

Abstract
Background
Females with the neurological disorder Rett syndrome are heterozygous for mutations in X-linked MECP2 that encodes methyl-CpG binding protein 2 (MeCP2) thought to act as a transcriptional repressor. To identify target genes for MeCP2 modulation, we studied global gene expression in single cell-derived wild-type and mutant MECP2 expressing fibroblast clones with four common mutations (R106W, R306C, 705delG, 1155del32) and in lymphoblastoid cell lines (LCLs) that included four mutant MeCP2 (T158M, 803delG, R168X and 1159del28) expressing, and five (1159del28, R106W, R255X, 803delG, 803delG) wild-type MeCP2 expressing lines.
Methods
Clonality and mutation status were verified by androgen receptor methylation assays for X-inactivation and by sequencing MECP2 transcripts. Expression studies were done with oligonucleotide microarrays (Affymetrix U95) and verified with real-time quantitative RT-PCR using Sybr Green.
Results
Expression of 49 transcripts was increased, and expression of 21 transcripts was decreased, in at least 3 of 4 mutant/wild-type fibroblast comparisons. Transcript levels of 11 genes, determined by quantitative RT-PCR, were highly correlated with the microarray data. Therefore, multiple additional clones from two Rett individuals were tested by RT-PCR only. Striking expression differences were found in both mutant and wildtype MeCP2 expressing clones. Comparing expression profiles of lymphoblastoid cell lines yielded 16 differentially expressed genes.
Conclusions
MeCP2 deficiency does not lead to global deregulation of gene expression. Either MeCP2's in vivo function does not involve widespread transcriptional repression, or its function is redundant in cell types that also express other methyl-CpG binding proteins. Our data suggest that clonal fibroblast strains may show substantial inter-strain variation, making them a difficult and unstable resource for genome-wide expression profiling studies.
Background
Rett syndrome (OMIM # 312750 http://www.ncbi.nlm.nih.gov/Omim/) is an X-linked dominant disorder that occurs with a prevalance of 1 in 10,000 to 15,000 births [1] and almost exclusively in females. Named after Andreas Rett who first recognized the disorder in 1966 [2], the syndrome is characterized by a constellation of clinical findings that have resulted in the establishment of clinical criteria for the diagnosis [3]. Common clinical features include early normal development until approximately 6–18 months of age followed by a period of developmental stagnation and subsequent regression, usually by 3 years of life. Characteristic findings include loss of speech and motor skills, deceleration of head growth, stereotypic hand movements, seizures, respiratory abnormalities, scoliosis and growth delay. Although the disorder is usually sporadic, due to new mutations, a few familial cases permitted exclusion mapping on the X chromosome with assignment of the responsible gene to band Xq28 [4-6].
Mutations in MECP2, the gene that encodes the DNA binding protein MeCP2 (methyl-CpG binding protein 2), have been found in approximately 65–77% of Rett syndrome patients [7-10]. The functional consequence of MECP2 mutations resulting in Rett syndrome is loss of function. Three of the four MECP2 exons encode a 486 amino acid protein that is believed to be a transcriptional repressor based on in vitro studies. MeCP2's functional structure consists of an 84 amino acid methyl-binding domain (MBD) [11] and a 104 amino acid transcriptional repression domain (TRD) [12]. The MBD is able to bind to 5-methyl cytosine residues of a single CpG dinucleotide [11]. Under the current model, MeCP2 binds to DNA, then complexes with Sin3A through the TRD and recruits histone deacetylases (HDAC). Deacetylation of core histones then alters chromatin structure and results in transcriptional repression [13,14].
While MeCP2 is a ubiquitously expressed protein, its expression is highest in the brain, lung and spleen and less abundant in the kidney and heart [15,16]. It's cellular localization is also specific, with expression in neurons but not in glia. Expression levels in brain increase postnatally [16]. These findings explain why the most prominent clinical features of Rett syndrome are related to central nervous system (CNS) dysfunction. The presence of growth retardation [17], osteopenia [18], and gastrointestinal abnormalities, however, suggest a potentially important role for MeCP2's function in the peripheral and autonomous nervous system and in other tissues. Alternatively, while MeCP2's expression is ubiquitous, other methyl-CpG binding proteins that exist within complexes, MBD1 and MBD2 [19] or Kaiso [20], may reduce the effect of mutations in MECP2 in peripheral organ systems while the lack of such redundancy may lead to a more pronounced phenotype in the brain. This theory remains speculative, however, since the tissue specificity and definitive identification of functional redundancy of methyl-CpG binding proteins have not been established. Regardless, the targets of MeCP2 modulation remain unknown in the brain or elsewhere.
To identify potential downstream targets of MeCP2 modulation, we have compared global gene expression patterns in four matched pairs of clonally derived mutant or wild-type MECP2-expressing fibroblast cell strains from girls with Rett syndrome, and in five wild-type and four mutant MeCP2-expressing lymphoblastoid cell lines, by hybridizing Affymetrix U95 (A-E) oligonucleotide microarrays. The use of clonally derived fibroblasts eliminated the mosaic X inactivation seen in tissues from most females with Rett syndrome. Furthermore, expression patterns in clones with the mutant MECP2 allele active were compared with those in clones with the wild-type allele on the active X chromosome. Because both types of clones are derived from the same individual, variability introduced by differences in autosomal genetic background is eliminated by our experimental strategy. The use of lymphoblastoid cell lines, while not all clonally derived, expanded the scope of genes to be assayed beyond those expressed in skin fibroblasts. Our interrogations identified a statistically significant number of genes with differential expression patterns between wild-type and mutant cell strains, however, the results across multiple clones from the same individual showed substantial variability, questioning the significance of some of the genes identified. The lymphoblasts, while not analysed in a matched comparison, yielded a relatively short list of genes with differential expression that includes some potentially interesting candidate genes.
