

Abstract
Background
Co-regulation of genes may imply involvement in similar biological processes or related function. Many clusters of co-regulated genes have been identified using microarray experiments. In this study, we examined co-regulated gene families using large-scale cDNA microarray experiments on the human transcriptome.
Results
We present a simple model, which, for each probe pair, distills expression changes into binary digits and summarizes the expression of multiple members of a gene family as the Family Regulation Ratio. The set of Family Regulation Ratios for each protein family across multiple experiments is called a Family Regulation Profile. We analyzed these Family Regulation Profiles using Pearson Correlation Coefficients and derived a network diagram portraying relationships between the Family Regulation Profiles of gene families that are well represented on the microarrays. Our strategy was cross-validated with two randomly chosen data subsets and was proven to be a reliable approach.
Conclusion
This work will help us to understand and identify the functional relationships between gene families and the regulatory pathways in which each family is involved. Concepts presented here may be useful for objective clustering of protein functions and deriving a comprehensive protein interaction map. Functional genomic approaches such as this may also be applicable to the elucidation of complex genetic regulatory networks.
Background
Recent progress in genomic sequencing has led to the rapid enrichment of protein sequence databases. Computational biology strives to extract the maximum possible information from these sequences by classifying them according to their homologous relationships. Classical protein families are distinguished by members which exhibit sequence similarity, a feature which can often be used to infer that the sequences are related evolutionarily as well as functionally. One of the first goals of any genome-sequencing project is to broadly classify as many genes and their products as possible into putative functional families.
Proteins are translated from their corresponding mRNAs. Cellular mRNA levels are immensely informative about cell state and the activity of genes, and in most cases, changes in mRNA abundance are positively correlated with changes in protein abundance. Co-regulation of proteins often reflects that these proteins are involved in similar biological processes and have related functions [1]. Therefore changes in the expression patterns of protein families under different experimental conditions can provide clues about regulatory mechanisms, the relationships between broader cellular functions and biochemical pathways, as well as interactions between different protein motifs [2-7].
The advent of microarray technology has made simultaneous analysis of the gene expression profiles of tens of thousands of genes a practical reality [8]. Availability of human genome sequences and massive high throughput data on their expression provides an opportunity to study regulatory relationships between gene families [9,10]. Incyte's LifeExpress RNA Database is a gene expression database that contains raw and normalized data from hundreds of Incyte microarray experiments. Using these large-scale mRNA expression data, we can infer the functional relationships between protein families by comparing the aggregated expression profiles of the members of each protein family.
Pfam is a functionally annotated database of protein domain families [11]. In this study, each member of Pfam families was mapped to Incyte clones which were presented in the Incyte microarray chips based on the sequence identity. We then generated mRNA expression profiles for these Pfam family members using Incyte's LifeExpress RNA (LE) database (Version 3.0, April 2001 release, Incyte Genomics, Inc.). We analyzed 135 Pfam families, whose family members are well represented on the Incyte microarrays. The expression data for various members of a single Pfam family in one experiment was first converted into binary digits and then summarized as the Family Regulation Ratio. The set of Family Regulation Ratios for a particular Pfam family across multiple experiments is called a Family Regulation Profile. By using Pearson Correlation, we analyzed the similarities between these Family Regulation Profiles to impute functional relationships between the different families. This method was validated by comparing Family Regulation Profiles generated based on two different randomly selected LE data subsets.
This study explores an approach to relate protein families based on quantitative comparison of the family mRNA expression profiles. Analysis of the profiles may be useful to address what protein domain families are co-regulated and possibly how they may interact physically or genetically with one another.
Results
Clones mapped to multiple Pfam families
Structural similarities between distinct proteins often involve only a portion of each protein sequence. These conserved sub-structures, which often have readily identifiable boundaries, may recur in a large number of different proteins in which they perform a similar function. In addition, many proteins consist of combinations of these recurrent substructures (domains) in which case the protein function can be economically annotated based on the presence of these domains. Pfam is a domain-based protein database. Multi-domain proteins can be classified into several Pfam families, which in turns means that several Pfam families can share the same corresponding proteins.
