).
A similar phenomenon has been described during continuous renal replacement therapy (CRRT), but the increment in lactate levels was less due to the lower rate of lactate administration [22]. It is important to note, however, that increments in lactate levels in patients on CRRT are not simply dependent on the rate of lactate administration, but also on the body's ability to handle a given lactate load. The administration of up to 200 mmol/hour lactate may thus lead to modest changes in lactate levels and the pH. However, the administration of the same amount or even less in a patient with pre-treatment lactate intolerance (liver failure, severe septic shock) will induce a dramatic increase in lactate concentration and a profound acidosis. Under such circumstances, lactate-buffered replacement solutions should be avoided [23]. Furthermore, in patients with lactic acidosis and acute renal failure receiving CRRT, the administration of bicarbonate-based replacement fluids is an effective way of avoiding any exacerbation of hyperlactatemia and of restoring acid–base homeostasis [24].
Some investigators have suggested that lactate removal during CRRT may lower plasma lactate levels and may participate in the correction of acidosis seen during bicarbonate-based CRRT. In response to this hypothesis, Levraut et al. conducted a careful analysis of lactate clearance during CRRT and compared it with endogenous clearance [25]. They found that the median endogenous lactate clearance was 1379 ml/min, while the median filter lactate clearance was 24.2 ml/min. CRRT-based lactate clearance thus accounted for <3% of total lactate removal (Fig. 3).
Figure 3.
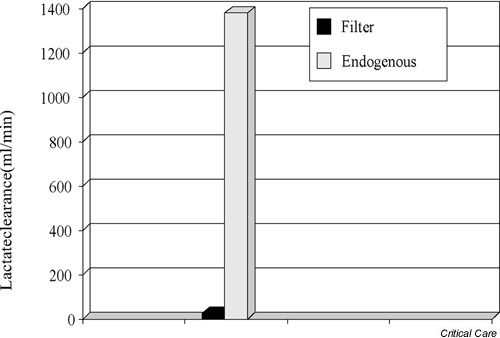
Diagram comparing endogenous lactate clearance with lactate clearance during hemofiltration (filter). There is very little contribution of hemofiltration to lactate clearance.
Finally, it may appear surprising that increases in plasma lactate concentration of up to 8 mmol/l would not induce a pronounced degree of acidification. These increases should do so by increasing the concentration of anions in plasma, and thus decreasing the strong ion difference and its effect on the dissociation of plasma water into hydrogen ions [26].
Some preliminary observations in fact suggest that several complex events may occur during the onset of such rapid iatrogenic hyperlactatemia. In particular, a marked decrease in chloride appears to occur despite the administration of chloride-rich replacement fluid (Fig. 4). This change in chloride is probably secondary to a shift into cells similar to that seen in venous blood when the CO2 increases (Hamburger shift). Such a shift in chloride rapidly attenuates the impact of hyperlactatemia on the pH and prevents the development of a progressive and sustained acidemia.
Figure 4.
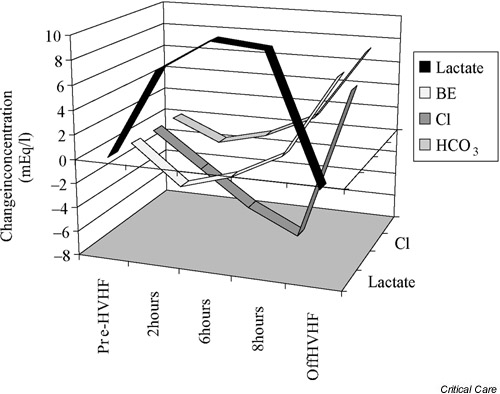
Changes in the concentration of lactate, bicarbonate (HCO3), base excess (BE) and chloride (Cl) after a patient with septic shock was placed on high-volume hemofiltration (HVHF) and received an infusion of 240 mmol/hour lactate. The acidifying effect of hyperlactatemia (fall in bicarbonate and in base excess) was markedly attenuated by a decrease in serum chloride concentration (chloride shift). At the end of 8 hours of HVHF, there was a rebound alkalosis.