Methods
Cell culture, single cell cloning and nucleic acid extraction for fibroblasts
Four fibroblast strains from females with clinically diagnosed Rett syndrome were obtained from Coriell Cell Repositories. MECP2 mutations were identified in our laboratory as previously reported [10] [GM07982: 705delG; GM11270: 916C>T (R306C); GM11272: 1155del32 and GM11273: 316C>T (R106W)]. From each of the four cultures, single-cell derived clonal cell strains were established. To do so, fibroblasts were trypsinized (0.05%) (GibcoBRL Life Technologies) and diluted to 100 cells/ml. By using a 125 μm Stripper™ Tip (MidAtlantic Diagnostics, Marlton, NJ) under direct microscopic visualization, individual cells were transferred to 96 well plates. Cells were grown with filtered (0.2 μm) conditioned medium that was prepared by culturing the primary cell strain for two days during the exponential growth phase in MEM-alpha (Sigma, St. Louis, MO) with 15% fetal calf serum (Gemini, Woodland, CA), penicillin (100 U/ml), streptomycin (100 μg/ml), and glutamine (290 ng/ml) (GibcoBRL Life Technologies). Upon confluency of the well, the cells were trypsinized and expanded. Growth rates and cellular morphology were closely monitored. Only clonal cell strains in the exponential growth phase with similar cell and clonal morphology were selected for further study. RNA was extracted using RNA STAT-60 (Tel-Test Inc., Friendswood, TX) from eight 150 mm plates of exponentially growing sub-confluent cells that were at similar passage numbers. DNA was extracted from two additional 150 mm plates using a standard extraction protocol to determine X chromosome inactivation status and verify monoclonality.
Cell culture, single cell cloning and nucleic acid extraction for lymphoblasts
Clonal cell lines with the mutant MECP2 allele on the active X chromosome were derived from lymphoblastoid cell lines (LCLs) from four females with clinically diagnosed Rett syndrome and known mutations (T158M, 803delG, R168X) [10] and 1159del28 (unpublished) using the technique described above. Individual cells were cultured in conditioned RPMI 1640 medium with 15% fetal calf serum, penicillin (100 U/ml), streptomycin (100 U/ml) and glutamine (290 ng/ml). Four cell lines from females with known MECP2 mutations (R106W, R255X, and two with 803delG) but completely skewed X chromosome inactivation with the wild-type MECP2 allele on the active X chromosome were also cultured. In addition, one clonal cell line with the wild-type MECP2 allele on the active X chromosome was established using the cloning technique described above. RNA was extracted using RNA STAT-60 (Tel-Test Inc., Friendswood, TX). DNA was extracted using a standard extraction protocol to determine X chromosome inactivation status and verify monoclonality.
X chromosome inactivation studies
Clonality was further confirmed and X chromosome inactivation status was determined by the length of the polymorphic CAG trinucleotide repeat on the inactive androgen receptor allele [21,22]. Restriction digestion of four restriction sites between the flanking primers and adjacent to the trinucleotide repeat were carried out with the methylation sensitive enzymes HpaII and HhaI. Amplification of the undigested allele that included the trinucleotide repeat region was performed using the fluorescent-labeled primer 5'-6FAM CTGTGAAGGTTGCTGTTCCTCAT-3' and the unlabeled primer 5'-TCCAGAATCTGTTCCAGAGCGTGC-3'. The lengths of the amplification products were identified using the ABI 377 sequencer with the GeneScan™ fragment analysis software (Applied Biosystems, Foster City, CA). Undigested DNA samples from each of the original cell cultures were amplified to determine the lengths of each allele.
Allele-specific MECP2 transcript analysis
Reverse transcription of 2 μg of total RNA was performed after Dnase I digestion (Roche Diagnostics, Indianapolis, IN) with the Superscript™ II RNase H-Reverse Transcriptase (GibcoBRL Life Technologies) protocol for cDNA synthesis using 500 ng of random hexamers. Absence of DNA contamination was verified by amplification of the intron containing region of the MECP2 gene between exons 3 and 4 using exon 3 based primer 5'-AGCCCGTGCAGCCATCAGCC-3' and exon 4 based primer 5'-CTTCCCAGGACTTTTCTCCA-3'. The regions containing the mutations were amplified by PCR using primers and conditions previously described [7] and later modified [23]. PCR products were treated with 1 U exonuclease and 1 U shrimp alkaline phosphatase for 15 min at 37°C. The enzymes were heat inactivated at 75°C for 15 minutes. Both strands of the treated PCR product were sequenced with fluorescent labelled terminator nucleotides on the ABI PRISM®377 DNA sequencer. Cell clones expressing only the mutant MECP2 allele are called "mutant clones", and cell clones or skewed cell lines expressing only the normal MCP2 allele are called "wild-type".
Immunoblot analysis with MeCP2 antibody
MeCP2 protein expression in fibroblast clones and lymphoblastoid cell lines was determined by immunoblot analysis. Cells were washed twice with PBS and lysed with RIPA buffer (1% Triton X-100, 1% NaDOC, 0.1% SDS, 158 mM NaCl, 10 mM Tris pH 7.2, 5 mM NaEDTA and 1% protease inhibitors, added just before use) (Sigma, St. Louis, MO). The homogenate was sonicated, followed by centrifugation at 4°C for 30 min. The supernatant was saved for immunoblot. Equal quantities of protein, as determined by a protein assay (BioRad, Hercules, CA), were separated by electrophoresis in a 12% SDS-polyacrylamide gel. The gel was then equilibrated in transfer buffer for 15 min and transferred to a polyvinylidine difluoride membrane (Novex, San Diego, CA), also equilibrated in transfer buffer for 15 min, for 1 hr at 100 V. The membrane was blocked overnight with 5% non-fat milk in PBS at 4°C, and incubated with a rabbit polyclonal antibody (1:2000) against a peptide (CPRPNREEPVDSRTP) at the C-terminal region of MECP2 (Upstate Biotechnology, Lake Placid, NY) at room temperature for 2 h in a western wash solution (PBS containing 2% non-fat milk, 0.1% Tween 20, and 350 mM NaCl). The membrane was washed in the same solution and then incubated with a horseradish peroxidase conjugated secondary antibody (1:5000) in the same solution for 30 min. The membrane was subsequently washed again, and bound antibody was detected with an ECL kit (Amersham, Piscataway, NY).
