

Abstract
The oncoprotein hdm2 ubiquitinates p53, resulting in the rapid degradation of p53 through the ubiquitin (Ub)–proteasome pathway. Hdm2-mediated destabilization and inactivation of p53 are thought to play a critical role in a number of human cancers. We have used an in vitro enzyme assay, monitoring hdm2-catalyzed Ub transfer from preconjugated Ub-Ubc4 to p53, to identify small molecule inhibitors of this enzyme. Three chemically distinct types of inhibitors were identified this way, each with potency in the micromolar range. All three types of compounds display selective inhibition of hdm2 E3 ligase activity, with little or no effect on other Ub-using enzymes. Most strikingly, these compounds do not inhibit the autoubiquitination activity of hdm2. Steady-state analysis reveals that all three classes behave as simple reversible inhibitors of the enzyme and that they are noncompetitive with respect to both substrates, Ub-Ubc4 and p53. Studies of the effects of combinations of two inhibitory molecules on hdm2 activity indicate that the three types of compounds bind in a mutually exclusive fashion, suggesting a common binding site on hdm2 for all of these inhibitors. These compounds establish the feasibility of selectively blocking hdm2-mediated ubiquitination of p53 by small molecule inhibitors. Selective inhibitors of hdm2 E3 ligase activity could provide a novel mechanism for the development of new chemotherapeutics for the treatment of human cancers.
Perturbation in concentration and/or function of the p53 tumor suppressor protein is one of the most common features associated with human cancers (1, 2). The p53 protein is a transcriptional activation factor for a variety of genes and is present at a very low concentration in normal cells. In response to certain stimuli, such as DNA damage and other stress signals, p53 is stabilized, leading to a net increase in protein level and transcriptional activity. The transcriptional activity of p53 results in growth arrest to allow the cell to undergo genetic repair; if, however, the damage is beyond repair, the increased activity of p53 leads instead to apoptotic cell death. By either mechanism, the propagation of genetic mutations is halted by the action of p53 (1, 2). Hence, loss of p53 function provides a common mechanism for the uncontrolled proliferation of mutant cells that is associated with many human cancers. Loss-of-function mutations in p53 exist in ≈50% of all human tumors. In the remaining 50% of tumors, wild-type p53 is commonly inactivated by a variety of other mechanisms. One such mechanism for the loss of p53 function involves alteration in the functional level of the oncoprotein hdm2, a negative regulator that interacts directly with the p53 protein.
The hdm2 oncoprotein plays an important role in regulating p53 concentration and function (3–5). By binding within the transactivation domain of p53, hdm2 inhibits the transcriptional activity of the tumor suppressor. Additionally, hdm2 catalyzes the ubiquitination of p53, thus promoting p53 degradation through the ubiquitin (Ub)/proteasome pathway. Transcription of the hdm2 gene itself is regulated by p53 in response to cellular stress; thus p53 and hdm2 form an autoregulatory feedback loop, providing a mechanism for tight control over the protein level, hence function of p53 (1, 2, 5).
By tagging p53 with Ub, hdm2 destines p53 for proteasome-mediated degradation. The Ub/proteasome pathway is an important general regulatory pathway that maintains the steady-state level of a variety of key enzymes, receptors, and other proteins (6–8). Proteins destined for proteasome-mediated hydrolysis are labeled by covalent attachment of Ub to produce a poly-Ub chain. Ub is transferred to target proteins by sequential transfer from the Ub-activating enzyme (E1) to a Ub-conjugating enzyme (E2) and then, in combination with an Ub ligase (E3), to the target protein. Multiple groups have demonstrated that hdm2 functions as an E3 Ub ligase for p53 and that the C-terminal RING finger domain is essential for this activity (9–11). hdm2 can also ubiquitinate itself, which leads to the degradation of hdm2 by the proteasome pathway (11, 12). This “autoubiquitination” of hdm2 could be an important mechanism to regulate the level of hdm2 in vivo.