Among 135 Pfam families discussed here, there are 1647 clones mapped to more than one Pfam family, and 346 Pfam families sharing more than one clone. Multi-domain proteins are the major reason for this observation, i.e. PF00043 and PF02798 representing C-domain and N-domain of Glutathione S-transferease respectively. Non-uniformity of Pfam classification and clone mapping may also cause clone sharing between Pfam families. A table listing pairs of Pfam families that share more than 50% of their family members is provided as an additional file (see Additional File 1: [supp1.doc]). These Pfam family pairings may suggest that the two domains are associated with a distinct functional class.
Correlations of Pfam Family Expression Profiles for Related Pfams
Our analysis includes 135 Pfam families, whose names are provided in an additional file (see Additional File 2: [supp2.doc]). Pearson Correlation Coefficients (PCC) were calculated between each pair of Pfam Family Regulation Profiles. Common clones shared by Pfam pairs were removed during PCC calculation to reduce this source of bias (See Methods). 9045 PCCs were computed based on the Family Regulation Profiles consisting of more than ten clone members. Of these, 781 PCCs were greater than 0.6 and 71 PCCs greater than 0.75. Table 1 shows the top hits (PCC — 0.75) with the highest PCC of 0.89 between PF00046 (homeobox domain) and PF00520 (ion transport protein). More detailed and completed Pfam-pairs (PCC — 0.6) are listed in the Additional File 3: [supp3.doc]. The line graphs of Family Regulation Profiles of PF00046 and PF00520 are shown as Fig 1.
Table 1.
List of the Pfam-pairs with the Pearson Correlation Coefficient greater than 0.75. More detailed and completed results are included in the Additional File 3: [supp3.doc].
| PfamID1 | PfamID2 | PCC | PfamID1 | PfamID2 | PCC | PfamID1 | PfamID2 | PCC |
| PF00046 | PF00520 | 0.89 | PF00008 | PF00560 | 0.78 | PF00096 | PF00168 | 0.76 |
| PF00001 | PF00046 | 0.84 | PF00046 | PF00105 | 0.78 | PF00096 | PF00169 | 0.76 |
| PF00008 | PF00041 | 0.83 | PF00046 | PF00065 | 0.78 | PF00028 | PF00069 | 0.76 |
| PF00069 | PF00096 | 0.83 | PF00028 | PF00046 | 0.78 | PF00036 | PF00041 | 0.76 |
| PF00008 | PF00069 | 0.82 | PF00046 | PF00104 | 0.78 | PF00041 | PF00560 | 0.76 |
| PF00001 | PF00069 | 0.81 | PF00096 | PF00105 | 0.78 | PF00008 | PF00168 | 0.76 |
| PF00001 | PF00520 | 0.81 | PF00105 | PF00520 | 0.78 | PF00028 | PF00105 | 0.76 |
| PF00046 | PF01094 | 0.81 | PF00046 | PF00096 | 0.78 | PF00028 | PF00560 | 0.76 |
| PF00069 | PF00169 | 0.81 | PF00147 | PF00520 | 0.78 | PF00001 | PF00104 | 0.76 |
| PF00069 | PF00560 | 0.81 | PF00104 | PF00520 | 0.78 | PF00036 | PF00560 | 0.76 |
| PF00041 | PF00096 | 0.8 | PF00036 | PF00096 | 0.78 | PF00046 | PF00168 | 0.76 |
| PF00069 | PF00105 | 0.8 | PF00001 | PF00096 | 0.77 | PF00008 | PF01391 | 0.75 |
| PF00036 | PF00069 | 0.8 | PF00102 | PF00520 | 0.77 | PF00041 | PF00069 | 0.75 |
| PF00069 | PF00104 | 0.8 | PF00520 | PF00595 | 0.77 | PF00001 | PF00047 | 0.75 |
| PF00096 | PF00595 | 0.8 | PF00520 | PF01094 | 0.77 | PF00069 | PF00168 | 0.75 |
| PF00046 | PF00595 | 0.8 | PF00001 | PF00168 | 0.77 | PF01391 | PF01462 | 0.75 |
| PF00096 | PF00520 | 0.8 | PF00096 | PF00104 | 0.77 | PF00069 | PF00097 | 0.75 |
| PF00008 | PF00047 | 0.79 | PF00076 | PF00096 | 0.77 | PF00001 | PF00169 | 0.75 |
| PF00008 | PF00096 | 0.79 | PF00008 | PF00036 | 0.77 | PF00046 | PF00102 | 0.75 |
| PF00069 | PF00076 | 0.79 | PF00001 | PF00105 | 0.77 | PF00001 | PF00008 | 0.75 |
| PF00001 | PF00065 | 0.79 | PF00028 | PF00041 | 0.76 | PF00028 | PF00520 | 0.75 |
| PF00065 | PF00520 | 0.79 | PF00003 | PF00595 | 0.76 | PF00028 | PF00096 | 0.75 |
| PF00595 | PF01094 | 0.79 | PF00003 | PF00520 | 0.76 | PF00046 | PF00067 | 0.75 |
| PF00008 | PF00169 | 0.79 | PF00001 | PF00102 | 0.76 | PF00041 | PF00105 | 0.75 |
| PF00169 | PF00560 | 0.79 | PF00028 | PF00104 | 0.76 | PF00096 | PF00560 | 0.75 |
| PF00003 | PF00046 | 0.79 | PF00046 | PF00069 | 0.76 | PF00018 | PF00069 | 0.75 |
Figure 1.