Affymetrix oligonucleotide array (GeneChip®) hybridization
Messenger RNA was purified from total RNA extract using theOligotex™ batch protocol (Qiagen, Valencia, CA). The SuperScript™ Choice System was used for (ds)-cDNA synthesis (GibcoBRL Life Technologies) using the T7-(dT)24 primer 5'-GGCCAGTGAATTGTAATACGACTCACTATAGGGAGGCGG-(dT)24-3' and 3 μg of mRNA for each reaction. Biotin-labeled in vitro transcription (IVT) was accomplished with the Enzo BioArray™ High Yield™ transcript labeling kit (Affymetrix, Santa Clara, CA) using 4 μl of (ds)-cDNA product. IVT cleanup was performed using Rneasy® Columns (Qiagen, Valencia, CA). The IVT product was chemically fragmented with 40 mM Tris-acetate, pH 8.1, 125 mM KOAc and 30 mM MgOAc at 94°C for 35 minutes. The fragmented product was hybridized to the Affymetrix GeneChip® Human Genome U95 Set (A-E) (Affymetrix, Santa Clara, CA).
Quality control parameters were assessed throughout the experimental process to assure the quality of the starting RNA, the efficiency of transcription, integrity of hybridization, and consistency of qualitative calls. Assessment during the synthesis of the hybridization transcript was accomplished by spectrophotometric analysis of the starting RNA and cRNA and gel electrophoresis following synthesis of the cDNA, cRNA and fragmentation of the cRNA. A 3'/5' ratio of GAPDH less than 2 was considered acceptable for efficiency of transcription. Spiked control transcripts were also monitored to verify hybridization integrity. Scaling factors for each sample ranged between 0.88 and 17.8 with the ratio of scaling factors between samples for a given chip ranging from 1.4 to 2.5. All quality control measures were consistent with the manufacturers recommended procedures and conformed to the recommended cut-off values.
Microarray data analysis for fibroblast clones
All GeneChips® were analyzed using MicroArray Suite 3.0™ (MAS 3.0) with target intensities normalized to 2500. Baseline genetic expression profiles of one clonal wild-type MeCP2-expressing fibroblast strain were compared to the expression profile of the matched clonal mutant MeCP2-expressing cell strain, for each of four different MECP2 mutations. Analyzed data were imported to MicroDB™ and queried using Affymetrix's Data Mining Tool (DMT). All four matched pair comparisons were queried for directional changes. For genes/ESTs with increased levels of expression, defined by MAS 3.0 as marginally increased or increased, wild-type baseline expression levels could be present or absent. For genes with decreased levels of expression, defined by MAS 3.0 as marginally decreased or decreased, wild-type baseline expression levels were required to be present. We identified genes that demonstrated the same directional change in at least three of four comparisons. We computed p values for genes identified by this criterion. We assumed that if a gene was not subject to a systematic change in expression level, an observed directional change in a given mutant strain would occur randomly and independently with probability equal to the overall proportion of directional changes observed in that mutant strain. We used one-sample, two-sided t-tests on the reported GeneChip® fold changes to assess whether the log fold changes of mutant cell strains differ from zero on average. We report the corresponding 95% confidence intervals (CI) after conversion back from the log scale. Although the assumptions of the t-test cannot be verified, there was insufficient data to carry out alternative tests.
Microarray data analysis for lymphoblastoid cell lines
All GeneChips® were analyzed using MAS 3.0 with target intensities normalized to 2500. Only one lymphoblast cell line yielded both a clonal wild-type and clonal mutant cell line (1159del28). Baseline expression profiles were derived from the clonal wild-type cell line (1159del28), and the four completely skewed wild-type cell line controls (R106W, R255X, 803delG, 803delG). Expression profiles from the mutant clonal cell line (1159del28) and three additional clonal mutant MeCP2 expressing cell lines (T158M, 803delG, R168X) were compared to the baseline expression profiles. Each mutant expression profile was compared to each wild-type control profile yielding 20 comparisons. Analyzed data was imported to MicroDB™ and queried using DMT. All 20 comparisons were queried to identify genes that demonstrated similar directional changes in at least 15 of 20 unmatched pair comparisons (four mutant samples compared individually to each of five wild-type samples). For genes with increased levels of expression, defined by MAS 3.0 as marginally increased or increased, wild-type baseline expression levels could be present or absent. For genes with decreased levels of expression, defined by MAS 3.0 as marginally decreased or decreased, wild-type baseline expression levels were required to be present. We used two-sided Wilcoxon rank-sum tests on the reported uncompared values for the two groups to test whether the expression profiles of mutant cell lines differ from those of wild-type cell lines.