Several lines of evidence suggest that the regulatory mechanisms of hdm2 can be exploited as potential targets for cancer therapies. Inhibition of these regulatory mechanisms could lead to activation of p53, resulting in cell-cycle arrest or apoptosis. Efforts to block the formation of the hdm2–p53 binary complex have been put forth as a potential mechanism for intervention in human cancers with functional p53. From these efforts, a number of small peptides have been found to bind to a specific pocket within an N-terminal domain of hdm2 with nanomolar affinity to hdm2 (13–16). Small molecular weight nonpeptidic molecules have also been identified that block hdm2–p53 complex formation (17, 18). Transfection of cells with an antisense oligonucleotide against hdm2 results in inhibition of hdm2 expression, diminution of hdm2 levels, and cell cycle arrest or apoptosis, depending on the identity of the transfected cell line (19, 20). Likewise, blockage of hdm2 function by its natural inhibitor, p19ARF, also leads to elevations of p53 level and transcriptional activity (21). The p19ARF protein binds to hdm2 and blocks hdm2 function in two ways. First, it inhibits hdm2 Ub ligase activity by a mechanism that has yet to be elucidated (22, 23). Second, p19ARF binding alters the nuclear localization of hdm2 (24–26). These results raise the possibility that small molecule inhibitors of hdm2 function might form an attractive pharmacological mechanism for inducing apoptotic cell death in human tumors containing wild-type p53.
We have focused our efforts on understanding the catalytic function of hdm2 as a Ub ligase for p53 (27), and on identifying small molecular weight inhibitors of this catalytic activity. By inhibiting the Ub ligase activity of hdm2, we hope to stabilize p53 in tumor cells and thus restore the ability of p53 to function as a tumor suppressor. A potential complication for this strategy is the autoubiquitination activity of hdm2. Hence, inhibitors of hdm2 Ub ligase activity may block both p53 and hdm2 ubiquitination, without a net stabilization of p53. Thus a critical issue for our proposed strategy of pharmacological intervention is whether the reaction mechanisms for hdm2-mediated ubiquitination of p53 and autoubiquitination are distinct, thus allowing selective inhibition of the former while sparing the latter activity.
In this paper, we report the discovery of small molecules that function as selective inhibitors of hdm2-mediated ubiquitination of p53. Three distinct types of molecules are reported that block ubiquitination of p53 with micromolar potency. These inhibitors display no ability to inhibit other Ub-dependent enzymes at concentrations as high as 100 μM. Most significantly, all three compound types are also unable to block hdm2 autoubiquitination. These compounds thus provide compelling evidence to suggest that the two catalytic functions of hdm2 can be differentiated by small molecule inhibitors.
Materials and Methods
Protein Expression and Purification.
The proteins E1 (28), Ubc4 (28), p53 (27), GST-hdm2 (27), Oregon green-labeled Ub, and Og-Ub-Ubc4 (27) were prepared as described. Soluble THP.1 extract and phosphorylated IκB/p50/p65 triple complex were prepared as described (29). Biotinylated Ub was prepared by using EZ-Link NHS-Biotin (Pierce) following the manufacturer's instruction. Bi-Ub-Ubc4 was prepared and purified following the same procedure as for Og-Ub-Ubc4 (27). A polyclonal antibody against IκB was purchased from Santa Cruz Biotechnology. Ub aldehyde was purchased from Boston Biochem (Boston).
GST-IκB and Nedd-4 were generous gifts from Thomas Parsons and Michael Boisclair of Mitotix (Boston).
In Vitro Ubiquitination of p53 Catalyzed by hdm2.
Reactions were carried out in 15 mM Hepes (pH 7.5)/5 mM NaCl/10 mM octyl glucoside in a 20-μl reaction volume. Final DMSO concentration was maintained at 2% (vol:vol). The potency of compounds as inhibitors of the hdm2-mediated ubiquitination of p53 was evaluated by preincubating compounds with hdm2 (final 1 nM) for 15 min. Premixed p53 and Og-Ub-Ubc4 (final concentration 1 μM each) were added to initiate the reaction. The reaction was quenched at 20 min (within the linear initial velocity phase of the reaction progress curve) with reducing SDS/PAGE sample buffer and resolved on a 4–20% tricine–glycine gel from NOVEX (San Diego). Fluorescence intensity of p53-(Og-Ub)n was quantified on a Storm 860 imager (Molecular Dynamics).
Steady-State Analysis of Compound Mechanism.
All reactions were stopped with reducing SDS/PAGE sample buffer 20 min after initiated with premixed p53 and Og-Ub-Ubc4 unless otherwise specified. The reversibility of inhibition by compounds was determined by preincubating compounds at 10 times the IC50 with 100 nM hdm2 for 15 min. The reaction was initiated by diluting the above enzyme–compound mixture 100-fold into reaction buffer containing 1 μM each of p53 and Og-Ub-Ubc4. The reaction was stopped with reducing SDS/PAGE sample buffer at various time points. Initial velocity was determined by fitting the product time course to a linear function.