Family Regulation Profiles for PF00046 and PF00520. Because of size limitation, only part of the line graph is shown here. The comparison between two profiles shows the high correlation. The Pearson Correlation Coefficients between PF00046 and PF00520 is 0.89.
The Pfam co-regulation network can be visualized using the Pajek software package, a program originally designed for social network analysis (12). The map is layed out with Kamada-Kawi methods where each node represents a Pfam family and each edge represents a correlation coefficient greater than 0.6 (Fig. 2A) and 0.75 (Fig. 2B) between Family Regulation Profiles of the connecting nodes. A subset of interconnected Pfams in which each Pfam has at least k interactions (where k is an integer) forms a k-core. These cores represent Pfams associated with one another by multiple interactions. The 20-core subgraph where each Pfam had at least 20 connections, was derived from Fig. 2A and displayed as Fig 2C. This graph may represent the core network of cellular regulation.
Figure 2.

(A) Pfam regulation network predicted by the correlation of Family Regulation Profiles. In total, 89 Pfams and 781 correlations with PCC — 0.6 are shown (a network with each Pfam ID labeled is showed as an additional file (see Additional File 4: [supp4.png]). (B) 27 Pfams and 156 correlations with PCC — 0.75. (C)The derived 20-core subgraph from (A). The 20-core subset, which might represent the core networks of cell regulation, contains 27 Pfams.
Data cross-validation
Two independent sets of Family Regulation Profiles were constructed from the randomly selected, non-overlapping subsets of expression data S1 and S2 (see Methods). Separate Pearson Correlation Coefficients, PCC1 and PCC2, were computed from these data subsets respectively. The correlation between PCC1 and PCC2 was determined by calculating an Enrichment Factor (EF) that expresses the degree to which calculations based on one data subset were corroborated by calculations based on the other data subset.
First, we selected Pfam pairs from S1 by screening for pairs where PCC1 is greater than 0.5. If PCC1 and PCC2 for a particular pair of Pfam models are positively correlated, an increase in PCC2 cutoff should result in a greater likelihood of PCC1 falling above 0.5, i.e. EF should increase. We calculated a series of EFs with the PCC2 cutoff increasing from 0 to 1. EF stayed around 1 for low PCC2 (0 to 0.2), and gradually increased to as high as 4.2 when PCC2 — 0.8 (Fig. 3).
Figure 3.

Enrichment factor analysis validates the correlation between PCC1 and PCC2. PCC1 — 0.5 is used as G1 cutoff. The EF is plotted for different PCC2 cutoffs. Please see Methods for the detail of EF calculation.
EFs were also computed to compare PCC1 and PCC2 in the same cutoff range from 0 to 1. An EF greater than 1 indicates higher than background correlation between PCC1 and PCC2. The representation ratio predicted to occur if there is no correlation between PCC1 and PCC2, and the observed representation ratios were calculated as described in Methods. The discrepancy between them was evaluated by determining the statistical variable chi-square (χ2) and the corresponding p value. The higher the chi-square and the lower the p value, the less likely the observed degree of correlation occurred by chance, which implies that the observed relationship is biologically significant. As shown in Table 2, both EF and χ2 analyses showed strong correlation between PCC1 and PCC2 with the p value less than 0.00001.
Table 2.