Real-time comparative quantitative PCR
Total RNA from the four matched pairs of clonal fibroblast cell strains and ten lymphoblast cell lines used on the GeneChips® was used as template for RT-PCR to independently assess the validity of Affymetrix GeneChip® data. Each RT reaction was performed with RNA from the same preparation used for the GeneChip® hybridization. Total RNA from eight additional clonal cell strains (four mutant and four wild-type) with the R106W mutation and fourteen additional clonal cell strains (seven mutant and seven wild-type) with the 1155del32 mutation were also used as template for RT-PCR to assess consistency of results across different clonal fibroblast cell strains. Reverse transcription of total RNA was performed after Dnase I digestion (Roche Diagnostics, Indianapolis, IN) with the Superscript™ II Rnase H-Reverse Transcriptase (GibcoBRL Life Technologies) protocol for cDNA synthesis using 2 μg of total RNA and 1 μg of random hexamers. Absence of DNA contamination was verified by amplifying the 756 bp intron containing region of the MECP2 gene between exons 3 and 4 using exon 3 based primer 5'-AGCCCGTGCAGCCATCAGCC-3' and exon 4 based primer 5'-CTTCCCAGGACTTTTCTCCA-3'. Minus RT samples tested simultaneously with experimental samples by quantitative RT-PCR with Sybr® Green consistently yielded no amplification below 33 cycles using the above protocol. The RT product was diluted 10 fold, aliquoted and stored at -80°C. Forward and reverse primers were designed using Applied Biosystem's Primer Express v1.0™ software (Applied Biosystems, Foster City, CA) from consensus sequences provided by Affymetrix http://www.netaffx.com for each gene of interest. Each 25 μl PCR mix contained 7.875 μl Sybr® Green Master Mix (1X Sybr® Green Buffer, 3 mM MgCl2, 200 nM dNTP blend, 0.6275 U Amplitaq Gold®, 0.25 U AmpErase® UNG), 200–400 nM forward and reverse primers and approximately 5 ng template. Cycle conditions for the ABI 7700 were 50°C for 2 minutes, 95°C for 10 minutes and 35 cycles of 95°C for 15 sec and 60°C for 1 minute. The average of four endogenous controls (GAPDH, β actin, TATA IID and transferrin) was used to normalize the amount of template for each wild-type and mutant pair. Each reaction was run in triplicate with the results averaged on the log (base 2) scale. Standard curves for each amplification product were generated from 10-fold dilutions of pooled cDNA to determine primer efficiency. Data was analyzed with Microsoft Excel 2000 to generate raw expression values.
We performed paired t-tests on the RT-PCR values for the original set of fibroblast clones to test whether the average log change across cell strains differs from zero. We used unpaired t tests for the second set of fibroblast clones from each of two cell strains to determine whether multiple clones from individuals undergo a systematic fold change. We used unpaired Wilcoxon rank-sum tests for the lymphoblast cell lines to test whether there is a systematic fold change across lines. All reported 95% confidence intervals are reported in terms of fold changes, but derived from t-tests computed on the log scale.
Results
Characterization of clonal cell strains
Clonal mutant and wild-type MECP2 expressing fibroblast cell strains were successfully established from four Rett females with different mutations, one missense in the MBD, one missense in the TRD and two frameshift mutations leading to premature stop codons. Clonality was established by three independent methods: visualization of each cell transferred for clonal culture, X inactivation pattern analysis of the polymorphic androgen receptor alleles using GeneScan™, and identification of single-allele MECP2 transcripts (Fig. 1).
Figure 1.

MECP2 transcript sequence and GeneScan™ results for the human androgen receptor (AR) polymorphism used to determine X chromosome inactivation patterns. A. Parental cell strain with 916C>T (R306C) mutation and mosaic X-inactivation with expression of 285 bp and 288 bp AR alleles. B. Fibroblast clone from R306C donor expresses MECP2 transcript with mutant T allele and carries the 288 bp AR allele on the inactive X chromosome. C. Fibroblast clone from R306C donor expresses MECP2 transcript with normal C allele and carries the 285 bp AR allele on the inactive X chromosome.
The levels of MECP2 transcripts were virtually identical by real-time quantitative RT-PCR analysis (less than 0.27 cycle thresholds difference) across all eight clones, one wild-type and one mutant from each individual, that were used for oligonucleotide array hybridization (data not shown). This result is not surprising. Given the fact that the premature termination codons resulting from the frameshift mutations are all located in the 3' final exon of the MECP2 gene, preferential degradation of nonsense codon-containing transcripts would not be expected [24].
To assess MeCP2 expression at the protein level, immunoblot analysis with an antibody against the C-terminal region of MeCP2 was carried out on total cell lysates from all eight fibroblast clones, and from some of the LCLs, that were used for array hybridization (Fig. 2). A reacting protein band (at the molecular weight of 75 kD, as indicated by the company, http://www.upstate.com) was seen in wild-type cell lines expressing a normal sized MeCP2 protein, and in cell lines with missense mutations (FB R106W, FB R306C and LCL T158M). Cell clones expressing an allele with a truncating mutation (FB 705delG, FB 1155del32, LCL R168X), do not produce a normal sized MECP2 protein. Due to the location of the epitope, the antibody would not react with putative truncated proteins. The bands seen at approximately 48 and 100 kD are unidentified proteins that cross-react with the MeCP2 antibody. The absence of a normal sized MeCP2 protein in mutant clones with truncating mutations confirms their purity, and, together with the presence of MeCP2 in the wild-type clones, validates these matched pairs of cell cultures as a useful experimental system for the search of downstream effects of MeCP2 deficiency.
Figure 2.

Immunoblots of total cell lysates from wild-type (WT) and mutant (M) fibroblast clones and lymphoblastoid cell lines used for the oligonucleotide expression array hybridization studies. MeCP2 expression is detected with a C-terminal antibody; the molecular weight of MeCP2 is shown as 75 kDa, consistent with the company's information http://www.upstate.com. In mutant cell lines with nonsense mutations (FB 705delG, FB 1155del32, and LCL R168X), an intact MECP2 protein is not seen. Brain lysate from mice and normal human male lymphoblastoid cells serve as positive controls.
Oligonucleotide array hybridization data of matched fibroblast clones
The percent "present" calls ranged from 13% (chip D) to 55% (chip A). Standard deviations for the percent "present" calls for a given chip were 1–3%. There were no significant differences in percent "present" calls between MeCP2-deficient and MeCP2-positive samples. A total of 252,700 comparisons (four Rett females × 63,175 interrogations on five GeneChips®) revealed 6,745 increased (or marginally increased) genes/ESTs and 6,966 decreased (or marginally decreased) genes/ESTs. The number of genes with increased (+) and decreased (-) levels of expression in mutant MeCP2 over wild-type expressing clones differed among the fibroblast strains studied (R106W: 889+, 3056-; R306C: 1700+, 1435-; 705delG: 1160+, 1240-; 1155del32: 2996+, 1235-). The unadjusted p value for any one gene that was increased (decreased) in three out of four matched pair comparisons was .000060 (.000075). The p value for any gene that increased (decreased) in all four comparisons was 3.29 E-07 (4.72 E-07).