Inhibitor modality was defined by incubating compounds at various concentrations with 3 nM hdm2 for 15 min. Premixed p53 and Og-Ub-Ubc4, varying one substrate concentration while keeping the other substrate constant, was then added to initiate the reaction. The data were fitted globally to models for competitive, noncompetitive (mixed type), and uncompetitive inhibition (30) by using grafit (Erithucas Software, Middlesex, U.K.). The choice of which model best described the data was made on the basis of criteria described by Cleland (31).
Mutual exclusivity studies were carried out by measuring the enzyme velocity while varying one compound concentration at several fixed concentrations of another compound. Data were fitted globally to the equation of Yonetani and Theorell (32) by using grafit:
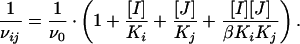 |
In this equation, v0 is the velocity when no inhibitor is present; Ki and Kj are the apparent dissociation constants (equal to IC50 values for the noncompetitive inhibitors) for inhibitors I and J, respectively; and β is a constant that defines the degree of interaction between the two inhibitors.
Evaluation of Effects of Compounds on hdm2 Autoubiquitination.
Compounds were premixed with 15 nM hdm2 for 15 min in 15 mM Hepes (pH 7.5)/5 mM NaCl/10 mM octyl glucoside. Og-Ub-Ubc4 (final concentration: 3 μM) was added to initiate the reaction. Reactions were quenched at 5 min (within the linear time course of the reaction) with reducing SDS/PAGE sample buffer. The amount of Og-Ub incorporated into hdm2 was quantified as described above.
In Vitro Ubiquitination of E1-Mediated Ub Conjugation to Ubc4.
The E1 reaction was carried out as described (28). Compounds at the indicated concentrations were added to the enzyme and substrate mixture without Mg-ATP (final DMSO: 1%, vol:vol) and allowed to incubate for 15 min. The reaction was initiated by adding MgCl2/ATP to a final concentration of 750 μM. The reaction was carried out at 25°C for 45 min and stopped by adding nonreducing SDS/PAGE sample buffer. Samples were separated on 4–20% tricine glycine SDS/PAGE gels from NOVEX. The fluorescence intensity of the Og-Ub-Ubc4 band was quantified on a Storm 860 fluorescence image reader from Molecular Dynamics.
In Vitro Ubiquitination of Phospho-IκB by SCFβ-TRCP.
The reaction was carried out in 50 mM Tris (pH 8.0) and DMSO at 1% (vol:vol) final concentration (27, 33). Compounds were incubated with THP.1 cell extract (final protein concentration, determined by the Bradford assay, was 2.4 mg/ml) for 15 min. A substrate mixture was added to initiate the reaction, resulting in the indicated final concentrations of substrates: 25 nM phosphorylated IκB-p50-p65 triple complex, 16 μM biotinylated Ub, 4 mM Mg2+-ATP. Ub aldehyde was added to a final concentration of 10 μg/ml to inhibit Ub hydrolase. The reaction was allowed to proceed at 37°C for 90 min and quenched with a 5-fold excess of TBST–casein [25 mM Tris (pH 7.2)/150 mM NaCl/0.1% Tween/1% casein]. This reaction mixture was captured for 60 min on Protein G plates (Pierce) coated with anti-IκB antibody. After three additional washes with TBST–casein, streptavidin–europium (Wallac) was added at a final concentration of 0.1 μg/ml and allowed to incubate for 40 min. After three washes with TBST-casein, enhancement solution (Wallac) was added 20 min before the time resolved fluorescence of Europium was read on a Victor-2 (Wallac, Gaithersburg, MD) fluorescence plate reader.
In Vitro Ubiquitination of GST-IκB by Nedd-4.
The reaction was carried out in 25 mM Tris (pH 8.0)/0.5 mM MgCl2/5 mM NaCl/10 mM octyl glucoside, final DMSO concentration, 2% (vol:vol). Compounds were premixed with Nedd-4 (40 nM final concentration) and GST-IκB (250 nM) for 15 min. Bi-Ub-Ubc4 was added to a final concentration of 2 μM. The reaction was incubated at room temperature for 60 min and stopped with premixed N-ethylmaleimide and NaCl solution (final concentration 20 and 300 mM, respectively). The reaction product was captured on Protein G plates (Pierce) coated with anti-IκB antibody. Streptavidin–europium binding and fluorescence measurements were carried out following the same procedure as described above.