Correlation between PCC1 and PCC2 in the cutoff range from 0.1 to 1. Enrichment Factors and Chi-square values are shown. (Note: p value is derived from Chi-square.)
| Range |
Observed Count (PCC2 v Range | PCC1 v Range) |
Expected Count (PCC2 v Range | PCC1 v Range) |
EF | χ2 | P value |
| Cutoff < 0.1 | 76 | 11.194 | 6.8 | 383.4 | <0.00001 |
| 0.1 < cutoff < 0.2 | 280 | 103.069 | 2.7 | 337.4 | <0.00001 |
| 0.2 < cutoff < 0.3 | 570 | 317.277 | 1.8 | 247.8 | <0.00001 |
| 0.3 < cutoff < 0.4 | 681 | 461.305 | 1.5 | 135.0 | <0.00001 |
| 0.4 < cutoff < 0.5 | 613 | 363.712 | 1.7 | 211.4 | <0.00001 |
| 0.5 < cutoff < 0.6 | 428 | 178.513 | 2.4 | 402.1 | <0.00001 |
| 0.6 < cutoff < 0.7 | 259 | 48.591 | 5.3 | 983.0 | <0.00001 |
| 0.7 < cutoff < 0.8 | 98 | 4.810 | 20.4 | 1834.7 | <0.00001 |
| cutoff — 0.8 | 10 | 0.067 | 149.5 | 1334.5 | <0.00001 |
The Pearson correlation coefficient between all PCC1 and PCC2 values was determined to be 0.76, showing that PCC1 and PCC2 were well correlated. Our approach is validated by this consistency of Pfam correlations derived from randomly selected LE expression experiments S1 and S2.
Discussion
Expression monitoring on a genome-wide scale was first successfully demonstrated in budding yeast [1,13,14]. Whole-genome expression profiles provide a rich source of information on protein function, protein-protein interactions, and gene pathways, and have been compared with data sets describing transcription factor binding sites, protein families, protein-protein interactions, and protein abundance [15-22] mainly in budding yeast. In this study we used large scale human genome-wide expression data to study the relationship between the expression patterns of different protein families. We summarized the change in gene expression level for a set of protein family members as a binary code of "regulated" and "unregulated". A two-fold change in expression level was chosen as a cutoff for categorizing a change in expression level as significant. This cutoff is arbitrary, but from our experience with Taqman confirmation (data not shown), a two-fold change in expression level reliably distinguishes real signals from background noise for most expression experiments. The gene regulation was then converted to a percentage for gene families to profile the regulation for each microarray experiment.
Since the mapping from Pfam families to Incyte microarray clones is through the Swissprot database, Pfam family members that are not represented in the Swissprot database cannot be mapped to Incyte clones, and are therefore not included in the Family Regulation Profile analysis. That may be one source of false negatives. Other factors such as sensitivity of the Incyte microarray technology, the arbitrary twofold cutoff, etc. may also contribute to false negative results. In order to reduce these types of errors, we included in our analysis only those Pfam families that map to at least 10 Incyte clones. Due to the fact that the domain hierarchy of Pfam classification is not perfectly reflected in the database, multi-domain proteins may fall into several Pfam members leading to a significant number of false positives. To address this potential source of error, we excluded from pair-wise correlation analysis those Incyte clones shared by both Pfam families in the pair.
To validate relationships that are revealed by microarray expression profiling, one direct way is to split all the experiments randomly into two non-overlapping pools and derive the Pfam relationships independently from each of these pools. The 555 LE experiment were divided into two data sets of S1 and S2 and the corresponding Pfam relationships were denoted as PCC1 and PCC2. The correlation coefficient for PCC1 and PCC2 of 0.76 indicates a high degree of correlation between two data sets. In order to ensure the independence of the data, we also performed another analysis where data were split into two sets based on the tissue type. The Pfam relationships (not shown) derived from these latter data sets are consistent with each other and with those derived from the complete data pool, further corroborating the observed relationships.
It is important to note that many of the protein family relationships (based on co-expression) identified here are supported by functional links. Taking the G protein-coupled receptors (GPCRs) as an example, after agonist action, GPCRs activate G proteins and become phosphorylated by G protein-coupled receptor kinases [23]. This event promotes activation of effector enzymes and ion channels by the activated Gα GTP. This GPCR-mediated regulation of ion channels also depends on the coordination and parallel regulation of protein tyrosine kinase and protein tyrosine phosphatase activities [24]. GPCRs can also interact with the growing family of PDZ domain-containing proteins [25]. All of these relationships are reflected in our study by the strong correlation between 7 transmembrane receptors (rhodopsin family) (PF00001), protein kinases (PF00069), protein tyrosine phosphatases (PF00102), ion transport proteins (PF00520), and PDZ domain proteins (PF00595).