Forty-six genes showed increased levels of expression in mutant cells in three out of four comparisons. Three genes were increased in all four comparisons. Of the 49 genes, 29 had been annotated as known genes and 20 as unknown transcripts. Of those, 7 were found to encode known genes by searching recent database releases and the Celera database (Table 1). Expression levels of two genes (VSHK1 and CA12), represented twice on the chips with different accession numbers, were increased in both cases. Of the 47 unique genes with increased transcript levels in MeCP2 deficient cells, 32 are known to be expressed in brain (Table 1).
Table 1.
Genes with increased levels of expression in at least 3 of 4 mutant MeCP2 expressing clonal fibroblast cell strains when compared to matched clonal wild type MeCP2 expressing cell strains
| GenBank Accession | Expression In Brain | Gene Symbol/UniGene No. | Gene Name | Function |
| J00123 | x | HUMENK2 | Human enkephalin gene | neuropeptide |
| AW007146 | x | SYTL2 | Synaptotagmin-like 2 | neurotransmission |
| AF051850 | SVIL | Supervillin | link between cytoskeleton and membrane | |
| AA447177 | Hs.202101 | Beta syntrophina | cytoskeletal protein | |
| M13509 | x | MMP1 | Matrix metalloproteinase 1 (interstitial collagenase) | breakdown of extracellular matrix |
| X05232 | MMP3 | Matrix metalloproteinase 3 (stromelysin 1, progelatinase) | breakdown of extracellular matrix | |
| X07820 | MMP10 | Matrix metalloproteinase 10 (stromelysin 2) | breakdown of extracellular matrix | |
| AB011153 | x | PLCB1 | Phospholipase C, beta 1 (phosphoinositide-specific) | phospholipase |
| M72393 | PLA2G4A | Phospholipase A2, group IVA (cytosolic, calcium-dependent) | phospholipase | |
| AW015590 | x | RGC32 | RGC32 protein | extracellular matrix protein |
| AW007426 | x | SPON2 | Spondin 2, extracellular matrix protein | extracellular matrix protein |
| AA401504 | VSHK1 | Orphan seven-transmembrane receptor, chemokine related | chemokine receptor | |
| AI131555 | VSHK1 | Orphan seven-transmembrane receptor, chemokine related | chemokine receptor | |
| AF037335 | x | CA12 | Carbonic anhydrase XII | zinc metalloenzyme |
| AI050855 | x | CA12 | Carbonic Anhydrase XIIa | zinc metalloenzyme |
| AI681917 | x | Hs.3321 | Iriquois related homeobox 3 related proteina | transcription factor |
| AF047419 | x | TCF21 | Transcription factor 21 | transcription factor |
| U01244 | x | FBLN1 | Fibulin 1 | secreted glycoprotein (extracellular matrix) |
| X60708 | x | DPP4 | Dipeptidylpeptidase IV | membrane glycoprotein |
| M14058 | x | C1R | Complement component 1, r subcomponent | serine protease (complement pathway) |
| X90392 | x | DNASE1L1 | Deoxyribonuclease I-like 1 | DNAse |
| Y17782 | HSBP3 | Heat shock 27 kD protein 3 | heat shock protein | |
| AF058056 | SLC16A7 | Solute carrier family 16 (monocarboxylic acid transporters), member 7 | monocarboxylate transport family member | |
| X04430 | IL6 | Interleukin 6 (interferon, beta 2) | induces maturation of B cells | |
| U81607 | x | AKAP12 | A kinase (PRKA) anchor protein (gravin) 12 | kinase anchor protein |
| D28915 | x | MTAP44 | Interferon-induced, hepatitis C-associated microtubular aggregate protein | member of interferon inducible proteins |
| M34057 | x | LTBP1 | Latent transforming growth factor beta binding protein 1 | assembly of TGF beta |
| AL050227 | x | Hs.27860 | Prostaglandin E2 receptor, EP3 Subtypea | prostaglandin receptor |
| W31022 | x | Hs.48376 | TRADEbetaa | tumor necrosis factor signaling |
| AI669308 | x | BID | BH3 interacting domain death agonist | cell death regulation |
| W95076 | Hs.271639 | RSK-like proteina | signal transduction | |
| AI752682 | x | NTN4 | Netrin 4 | leukocyte adhesion |
| M97935 | x | STAT1 | Signal transducer and activator of transcription 1, 91 kD | signal transduction |
| AI338491 | Hs.209209 | CALD 1 – caldesmon 1a | cytoskeletal regulator | |
| M33197 | x | GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | carbohydrate metabolism |
| AA625449 | x | Hs.238532 | N-acetylgalactosaminyltransferase 7a | glycosylation |
| AI189112 | x | Hs.65366 | ||
| AI809872 | x | Hs.19669 | ||
| AI949749 | Hs.44441 | |||
| AI884906 | x | Hs.111164 | ||
| AI984197 | x | Hs.16064 | ||
| AI307802 | x | Hs.135560 | ||
| AI806221 | x | Hs.38022 | ||
| AI018671 | Hs.93701 | |||
| N48346 | x | Hs.125489 | ||
| AA426500 | Hs.85989 | |||
| N40767 | Hs.6786 | |||
| AB002365 | x | Hs.23311 | ||
| AL080058 | x | Hs.6909 |
aIdentified in Celera database
Twenty-one genes demonstrated decreased levels of expression in three out of four matched comparisons. Of the 21 genes, 13 had been annotated as known genes and 8 as unknown transcripts, of which one was found to encode a known gene by searching recent database releases and the Celera database (Table 2). Oligonucleotide probes for two of the genes (COMP and IGFBP5) were represented twice on the chips, and both genes were detected as showing differential expression. Of the 19 unique genes with decreased expression in MeCP2 deficient cells, 12 are known to be expressed in brain (Table 2).
Table 2.