Effect of Compounds on the Physical Interaction of hdm2 and p53.
Binding of hdm2 (1–126) to Oregon green-labeled p53 (1–35) peptide was carried out as described with minor modifications (16). The binding of full length GST-hdm2 and p53 was carried out following the protocol of Chen et al. (34). Mouse monoclonal anti-p53 antibody Ab-6 (Calbiochem) was used to assess the effects of compounds on hdm2-p53 interaction.
Results
Inhibitor Identification.
Three chemically distinct types of inhibitors were identified through screening (at Mitotix) of a chemical library for inhibitors of the hdm2-mediated p53 ubiquitination: the benzsulfonamides (Fig. 1 Top), the ureas (Fig. 1 Middle), and the imidazolones (Fig. 1 Bottom), exemplified by compounds 1, 2, and 3, respectively. Each of these compounds inhibited hdm2 in a concentration-dependent fashion (Fig. 2) with IC50s in the micromolar range, as summarized in Table 1.
Fig 1.

Chemical structures of compounds 1–3: (Top) benzsulfonamide; (Middle) urea; and (Bottom) imidazolone.
Fig 2.

Concentration-dependent inhibition of hdm2-catalyzed p53 ubiquitination by compounds 1–3. A is a representative fluorescent image of p53-(Og-Ub)n captured by a Storm 860 digital imager in the presence of compound 1 from 200–0.16 μM (as a 2.5-fold serial dilution). The rightmost lanes represent reactions run in the absence of inhibitor. The integrated intensities of the p53-(Og-Ub)n bands in each lane were quantified, plotted, and fit to an inhibition isotherm to produce the concentration-response plot illustrated in B. The symbols in B represent the data for compounds 1 (•), 2 (○), and 3 (□).
Table 1.
IC50 determination of compounds 1–3 in various ubiquitination assays
| Enzyme
|
IC50, μM | ||
|---|---|---|---|
| Compound 1 | Compound 2 | Compound 3 | |
| Hdm2 (p53 ubiquitination) | 12.7 ± 2.5 | 14.2 ± 4.8 | 3.2 ± 0.5 |
| E1 | >100 | >100 | >100 |
| Nedd-4 | >100 | >100 | >100 |
| SCFβ-TRCP | >100 | >100 | >100 |
| Hdm2 (autoubquitination) | >100 | >100 | >100 |
In all cases, the inhibitor displayed ≤20% inhibition at concentrations up to 100 μM.
Inhibitor Selectivity.
To avoid untoward side effects in vivo, an hdm2 ligase inhibitor would need to demonstrate selectivity for hdm2 inhibition over inhibition of other E3-ligases or other members of the Ub transfer cascade. We therefore tested compounds 1–3 in activity assays for representatives of E3 ligases distinct from hdm2: (i) a HECT domain E3 ligase, Nedd-4; and (ii) a modular E3 ligase containing a RING finger domain, SCFβ−TRCP. We also tested the compounds for inhibition of the common Ub activating enzyme E1. All of these enzymes were refractory to inhibition by compounds 1–3 at concentrations up to 100 μM (Table 1). Thus all three compounds demonstrated selectivity for hdm2 compared with other representative Ub ligases. Because a common Ub-E2 (i.e., Ub-Ubc4) was used in the assays of the various E3 ligases, inhibition at the level of the E2 component can be excluded for these compounds.
Specificity for hdm2 Ligase Activity.