Our study revealed many intriguing links between protein families. For example, the link between homeobox proteins (PF00046) and other protein families. Homeobox proteins are a very important family of transcription factors. According to our analysis, homeobox proteins are involved in the regulation of or are regulated by many protein families such as GPCRs, ion channels, tyrosine phosphatases, nuclear hormone receptors, and protein kinases. For example the cone rod homeobox (Crx) binds and transactivates the rhodopsin promoter [26]. However, it has not been determined whether other rhodopsin GPCRs are also regulated by homeobox proteins. In C. elegans, it was reported that the homeobox gene, lim-6, is required for distinct chemosensory representations (ion sensing), which is related to ion channel/transportation mechanisms. Again, it has not been studied systematically whether homeobox proteins are important regulators of ion channel/transporter. Another example of an intriguing relationship revealed in the present study is between the PH domain, the EGF domain and the immunoglobulin superfamily. Pleckstrin-homology (PH) domains are protein modules of approximately 120 amino acids found in a wide variety of signaling proteins in organisms ranging from yeasts to humans. The EGF domain, which often occurs as multiple tandem repeats, is widely distributed among extracellular proteins involved in adhesion, receptor-ligand interactions, extracellular matrix structure, determination of cell fate, and blood coagulation [27]. Cell adhesion usually activates PI 3-kinase activity. Many PH domain proteins bind with high affinity to specific phosphoinositides such as PI-4,5-P2, PI-3,4-P2 or PI-3,4,5-P3 which are generated by PI-3 kinase [28]. This relationship was reflected by the high correlation coefficient (0.79) between the PH domain and the EGF domain seen in our analysis. Furthermore, another major cell adhesion molecular family is the immunoglobulin superfamily (PF00047). Two immunoglobulins which are particularly important in the cell adhesion cascade are Intercellular Adhesion Molecule-1 (ICAM-1) or CD54 and Vascular Cell Adhesion Molecule-1 (VCAM-1). The co-expression that we observed in our analysis for the EGF-like domains and immunoglobin superfamily suggested that they may cooperate with each other in the cell adhesion process. Links between Pfam families like these suggest novel hypotheses about family interactions that may be further explored.
Conclusions
Integrating genome-wide expression information with protein functional classifications and genetic networks is a huge challenge. Our model is based on the binary idealization of gene expression change to generate a Family Regulation Profile. Although this model is simplified, the abstraction may be useful for conceptualizing the nature of functional relationships between families of protein domains.
Methods
Database resources
Pfam is a large collection of multiple protein sequence alignments and Profile Hidden Markov Models covering many protein domains. Pfam Version 6.3 (released on May, 2001) contains alignments and models for 2847 protein families, based on the Swiss-Prot 39 and SP-TrEMBL 14 protein sequence databases [29]. Pfam was downloaded from ftp://genetics.wusti.edu/pub/eddy/pfam-6.3/Pfam-A.full.gz. Pfam family ID and family members (represented by Swiss-Prot ID or TrEMBL ID) were extracted and organized in a relational database (Sybase, Adaptive Server Enterprise Release 11.9.3, CA, Sybase Inc.). A non-redundant protein sequence database comprised of Swiss-Prot and TrEMBL was also downloaded from ftp://ftp.expasy.org/databases/sp_tr_nrdb and the data were parsed and stored in Sybase.
The Incyte microarray is manufactured by depositing DNA onto a glass surface in an array format at a density of up to 10,000 array elements per chip. LifeExpress RNA is a gene expression database which, in Version 3.0 (April 2000 release), includes 6307 expression experiments performed using Incyte's Human Genome chip 1~5. (Incyte Human Genome chip 1 (HG1) contains 9766 cDNAs, while HG2 containing 9612 cDNAs, HG3 containing 9686 cDNAs, HG4 containing 9249 cDNAs, HG5 containing 9219 cDNAs. In total the five arrays contain 46909 cDNAs which represent about 41296 unique genes.). These experiments encompass the therapy areas of Cancer Biology, the Cardiovascular System, Immunology and Inflammation, Metabolism, Neurobiology, Toxicology, and Body Map.