Conclusions
The native kidney profoundly affects lactate metabolism by its uptake and utilization in the cortex. Its cortex uses lactate from medullary and systemic sources to obtain energy through oxidation and to form glucose for systemic and medullary use. Such metabolic pathways account for about 30% of total lactate disposal, and lactate-based gluconeogenesis contributes to systemic glucose homeostasis. These pathways are increased by acidosis and hypoglycemia, they continue to function during endotoxemia and they only fail during gross reductions in renal blood flow.
If renal replacement becomes necessary because of renal failure, lactate clearance is probably limited and contributes little to lactate removal. If large amounts of lactate-based dialysate or replacement fluids are administered, however, iatrogenic hyperlactatemia occurs, which can significantly contribute to the aggravation of metabolic acidosis. No complete understanding of the pathogenesis of hyperlactatemia can be achieved without a full appreciation of the 'renal' side of the lactate balance equation.
Competing interests
None declared.
Abbrevations
CRRT = continuous renal replacement therapy.
Acknowledgments
Acknowledgement
This work was supported by the Austin Hospital Anaesthesia and Intensive Care Trust Fund.
See related Commentary: http://ccforum.com/content/6/4/284. This article is based on a presentation at the Lactate Satellite Meeting held during the 8th Indonesian-International Symposium on Shock and Critical Care, Bali, Indonesia, 24 August 2001.
References
- Mizock BA, Falk JL. Lactic acidosis in critical illness. Crit Care Med. 1992;20:80–83. doi: 10.1097/00003246-199201000-00020. [DOI] [PubMed] [Google Scholar]
- Madias NE. Lactic acidosis. Kidney Int. 1986;28:752–774. doi: 10.1038/ki.1986.62. [DOI] [PubMed] [Google Scholar]
- Guitierrez G, Wulf ME. Lactic acidosis in sepsis: a commentary. Intensive Care Med. 1996;22:2–16. doi: 10.1007/BF01728325. [DOI] [PubMed] [Google Scholar]
- Yudkin J, Cohen RD. The contribution of the kidney to the removal of lactic acid load under normal and acidotic conditions in the conscious rat. Clin Sci Mol Med. 1975;48:121–131. doi: 10.1042/cs0480121. [DOI] [PubMed] [Google Scholar]
- Leal-Pinto E, Park HC, King F, MacLeod M, Pitts RF. Metabolism of lactate by the intact functioning kidney of the dog. Am J Physiol. 1973;224:1463–1467. doi: 10.1152/ajplegacy.1973.224.6.1463. [DOI] [PubMed] [Google Scholar]
- Brand PH, Cohen JJ, Bignall MC. Independence of lactate oxidation from net Na+ reabsorption in dog kidney in vivo. Am J Physiol. 1981;240:F343–F351. doi: 10.1152/ajplegacy.1974.227.6.1255. [DOI] [PubMed] [Google Scholar]
- Nelimarkka O, Halkola L, Ninikoski J. Renal hypoxia and lactate metabolism in hemorrhagic shock in dogs. Crit Care Med. 1984;12:656–660. doi: 10.1097/00003246-198408000-00011. [DOI] [PubMed] [Google Scholar]
- Dawson AG. Contribution of pH sensitive metabolic processes to pH homeostasis in isolated rat kidney tubules. Biochim Biophys Acta. 1977;499:85–98. doi: 10.1016/0304-4165(77)90231-8. [DOI] [PubMed] [Google Scholar]
- Yudkin J, Cohen RD. The effect of acidosis on lactate removal by the perfused rat kidney. Clin Sci Mol Med. 1976;50:185–194. doi: 10.1042/cs0500185. [DOI] [PubMed] [Google Scholar]