Genes with decreased levels of expression in at least 3 of 4 mutant MeCP2 expressing clonal fibroblast cell strains when compared to matched clonal wild type MeCP2 expressing cell strains
| GenBank Accession | Expression In Brain | Gene Symbol/UniGene No. | Gene Name | Function |
| AC003107 | COMP | Cartilage oligomeric matrix protein | extracellular matrix protein | |
| L32137 | COMP | Cartilage oligomeric matrix protein | extracellular matrix protein | |
| U59289 | x | CDH13 | Cadherin 13, H-cadherin | cell adhesion |
| X53586 | x | ITGA6 | Integrin, alpha 6 | cell adhesion |
| AI815057 | KLF5 | Kruppel-like factor 5 | transcriptional regulation of cellular proliferation | |
| D43969 | x | RUNX1 | Runt-related transcription factor 1 | transcription factor |
| L27560 | x | IGFBP5 | insulin-like growth factor binding protein 5 | growth homeostasis |
| AW022852 | x | IGFBP5 | insulin-like growth factor binding protein 5 | growth homeostasis |
| AI433710 | x | Hs.25557 | plasma-cell membrane glycoprotein PC-1a | nucleotide phosphatase |
| AA142891 | x | P4HA2 | prolyl 4-hydroxylase (alpha subunit)a | hydroxylase |
| U38810 | x | MAB21L1 | Mab-21-like | eye and cerebellum development |
| Y00815 | x | PTPRF | protein tyrosine phosphatase, receptor type, F | cell signaling |
| AI017574 | x | CRIP1 | Cysteine-rich protein 1 | zinc carrier protein |
| AI862775 | x | Hs.66915 | sphingolipid G-protein-coupled receptor, 5a | endothelial differentiation |
| AL035494 | Hs.184736 | |||
| AI125670 | x | Hs.16346 | ||
| AI718385 | x | Hs.302738 | ||
| AI818579 | Hs.121520 | |||
| AI597757 | Hs.207407 | |||
| AI935827 | Hs.169758 | |||
| AB018289 | Hs.49500 |
aIdentified in Celera database
Quantitative real-time RT-PCR analyses of fibroblast clones
To assess the reliability of the array hybridization results, transcript levels of 11 differentially expressed genes (8 with increased and 3 with decreased expression) were quantified using real-time quantitative RT-PCR analysis with Sybr Green®. The same RNA samples used for making array hybridization probes were used as templates for RT-PCR. The Pearson correlation coefficient was 0.96 for the RT-PCR values from triplicate assays. The fold change data from the GeneChips® and RT-PCR analyses (Table 3) were converted to log scale for analysis and comparison. The correlation of the combined RT-PCR fold change data (based on average of log of the three measurements from each wild-type and each mutant sample) with the GeneChip® log fold change data was .92 (95% CI = 0.86, 0.96) (Fig. 3).
Table 3.
Comparisons of GeneChip® and RT-PCR expression fold changes in mutant versus wild-type MECP2 expressing clones for 11 differentially expressed genes
| Fold Changes for Paired Comparisons | ||||||
| Gene Symbol/Unigene No. | Method | 705delG | R306C | 1155del32 | R106W | p value |
| INCREASED | ||||||
| MMP3 | GC | 0.91 | 5.80 | 31.50 | 3.90 | 0.113 |
| RT | 0.89 | 4.81 | 11.63 | 3.49 | 0.094 | |
| FBLN1 | GC | 2.00 | 11.80 | 2.00 | 0.37 | 0.228 |
| RT | 2.03 | 15.87 | 2.97 | 0.72 | 0.199 | |
| SLC16A7 | GC | 1.80 | 1.40 | 2.30 | 0.77 | 0.210 |
| RT | 1.30 | 1.69 | 1.76 | 0.94 | 0.112 | |
| MTAP44 | GC | 1.00 | 2.40 | 6.10 | 3.50 | 0.081 |
| RT | 1.36 | 4.77 | 20.10 | 24.15 | 0.058 | |
| LTBP1 | GC | 1.20 | 1.30 | 2.10 | 2.00 | 0.047 |
| RT | 1.36 | 3.25 | 3.59 | 2.16 | 0.029 | |
| NTN4 | GC | 3.20 | 5.50 | 15.10 | 3.80 | 0.016 |
| RT | 2.30 | 2.10 | 9.80 | 3.10 | 0.039 | |
| Hs.19669 | GC | 2.00 | 2.20 | 1.90 | 1.40 | 0.008 |
| RT | 1.22 | 1.60 | 2.07 | 1.30 | 0.040 | |
| Hs.23311 | GC | 5.90 | 0.12 | 2.60 | 4.40 | 0.609 |
| RT | 3.00 | 0.23 | 1.86 | 11.79 | 0.467 | |
| DECREASED | ||||||
| COMP | GC | 0.28 | 0.37 | 0.02 | 0.10 | 0.045 |
| RT | 0.18 | 0.25 | 0.05 | 0.09 | 0.011 | |
| KLF5 | GC | 0.53 | 0.42 | 0.56 | 0.27 | 0.014 |
| RT | 0.63 | 0.29 | 1.25 | 0.42 | 0.157 | |
| CRIP1 | GC | 0.32 | 2.80 | 0.11 | 0.50 | 0.347 |
| RT | 0.20 | 2.81 | 0.10 | 0.42 | 0.284 | |
P-values correspond to t-tests of whether average log fold change across the 4 different sets of matched clones differs from zero GC = GeneChip, RT = RT-PCR
Figure 3.
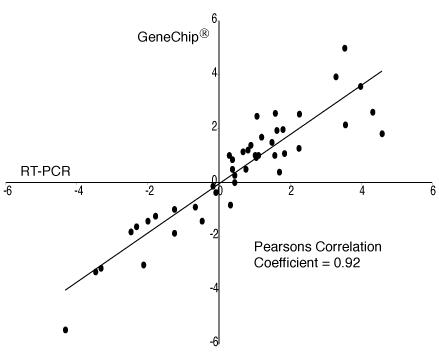
Correlation between GeneChip® and RT-PCR data for fibroblast clones. Data are log base 2 fold changes for each mutant/wild-type pair. RT-PCR values are average log expression levels from triplicate reactions for each clone. X and Y axes are log fold change values.