In addition to ubiquitination of p53, hdm2 can also catalyze its own ubiquitination in a process referred to as autoubiquitination (10, 11). Whether autoubiquitination of hdm2 occurs through an intra- or intermolecular mechanism (i.e., cis or trans activity, respectively) remains ill defined. In either case, an inhibitor that blocks both hdm2 ligase and autoubiquitination activity would be less than ideal for pharmacological use, because the inhibition of autoubiquitination might stabilize hdm2 molecules along with p53 molecules, hence negating the positive effects of such compounds on p53 stabilization. We therefore tested the ability of compounds 1–3 to inhibit the autoubiquitination activity of hdm2. Remarkably, these compounds showed little effect on hdm2 autoubiquitination at concentrations as high as 100 μM, a concentration at which the E3 ligase activity of hdm2 for p53 is almost completely abrogated (Table 1). Thus, the three representative compounds described here specifically inhibit the E3 ligase activity of hdm2 without effect on hdm2 autoubiquitination or on the activities of other ubiquitination cascade enzymes. As one might expect, this selectivity is not a common feature of all of the inhibitors that we have discovered. Indeed, we have also found a number of compounds that inhibit the E3 ligase activity of hdm2 toward both itself and p53 (data not shown). Therefore, the three compound types described here are unique in their ability to differentiate between the two catalytic functions of hdm2.
Mode of Inhibition.
Titration of hdm2 with compounds 1–3 led to a concentration-dependent diminution of the initial velocity of the enzymatic reaction. The reaction progress curves were linear at all concentrations of these inhibitors, suggesting that inhibitor binding was rapid. For all three compounds, essentially complete recovery of enzymatic activity was realized after dilution of the enzyme–inhibitor complexes (data not shown). Hence, all three compounds behaved as simple rapidly reversible inhibitors of hdm2.
The inhibitor modality for compounds 1–3 was determined through steady-state analysis of the effect of each compound on the apparent values of kcat and Km for p53 ubiquitination. Fig. 3 illustrates the results of such experiments for compound 3, the representative imidazolone. Double reciprocal plots of the reaction velocity as a function of Ub-Ubc4 concentration (Fig. 3A) or of p53 concentration (Fig. 3B) at varying inhibitor concentration both yield patterns of intersecting lines, eliminating uncompetitive inhibition with respect to either substrate as a reasonable model. For both varied substrates, global analysis of the entire data set indicated that the data were best described by the velocity equation for noncompetitive inhibition. The value of Ki for the free enzyme (3.5 ± 0.8 μM) was equal to the values of αKi for the binary enzyme/Ub-Ubc4 complex (3.8 ± 0.6 μM) and for the binary enzyme/p53 complex (3.9 ± 0.4 μM) obtained from this fitting for compound 3. Similar data were obtained for compounds 1 and 2, and these inhibitors were also found to be noncompetitive with respect to both Ub-Ubc4 and p53 (data not shown). For all three compounds, the inhibitor dissociation constants for the free enzyme and for the enzyme–substrate complexes were of similar magnitude (in all cases within 3-fold of each other), indicating that all three inhibitors bind to hdm2 at a site or sites that are distinct from the binding sites for p53 and for Ub-Ubc4.
Fig 3.

Double reciprocal plots of hdm2 E3 ligase activity for p53 in the presence of compound 3 at 0 (○), 1 (•), 3 (□), and 10 (▪) μM, (A) velocity as a function of Og-Ub-Ubc4 concentration at 1 μM p53 and (B) velocity as a function of p53 concentration at 1 μM Og-Ub-Ubc4. The fitted lines represent global fitting of the untransformed data to a noncompetitive model.
Effect of Compounds on the Physical Interaction Between hdm2 and p53.
The lack of effect on p53 Km suggests that it is unlikely that the inhibition is due to disruption of the physical interactions between hdm2 and p53. To confirm this observation, we examined the effects of two of these compounds (2 and 3) on hdm2–p53 interactions through two types of equilibrium-binding experiments. In the first set of experiments, compounds were tested for their ability to block formation of a binary complex between the N-terminal domain of hdm2 (residues 1–126) and a fluorescently labeled peptide representing the interaction sequence of p53 (16). In the second set of experiments, the ability of the compounds to inhibit complex formation between GST-hdm2 and full length p53 was evaluated by glutathione-bead pull down and Western blot analysis. In both types of experiments, no effects on complex formation were observed at concentrations of compound sufficient to achieve ≥90% inhibition of p53 ubiquitination (data not shown). Thus, the lack of effect on complex formation in these equilibrium-binding studies is consistent with a noncompetitive inhibition modality with respect to p53 binding.
Compounds 1–3 Bind in a Mutually Exclusive Fashion to hdm2.