- Alleyne GAO, Flores H, Robool A. The interrelationship of the concentration of hydrogen ions, bicarbonate ions, carbon dioxide and calcium ions in the regulation of renal gluconeogenesis in the rat. Biochem J. 1973;136:445–453. doi: 10.1042/bj1360445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellomo R, Kellum JA, Pinsky MR. Transvisceral lactate uptake fluxes during early endotoxemia. Chest. 1996;110:198–204. doi: 10.1378/chest.110.1.198. [DOI] [PubMed] [Google Scholar]
- Hohmann B, Frohnert PP, Kinne R, Baumann K. Proximal tubular lactate transport in rat kidney: a micropuncture study. Kidney Int. 1974;261:5–11. doi: 10.1038/ki.1974.35. [DOI] [PubMed] [Google Scholar]
- McKelvie RS, Lindiger MI, Heigenhauser GJ, Sutton JR, Jones NL. Am J Physiol. 1989;257:R102–R108. doi: 10.1152/ajpregu.1989.257.1.R102. [DOI] [PubMed] [Google Scholar]
- Bartlett S, Espinal J, Janssens P, Ross BD. The influence of renal function on lactate and glucose metabolism. Biochem J. 1984;219:73–78. doi: 10.1042/bj2190073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JJ. Is the function of the renal papilla coupled exclusively to an anaerobic pattern of metabolism? Am J Physiol. 1979;236:F423–F428. doi: 10.1152/ajprenal.1979.236.5.F423. [DOI] [PubMed] [Google Scholar]
- Levy MN. Uptake of lactate and pyruvate by intact kidney of the dog. Am J Physiol. 1962;202:302–308. doi: 10.1152/ajplegacy.1962.202.2.302. [DOI] [PubMed] [Google Scholar]
- Cerosimo E, Molina PE, Abumrad NN. Renal lactate metabolism and gluconeogenesis during insulin-induced hypoglycemia. Diabetes. 1998;47:1101–1106. doi: 10.2337/diabetes.47.7.1101. [DOI] [PubMed] [Google Scholar]
- Cerosimo E, Garlick P, Ferretti J. Renal substrate metabolism and gluconeogenesis during hypoglycemia in humans. Diabetes. 2000;49:1186–1193. doi: 10.2337/diabetes.49.7.1186. [DOI] [PubMed] [Google Scholar]
- Stumvoll M, Meyer C, Perriello G, Kreider M, Welle S, Gerich J. Human kidney and liver gluoconeogenesis: evidence for organ subselectivity. Am J Physiol. 1998;274:E817–E826. doi: 10.1152/ajpendo.1998.274.5.E817. [DOI] [PubMed] [Google Scholar]
- Feriani M, Ronco C, Biasioli S, Bragantini L, La Greca G. Effect of dialysate and substitution fluid buffer on buffer flux in hemodiafiltration. Kidney Int. 1991;39:711–717. doi: 10.1038/ki.1991.86. [DOI] [PubMed] [Google Scholar]
- Davenport A, Will EJ, Davison AM. Hyperlactatemia and metabolic acidosis during hemofiltration using lactate-buffered fluids. Nephron. 1991;59:461–465. doi: 10.1159/000186609. [DOI] [PubMed] [Google Scholar]
- Thomas AN, Guy JM, Kishen R, Geraghty IF, Bowles BJM, Vadgama P. Comparison of lactate and bicarbonate buffered haemofiltration fluids: use in critically ill patients. Nephrol Dial Transplant. 1997;12:1212–1217. doi: 10.1093/ndt/12.6.1212. [DOI] [PubMed] [Google Scholar]
- Davenport A, Will EJ, Davison AM. The effect of lactate-buffered solution on the acid–base status of patients with renal failure. Nephrol Dial Transplant. 1989;4:800–804. [PubMed] [Google Scholar]
- Hilton PJ, Taylor J, Forni LG, Treacher DF. Bicarbonate-based haemofiltration in the management of acute renal failure with lactic acidosis. Q J Med. 1998;91:279–283. doi: 10.1093/qjmed/91.4.279. [DOI] [PubMed] [Google Scholar]
- Levraut J, Ciebiera J-P, Jambou P, Ichai C, Labib Y, Grimaud D. Effect of continuous veno-venous hemofiltration with dialysis on lactate clearance in critically ill patients. Crit Care Med. 1997;25:58–62. doi: 10.1097/00003246-199701000-00013. [DOI] [PubMed] [Google Scholar]
- Stewart PA. Modern quantitative acid–base chemistry. Can J Physiol Pharmacol. 1983;61:1444–1461. doi: 10.1139/y83-207. [DOI] [PubMed] [Google Scholar]