To assess the consistency of differential gene expression among a larger number of independently derived cell clones from the same individual, we selected five genes with large fold change values that had been identified by microarray analysis and confirmed by RT-PCR. Three genes with increased expression in mutant clones (MTAP44, MMP3 and NTN4) and two with decreased expression (CRIP1 and COMP) were quantitatively tested in multiple clonal cell strains from two Rett females (with MECP2 mutations 1155del32 and R106W) using RT-PCR. Expression levels varied significantly from clone to clone and between cell strains (Fig. 4). Fold change values for the multiple clone comparisons were mostly inconsistent with the original GeneChip/RT-PCR data. Furthermore, the 95% confidence interval for fold change included 1.0 (no change) in six of the ten comparisons (Fig. 5). Expression of CRIP1 was significantly different between wild-type and mutant clones for both Rett individuals tested (p = 0.004 and p = 0.035), however, the direction of change was different. Increased expression of only one gene, MTAP44, encoding the interferon-inducible microtubular aggregate protein 44, remained significant in comparison of the mutant and wild-type clones for both individuals tested (p = 0.027 and p = 0.01) (Fig. 5).
Figure 4.

Expression levels of five genes in additional fibroblast clones, not used for GeneChip studies, from two Rett individuals. Left: seven mutant and seven wild-type MeCP2 expressing clonal fibroblast cell strains with the 1155del32 mutation. Right: four mutant and four wild-type MeCP2 expressing clonal fibroblast cell strains with the R106W mutation. Expression values are raw numbers generated from RT-PCR and are for comparative purposes only.
Figure 5.

Ranges of fold changes, determined by quantitative RT-PCR for five genes, in multiple clonal cell strains from two Rett individuals (Statistical analysis of the data shown in Fig. 4). Dark bars represent comparisons of 7 wild-type and 7 mutant MeCP2 expressing clones from the 1155del32 mutation carrier. Open bars represent comparisons of 4 wild-type and 4 mutant MeCP2 expressing clones from the R106W mutation carrier. The lengths of the bars indicate the 95% confidence interval of the fold change in the log scale. The average estimated fold changes are identified by vertical lines (1.0 represents no change). P values are unpaired for the difference between the wild-type and mutant clones. Numbers at the bottom indicate log scale.
Oligonucleotide array hybridization data of lymphoblasts
The global gene expression pattern in fibroblasts is limited, and single cell cloning may amplify stochastic events in culture that may be responsible for the wide range of transcript levels in fibroblast clones. We, therefore, extended the microarray analyses to lymphoblastoid cell lines (LCLs) and studied four clonal mutant MeCP2 (T158M, 803delG, R168X and 1159del28) expressing and one clonal (1159del28) and four completely skewed (R106W, R255X, 803delG, 803delG) wild-type MeCP2 expressing LCLs. The percent "present" calls ranged from 13% (chip D) to 50% (chip A) and, as for the fibroblast data, standard deviations for the percent "present" calls for a given chip were 1–3%, and differences in percent "present" calls between wild type samples compared to mutant samples were not significant. When the expression profile of each mutant was compared to that of each wild-type LCL, 16 genes were found to be differentially expressed in at least 15 of 20 comparisons (Table 4). Eleven genes showed increased levels of expression, and 5 showed decreased expression. Of those 16 genes, 12 are annotated as known genes and 9 are expressed in brain. Quantitative RT-PCR was performed on four genes (three with increased and one with decreased expression) to assess the accuracy of GeneChip results. In a plot similar to that shown in Figure 3, the correlation of the combined RT-PCR fold change data with the GeneChip® log fold change data was 0.91 (data not shown).
Table 4.
Genes with increased and decreased levels of expression in at least 15 of 20 comparisonsbetween 4 mutant MeCP2 expressing LCLs and 5 wild type MeCP2 expressing LCLs
| INCREASED | ||||
| GenBank Accession | Expression In Brain | Gene Symbol/UniGene No. | Gene Name | Function |
| X15306 | x | NF-H | Neurofilament H | intermediate filament |
| J03600 | x | ALOX5 | Arachidonate 5-lipoxygenase | synthesis of leukotrienes |
| M38690 | CD9 | CD9 antigen (p24) | transmembrane protein | |
| J04076 | x | EGR2 | Early growth response 2 | transcription factor |
| AA873858 | x | H2BFD | H2B histone family, member D | compaction of DNA |
| M81750 | MNDA | Myeloid cell nuclear differentiation antigen | cell specific response to interferon | |
| S81914 | IER3 | Immediate early response 3 | inhibits apoptosis | |
| M33197 | x | GAPDH | Glyceraldehyde-3-phosphate dehydrogenase | carbohydrate metabolism |
| AI761588 | Hs.359719 | |||
| AF052145 | x | Hs.159528 | ||
| AB020695 | x | Hs.91662 | ||
| DECREASED | ||||
| GenBank Accession | Expression In Brain | Gene Symbol/UniGene No. | Gene Name | Function |
| L08177 | EBI2 | Epstein-Barr virus induced gene 2 | G-protein coupled receptor | |
| AW026672 | TXNIP | Thioredoxin interacting protein | upregulated by 1,25-dihydroxyvitamin D-3 | |
| AI239716 | Hs.344976 | Chloride intracellular channel 2 proteina | anion transport | |
| AI290954 | x | Hs.110080 | Alpha-2-macroglobina | cell growth |
| AI431931 | x | Hs.128797 | ||
aIdentified in Celera database
Discussion
Random X inactivation patterns are characteristic for Rett syndrome that is caused by heterozygosity for mutations in the X-linked locus MECP2. Our study design was intended to take advantage of the mosaic X chromosome inactivation in cell strains of Rett syndrome females. From four original fibroblast samples, we generated clonally derived cell strains that expressed only the mutant MeCP2 or only the wild-type MECP2 allele, but not both. We hypothesized that the effects of MeCP2 dysfunction in the mutant clonal cell strain would be more pronounced than in the original cell strain with mosaic X chromosome inactivation. Moreover, we were able to establish the ideal matched control for each clonal mutant cell strain, a clonal cell strain with the wild-type MECP2 allele on the active X chromosome from the same individual. Comparative transcription profiling of matched pairs of fibroblast clones should eliminate any noise contributed by polymorphisms or mutations of autosomal genes.