Selection of probe pairs in LifeExpress
The LE database contains data points from a total of 976 probe pairs (Note: here the term of "probe" refers to "fluorescent-labeled total RNA sample used for microarray hybridization") that have been hybridized to one or more Human Genome chips 1~5. 266 of these probe pairs have been hybridized to all five chips. Using data from all available probe pairs would reduce the number of genes that could be compared, since many genes are not represented in all 976 experiments. By contrast, limiting oneself to only those probe pairs that have been run against all the chips would greatly reduce the number of experiments from which the data is drawn. In order to optimize the amount of data that could be compared, 555 probe pairs that were hybridized to the first 4 Human Genome chips (HG1, HG2, HG3, and HG4) were used in this study.
Mapping Pfam to LifeExpress
A total of 4935 Swiss-Prot human sequences and 22208 TrEMBL human sequences are included in 1427 protein families covered by Pfam Version 6.3. Corresponding GenBank mRNA sequences for those protein records were identified from annotation present in Swiss-Prot and TrEMBL sequence records, and were assembled together with the clone sequences from LifeExpress Human Genome chips 1~4 using the PHRAP assembler (threaded version 3.01 licensed from Southwest Parallel Software, Inc. http://www.spsoft.com. 4177 Incyte clones belonging to 1069 Pfam families were mapped to these sequences. Pfam family PF00069 is most highly represented on the chips with 231 Incyte clones while 343 Pfam families are represented on the microarrays by only one family member. In total 135 Pfam families are represented by more than ten clones on the microarrays. There were 2644 clones that belonged to these largest families.
Data Processing
One probe pair in LE could have been hybridized to the same Incyte Human Genome chip several times. In these cases, the average of the differential expression values (fold difference) for the different hybridizations was used in subsequent calculations. For each gene, the expression ratio for a particular probe pair was reduced to a binary variable ("regulated" or "unregulated") rather than a continuous variable. For each pair of experimental conditions, a gene was considered to be "regulated" if it showed at least a two-fold change in expression level (either up- or down-regulated) between conditions, and "unregulated" if the expression ratio was less than two-fold.
In order to analyze the expression profile for each Pfam family, we calculated the Family Regulation Ratio (FRR) for each Pfam family as the percentage of its members which were "regulated" in a pair-wise comparison and assigned the ratio to the Pfam ID for this probe pair. For example, 140 clones in the PF00001 family had been hybridized with the probe pair of "PBMC Cells, Untx, 24 hr, Dn4625 vs t/2 4 hr", of which 28 clones (20%) showed greater than two fold difference in the pair-wise comparison. This meant that 20% members in the family of PF00001 were "regulated" with this probe pair. So the Family Regulation Ratio of 0.2 was assigned to PF00001 for this probe pair.
In order to reduce random noise, we only studied the 135 Pfam families represented by more than ten clones on the microarrays. Family Regulation Profiles were generated for each of these largest Pfam families by calculating the FRRs corresponding to 555 probe pairs.
Correlation measure
Correlations between Family Regulation Profiles are measured with the standard Pearson Correlation Coefficient (PCC). The PCC between two profiles a, b with k dimensions is calculated as
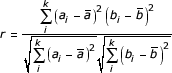
where ai and bi represent the Family Regulated Ratios of Pfam a and Pfam b for the sample i, and 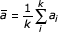 and
and 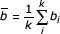 indicate the respective means. If a clone was mapped to both Pfam a and b, the Family Regulation Profiles of a and b would be modified with the clone excluded. In other words, common clones between Pfam pairs made no contribution to the PCC calculation.
indicate the respective means. If a clone was mapped to both Pfam a and b, the Family Regulation Profiles of a and b would be modified with the clone excluded. In other words, common clones between Pfam pairs made no contribution to the PCC calculation.
Cross-validation with two independent subsets
One way to validate our approach is to verify that the Pfam relationships based on our Family Regulation Profiles would be ubiquitous. In other words, different microarray experiment sets, as long as the experiment resource is rich, diverse and well-represented enough, should generate a similar result. The 555 LE probe pairs were randomly divided into two data sets with one data set containing 278 probe pairs (S1) and the other one containing 277 probe pairs (S2). Based on these two data sets, two Family Regulation Profiles for each Pfam were computed respectively. Pearson Correlations among Family Regulation Profiles PCC1 and PCC2 were calculated using S1 and S2 respectively. PCC1 and PCC2 should be positively correlated if the derived Pfam relationships are independent of the expression experiment selection.