The data presented above indicate that all three compound classes demonstrate similar selectivity for inhibition of hdm2 ligase activity and conform to a common mode of inhibition (noncompetitive). These similarities raise the question of whether the three classes of compounds share a common binding site on hdm2. To address this issue, we tested whether combinations of compound 1 or 2 with compound 3 would show additive or synergistic inhibition effects on hdm2. As first described by Yonetani and Theorell (32), the reaction velocity in the presence of a combination of two inhibitors I and J is described by Eq. 1. The term β in this equation is a constant that defines the degree of interaction between the two inhibitors. If the two inhibitors bind to the enzyme in a mutually exclusive fashion, the value of β is infinite; hence the last term in Eq. 1 is eliminated. Therefore, a plot of 1/vij as a function of the concentration of one of the inhibitors will yield a series of parallel lines for different concentrations of the second inhibitor. If the two inhibitors bind in a nonexclusive fashion, the lines in such a plot will intersect, the point of intersection depending on the value of β. Fig. 4 displays the results of such analysis for combinations of compounds 1 and 3 (Fig. 4A), and compounds 2 and 3 (Fig. 4B). In both cases, the plots display parallel lines, indicating that all three compounds bind to hdm2 in a mutually exclusive fashion with respect to one another. The simplest explanation for this observation is that the three compounds share a common binding pocket on the enzyme. The current data do not, however, exclude the alternative possibility that the compounds bind to separate sites on the enzyme that are somehow in allosteric anticooperative communication with one another.
Fig 4.

Exclusivity studies of compounds 1–3. The fitted lines represent global fitting of the untransformed data to Eq. 1. (A) 1/v is plotted as a function of compound 1 concentration at 0 (○), 1 (•), and 4 (□) μM of compound 3. (B) 1/v is plotted as a function of compound 2 concentration at 0 (○), 1 (•), and 4 (□) μM of compound 3.
Discussion
The strong association between hdm2 and cancer has been recognized for some time. It was determined early on that hdm2 binds p53 and inhibits the transcriptional activity of the latter protein (5, 34, 35). More recently, it has been recognized that hdm2 not merely serves as a p53-binding protein but also performs the catalytic function of an E3 ligase, facilitating the covalent attachment of Ub molecules to p53 (36, 37).
The hdm2-mediated ubiquitination of p53 has several potential physiological consequences. First, ubiquitination may target p53 for proteasome-mediated degradation, thus leading to a destabilization of p53 levels in cell expressing elevated levels of hdm2 (9–11). Second, recent cellular data suggest that ubiquitination may influence intracellular trafficking of hdm2 between nuclear and cytosolic compartments, thus further attenuating the transcriptional activity of p53 (38, 39). The multiple monoubiquitination of p53 may be consistent with a more complex role of the E3 ligase activity of hdm2 than simply tagging p53 for degradation (27). With the recognition of the catalytic function of hdm2, we and others have sought to identify small molecule inhibitors of this enzymatic activity as an alternative means of abrogating the negative regulation of p53 by hdm2 in cancer cells. Toward this ultimate goal, we have identified, and report here, examples of such small molecule inhibitors of hdm2 catalysis.
As described above, our screening efforts have identified three structurally distinct types of hdm2 E3 ligase inhibitors. Despite their structural diversity, these compounds share some striking mechanistic similarities. All three compounds inhibit hdm2 selectively, showing little effect on the other Ub-using enzymes: E1, Nedd-4, and SCFβ−TRCP. All three compound types conform to a noncompetitive mode of inhibition with respect to both substrates of the reaction, Ub-Ubc4 and p53. Mutual exclusivity studies suggest that the three compounds likely share a common binding site on the hdm2 molecule. Most surprisingly, all three compounds selectively inhibit the E3 ligase activity of hdm2 for p53 without concomitant effect on the alternative autoubiquitination activity of this enzyme.
The noncompetitive nature of the inhibition by these molecules suggests that they bind to the enzyme at a site distinct from the site or sites of substrate binding. Thus the molecules block catalysis by a mechanism other than competition with substrate for active site binding. This inference is confirmed by the lack of effects of these compounds on the physical interaction of hdm2 and p53 from the fluorescence polarization and the GST pulldown experiment.