The Enrichment Factor (EF) is a parameter that represents the extent to which the Pfam pairs identified by PCC1 within the cutoff range specified by C1 are over-represented in pools of Pfam pairs that could be identified by PCC2 within the cutoff range specified by C2. The number of total Pfam pairs are indicated as Gtotal while the number of Pfam pairs selected by PCC1 — C1 and PCC2 — C2 are indicated as G1 and G2 respectively. The number of Pfam pairs common to both PCC1 — C1 and PCC2 — C2 is indicated as Gboth. Therefore the expected background representation ratio, or the random representation ratio for G1 within G2 is,
![]()
and the observed representation ratio for G1 within G2 is,
![]()
EF is then defined as,
![]()
with a value for EF > 1 indicating higher than background representation ratio for G1 within G2.
Take an example where G1 is selected by PCC1 — 0.5 and G2 is selected by PCC2 — 0.8. Out of total 8968 Pfam pairs there are 2185 Pfam pairs with PCC1 — 0.5, so the ratio of R0 can be calculated as 2185/8968 = 0.24. There are 20 Pfam pairs with PCC2 — 0.8, of which all 20 also have PCC1 — 0.5. The ratio of R1, equals 20/20 = 1, therefore, EF = 1/0.24 = 4.2
Authors' contributions
WD, LW, PQ carried out the data analysis and drafted the manuscript. JG, MH and MK participated in the design of study. All authors read and approved the final manuscript.
Supplementary Material
The list of the Pfam pairs which share more than 50% family members
The list of these 135 Pfam family IDs and names
List of the Pfam-pairs with the Pearson Correlation Coefficient greater than 0.6.
Figure. Pfam regulation network predicted by the correlation of Family Regulation Profiles. In total, 89 Pfams (labeled with Pfam Ids) and 781 correlations with PCC — 0.6 are shown Table. The list of the Pfam pairs which share more than 50% family members
{kind=link}
Contributor Information
Wei Ding, Email: wei.ding@spcorp.com.
Luquan Wang, Email: luquan.wang@spcorp.com.
Ping Qiu, Email: ping.qiu@spcorp.com.
Mitchel Kostich, Email: mitchell.kostich@spcorp.com.
Jonathan Greene, Email: jonathan.greene@spcorp.com.
Marco Hernandez, Email: marco.hernandez@spcorp.com.
References
- DeRisi JL, Iyer VR, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- Bucher P. Regulatory elements and expression profiles. Curr Opin Struct Biol. 1999;9:400–407. doi: 10.1016/S0959-440X(99)80054-2. [DOI] [PubMed] [Google Scholar]
- Brazma A, Jonassen I, Vilo J, Ukkonen E. Predicting gene regulatory elements in silico on a genomic scale. Genome Res. 1998;8:1202–1215. doi: 10.1101/gr.8.11.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth FP, Hughes JD, Estep PW, Church GM. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat Biotechnol. 1998;16:939–945. doi: 10.1038/nbt1098-939. [DOI] [PubMed] [Google Scholar]
- Drawid A, Jansen R, Gerstein M. Genome-wide analysis relating expression level with protein subcellular organization. Trends Genet. 2000;16:426–430. doi: 10.1016/S0168-9525(00)02108-9. [DOI] [PubMed] [Google Scholar]
- Jansen R, Gerstein M. Analysis of the yeast transcriptome with structural and functional categories: characterizing highly expressed proteins. Nucleic Acids Res. 2000;28:1481. doi: 10.1093/nar/28.6.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein M, Jansen R. The current excitement in bioinformatics-analysis of whole-genome expression data: How does it relate to protein structure and function? Opin Struct Biol. 2000;10:574–584. doi: 10.1016/S0959-440X(00)00134-2. [DOI] [PubMed] [Google Scholar]
- Shalon D, Smith SJ, Brown PO. A DNA microarray system for analyzing complex DNA samples using two color flourescent probe hybridization. Genome Res. 1996;6:639–645. doi: 10.1101/gr.6.7.639. [DOI] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–70. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Ramsay G. DNA chips: state-of-the art. Nat Biotech. 1998;16:40–4. doi: 10.1038/nbt0198-40. [DOI] [PubMed] [Google Scholar]
- Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer ELL. The Pfam protein families database. Nucleic Acids Research. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batagelj V, Mrvar A. Pajek-Program for Large Network Analysis. Connections. 1998;21:247–257. [Google Scholar]
- Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown PO, Herskowitz I. The transcriptional program of sporulation inbudding yeast. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein M, Lan N, Jansen R. Interating Interactomes. Science. 2002;295:284–285. doi: 10.1126/science.1068664. [DOI] [PubMed] [Google Scholar]
- Greenbaum D, Luscombe NM, Jansen R, Qian J, Gerstein M. Interrelating different types of genomic data, from proteome to secretome: 'oming in on function. Genome Res. 2001;11:1463–1468. doi: 10.1101/gr.207401. [DOI] [PubMed] [Google Scholar]
- Qian J, Dolled-Filhart M, Lin J, Yu H, Gerstein M. Beyond synexpression relationships: local clustering of time-shifted and inverted gene expression profiles identifies new, biologically relevant interactions. J Mol Biol. 2001;314:1053–1066. doi: 10.1006/jmbi.2000.5219. [DOI] [PubMed] [Google Scholar]
- Jansen R, Greenbaum D, Gerstein M. Relating whole-genome expression data with protein-protein interactions. Genome Res. 2002;12:37–46. doi: 10.1101/gr.205602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge H, Liu Z, Church GM, Vidal M. Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nature Genet. 2001;29:482–486. doi: 10.1038/ng776. [DOI] [PubMed] [Google Scholar]
- Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol. 1999;19:1720–1730. doi: 10.1128/mcb.19.3.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futcher B, Latter GI, Monardo P, McLaughlin CS, Garrels JI. A sampling of the yeast proteome. Mol Cell Biol. 1999;19:7357–7368. doi: 10.1128/mcb.19.11.7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drawid A, Gerstein M. A Bayesian system integrating expression data with sequence patterns for localizing proteins: comprehensive application to the yeast genome. J Mol Biol. 2000;301:1059–1075. doi: 10.1006/jmbi.2000.3968. [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ. G protein-coupled receptors. III. New roles for receptor kinases and beta-arrestins in receptor signaling and desensitization. J Biol Chem. 1998;273:18677–18680. doi: 10.1074/jbc.273.30.18677. [DOI] [PubMed] [Google Scholar]
- Tsai W, Morielli AD, Cachero TG, Peralta EG. Receptor protein tyrosine phosphatase alpha participates in the m1 muscarinic acetylcholine receptor-dependent regulation of Kv1.2 channel activity. EMBO J. 1999;18:109–118. doi: 10.1093/emboj/18.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornau H, Seeburg P, Kennedy M. Interaction of ion channels and receptors with PDZ domain proteins. Curr Opin Neurobiol. 1997;7:368–373. doi: 10.1016/S0959-4388(97)80064-5. [DOI] [PubMed] [Google Scholar]
- Chen S, Wang QL, Nie Z, Sun H, Lennon G, Copeland NG, Gilbert DJ, Jenkins NA, Zack DJ. Crx, a novel Otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genes. Neuron. 1997;19:1017–1030. doi: 10.1016/s0896-6273(00)80394-3. [DOI] [PubMed] [Google Scholar]
- Lin H, Stacey M, Saxby C, Knott V, Chaudhry Y, Evans D, Gordon S, McKnight AJ, Handford P, Lea S. Molecular Analysis of the Epidermal Growth Factor-like Short Consensus Repeat Domain-mediated Protein-Protein Interactions. J Biol Chem. 2001;276:24160–24169. doi: 10.1074/jbc.M101770200. [DOI] [PubMed] [Google Scholar]
- Funamoto S, Milan K, Meili R, Firtel RA. Role of Phosphatidylinositol 3' Kinase and a Downstream Pleckstrin Homology Domain-containing Protein in Controlling Chemotaxis in Dictyostelium. J Cell Biol. 2001;153:795–810. doi: 10.1083/jcb.153.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The list of the Pfam pairs which share more than 50% family members
The list of these 135 Pfam family IDs and names
List of the Pfam-pairs with the Pearson Correlation Coefficient greater than 0.6.
Figure. Pfam regulation network predicted by the correlation of Family Regulation Profiles. In total, 89 Pfams (labeled with Pfam Ids) and 781 correlations with PCC — 0.6 are shown Table. The list of the Pfam pairs which share more than 50% family members