What mechanistic rationale can be offered for these observations? In a recent study of hdm2 E3 ligase activity, we have presented evidence to suggest that the catalytic mechanism of hdm2 E3 ligase activity involves a requisite conformational transition (i.e., enzyme isomerization) of hdm2 subsequent to Ub-Ubc4 and/or p53 binding (27). We noted in that report that the conformational transition was required for ligase activity toward p53, but that it is not clear whether this conformational transition was also required for autoubiquitination. If the enzyme isomerization step is uniquely required for E3 ligase activity for p53, a possible inhibition mechanism for the current compounds would be to block this conformational change. This speculative suggestion is consistent with all of the data obtained to date concerning hdm2 E3 ligase activity. Further studies would be required to clarify this point. It is clear, however, that the selectivity demonstrated by compounds 1–3 for inhibition of E3 ligase activity for p53 over autoubiquitination reflects fundamental mechanistic differences between the reaction pathways for these two catalytic activities of hdm2.
In summary, the data presented here indicate that it is possible to selectively inhibit hdm2 E3 ligase activity with small molecule inhibitors while sparing the autoubiquitination activity of this enzyme. Pharmacological optimization of compounds with this type of selectivity profile may provide a basis for new chemotherapeutic agents for the treatment of human cancers.
Acknowledgments
We thank Dr. Katherine V. Ferry and Sandra L. Kumpf for help with the development of the autoubiquitination assay and Drs. Thomas Parson and Michael Boisclair for help with the development of the initial screening assay for hdm2. We are indebted to Drs. Andrew Stern and Ross Stein for helpful discussions.
Abbreviations
Ub, ubiquitin
Ubc, Ub-conjugating enzyme
E1, Ub-activating enzyme
Og-Ub, Oregon green-labeled Ub
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Levine A. J., Chang, A., Dittmer, D., Notterman, D. A., Silver, A. & Thorn, K. (1994) J. Lab. Clin. Med. 123, 817-823. [PubMed] [Google Scholar]
- 2.Prives C. & Hall, P. A. (1999) J. Pathol. 187, 112-126. [DOI] [PubMed] [Google Scholar]
- 3.Oren M. (1999) J. Biol. Chem. 274, 36031-36034. [DOI] [PubMed] [Google Scholar]
- 4.Perry M. E. & Levine, A. J. (1994) Mt. Sinai J. Med. 61, 291-299. [PubMed] [Google Scholar]
- 5.Freedman D. A., Wu, L. & Levine, A. J. (1999) Cell. Mol. Life Sci. 55, 96-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scheffner M., Nuber, U. & Huibregtse, J. M. (1995) Nature 373, 81-83. [DOI] [PubMed] [Google Scholar]
- 7.Ciechanover A. & Schwatz, A. L. (1998) Proc. Natl. Acad. Sci. USA 95, 2727-2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hershko A. & Ciechanover, A. (1998) Annu. Rev. Biochem. 67, 425-479. [DOI] [PubMed] [Google Scholar]
- 9.Honda R., Tanaka, H. & Yasuda, H. (1997) FEBS Lett. 420, 25-27. [DOI] [PubMed] [Google Scholar]
- 10.Honda R. & Yasuda, H. (2000) Oncogene 19, 1473-1476. [DOI] [PubMed] [Google Scholar]
- 11.Fang S., Jensen, J. P., Ludwig, R. L., Vousden, K. H. & Weissman, A. M. (2000) J. Biol. Chem. 275, 8945-8951. [DOI] [PubMed] [Google Scholar]
- 12.Chang Y. C., Lee, Y. S., Tejima, T., Tanaka, K., Omura, S., Heintz, N. H., Mitsui, Y. & Magae, J. (1998) Cell Growth Differ. 9, 79-84. [PubMed] [Google Scholar]
- 13.Bottger V., Bottger, A., Howard, S. F., Picksley, S. M., Chene, P., Garcia-Echeverria, C., Hochkeppel, H. K. & Lane, D. P. (1996) Oncogene 13, 2141-2147. [PubMed] [Google Scholar]
- 14.Wasylyk C., Salvi, R., Argentini, M., Dureuil, C., Delumeau, I., Abecassis, J., Debussche, L. & Wasylyk, B. (1999) Oncogene 18, 1921-1934. [DOI] [PubMed] [Google Scholar]
- 15.Chène P., Fuchs, J., Bohn, J., García-Echeverría, C., Furet, P. & Fabbro, D. (2000) J. Mol. Biol. 299, 245-253. [DOI] [PubMed] [Google Scholar]
- 16.Lai Z., Auger, K. R., Manubay, C. & Copeland, R. A. (2000) Arch. Biochem. Biophys. 381, 278-284. [DOI] [PubMed] [Google Scholar]
- 17.Luke, R. W. A., Jewsbury, P. J. & Cotton, R. (2000) International Patent Application WO-00015657.
- 18.Stoll R., Renner, C., Hansen, S., Palme, S., Klein, C., Belling, A., Zeslawski, W., Kamionka, M., Rehm, T., Muhlhahn, P., et al. (2001) Biochemistry 40, 336-344. [DOI] [PubMed] [Google Scholar]
- 19.Chen L., Agrawal, S., Zhou, W., Zhang, R. & Chen, J. (1998) Proc. Natl. Acad. Sci. USA 95, 195-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang H., Zeng, X., Oliver, P., Le, L. P., Chen, J., Chen, L., Zhou, W., Agrawal, S. & Zhang, R. (1999) Int. J. Oncol. 15, 653-660. [PubMed] [Google Scholar]
- 21.Zhang Y. & Xiong, Y. (2001) Cell Growth Differ. 12, 175-186. [PubMed] [Google Scholar]
- 22.Midgley C. A., Desterro, J. M. P., Saville, M. K., Howard, S., Sparks, A., Hay, R. T. & Lane, D. P. (2000) Oncogene 19, 2312-2323. [DOI] [PubMed] [Google Scholar]
- 23.Honda R. & Yasuda, H. (1999) EMBO J. 18, 22-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weber J. D., Taylor, L. J., Roussel, M. F., Sherr, C. J. & Bar-Sagi, D. (1999) Nat. Cell. Biol. 1, 20-26. [DOI] [PubMed] [Google Scholar]
- 25.Tao W. & Levine, A. J. (1999) Proc. Natl. Acad. Sci. USA 96, 3077-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roth J., Dobbelstein, M., Freedman, D. A., Shenk, T. & Levine, A. J. (1998) EMBO J. 17, 554-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lai Z., Ferry, K. V., Diamond, M. A., Wee, K. E., Kim, Y. B., Ma, J., Benfield, P. A., Copeland, R. A. & Auger, K. R. (2001) J. Biol. Chem. 276, 31357-31367. [DOI] [PubMed] [Google Scholar]
- 28.Wee K. E., Lai, Z., Auger, K. R., Ma, J., Horiuchi, K. Y., Dowling, R. L., Dougherty, C. S., Corman, J. I., Wynn, R. & Copeland, R. A. (2000) J. Protein Chem. 19, 489-498. [DOI] [PubMed] [Google Scholar]
- 29.Winston J. T., Strack, P., Beer-Romero, P., Chu, C. Y., Elledge, S. J. & Harper, J. W. (1999) Genes Dev. 13, 270-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Copeland R. A., (2000) Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis (Wiley–VCH, New York).
- 31.Cleland W. W. (1967) Adv. Enzymol. 29, 1-65. [DOI] [PubMed] [Google Scholar]
- 32.Yonetani T. & Theorell, H. (1964) Arch. Biochem. Biophys. 106, 243-255. [DOI] [PubMed] [Google Scholar]
- 33.Strack P., Caligiuri, M., Pelletier, M., Boisclair, M., Theodoras, A., Beer-Romero, P., Glass, S., Parsons, T., Copeland, R. A., Auger, K. R., et al. (2000) Oncogene 19, 3529-3536. [DOI] [PubMed] [Google Scholar]
- 34.Chen J., Marechal, V. & Levine, A. J. (1993) Mol. Cell. Biol. 13, 4107-4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J., Lin, J. & Levine, A. J. (1995) Mol. Med. 1, 142-152. [PMC free article] [PubMed] [Google Scholar]
- 36.Haupt Y., Maya, R., Kazaz, A. & Oren, M. (1997) Nature 387, 296-299. [DOI] [PubMed] [Google Scholar]
- 37.Fuchs S. Y., Adler, V., Buschman, T., Wu, X. & Ronai, Z. (1998) Oncogene 17, 2543-2547. [DOI] [PubMed] [Google Scholar]
- 38.Boyd S. D., Tsai, K. Y. & Jacks, T. (2000) Nat. Cell. Biol. 2, 563-568. [DOI] [PubMed] [Google Scholar]
- 39.Geyer R. K., Yu, Z. K. & Maki, C. G. (2000) Nat. Cell. Biol. 2, 569-573. [DOI] [PubMed] [Google Scholar]