Although MeCP2 expression is higher in other tissues such as brain, spleen and lung [16], the value gained by the matched pair comparisons of clonal skin fibroblast strains cultured under identical environmental conditions, should overcome the drawbacks of evaluating a tissue with a lower level of expression. Previously reported microarray studies of female Rett and unrelated control brain samples revealed expression changes that were interpreted as secondary to the disease process, such as increased expression of glial-specific genes and decreased expression of neuronal genes [25]. Therefore, when searching for the primary targets of MeCP2 modulation, it might be advantageous to study a tissue that is less obviously involved in the pathologic processes of the disease.
To identify potential target genes of MeCP2 regulation, we looked for genes with increased transcript levels in at least three out of four mutant/wild-type MECP2 expressing fibroblast comparisons. Similarly, genes with decreased expression in at least three out of four matched pair comparisons were also considered potential targets of MeCP2 regulation. If the calls made did not reflect real increases or decreases, we would expect expression level increases and decreases to be scattered randomly throughout the 63,175 transcripts interrogated. By random chance alone, we would expect to see fewer than four genes increased in three of four comparisons and fewer than one (0.03 genes) increased in four of four comparisons. The number of differentially regulated genes identified by these criteria, 49 with increased and 21 with decreased expression, is significantly higher than expected by random chance, but it is much lower than one would expect if MeCP2 had a crucial role as transcriptional repressor of all genes with methylated promoters.
To validate the microarray expression data with an independent method, we carried out real-time quantitative RT-PCR analyses for a sub-set of 11 genes on all 8 samples that were used on the GeneChips®. The results were amazingly consistent for fold change and direction of change in almost all instances. We, therefore, chose to use quantitative RT-PCR alone to assess whether the increased or decreased expression patterns observed in the initial four matched pairs of clones were consistent in other clones and, therefore, might be biologically meaningful. Much to our surprise, expression levels of 5 genes in 11 additional MeCP2-deficient and 11 additional MeCP2-positive clones from two Rett individuals, one with a missense and one with a truncating mutation, showed tremendous variation. In the multiple clone comparisons, the fold change values trended away from the significant fold changes seen on the GeneChips® and RT-PCR of the original matched pair comparisons toward 1.0 (no change). CRIP1 expression generated significant p values, but the direction of change differed in the two individuals tested. In the truncating mutation 1155del32, expression was decreased and in the missense mutation R106W, it was increased. Although individual background genetic differences between the two Rett females might be responsible for these results, the possibility that the opposite type changes are mutation-type specific needs to be evaluated by studies of clones from additional patients.
Only MTAP44, encoding the interferon-inducible microtubular aggregate protein 44, remained significant in comparison of the mutant and wild-type clones, with average increases of 3.12 and 5.58 fold, for the two individuals tested. The product of this gene was first identified by a monoclonal antibody against microtubular aggregates in hepatocytes of chimpanzees infected with hepatitis C virus [26]. The production of p44 mRNA is markedly induced in the liver of chimpanzees infected with hepatitis C or hepatitis D virus [27]. Inducibility by interferon-alpha/beta, but not by interferon-gamma, is mediated by an interferon-stimulated response element in the promoter region [27]. The gene was independently identified in a search for novel interferon-inducible genes in the mouse (Dron, M., Meritet, J.F. and Tovey, M.G. unpublished, GenBank # AJ299405). Also known as IFI44 (for Interferon-inducible protein 44), the human gene is located on chromosome 1p31.1. Although predominantly expressed in uterus and other internal organs, transcripts have been isolated from brain and hypothalamus cDNA libraries (see dbEST). The putative function of this protein in the brain is still a mystery.
A different design was used for the microarray study of Rett syndrome LCLs. We initiated the LCL study because MeCP2 protein expression was higher in lymphoblasts than in fibroblasts, and LCLs have an overlapping expression pattern that includes genes not expressed in fibroblasts. Because of skewed X inactivation in the available LCLs, however, we were only able to obtain one matched pair of mutant versus wild-type expressing clones (for 1159del28). For the 20 different comparisons, we included three mutant-allele expressing clones (T158M, 803delG, R168X) and four completely skewed wild-type MeCP2 expressing LCLs (R106W, R255X, 803delG, 803delG) from different donors. Nevertheless, only 16 differentially expressed genes were identified, with a sub-set confirmed by quantitative RT-PCR. None of these genes were identical to any candidates found in the fibroblast study. Gene expression patterns in primary fibroblasts and transformed lymphoblasts are different and/or may respond differently to MeCP2 deficiency. Several of the candidate targets identified in lymphoblasts that are also expressed in brain (Table 4) may warrant further evaluation.
While our initial matched fibroblast clonal comparisons identified numerous genes with consistent results that could be targets of MeCP2 modulation, verification of a small subset of these data by comparing additional clones not used on the GeneChip® analysis revealed amazing inconsistency. The variability of gene expression across clonal cell strains with the same genotype and the same X chromosome active was similar among clones expressing the mutant or the wild-type MECP2 allele. Therefore, it is not likely to result from MeCP2 dysfunction and we can exclude the possibility that MECP2 mutations lead to transcript instability. More likely, fibroblasts in culture undergo random modification of gene expression, by somatic mutations or epigenetic regulation, and such rare events may be amplified in single-cell derived clones. In uncloned fibroblast populations, RNA samples from replicate cultures revealed consistent results in comparative large-scale gene expression studies [28]. Cell lines, i.e. transformed, immortalized or tumor-derived cell lines, are well known to be heteroploid i.e. chromosomally unstable, and any single-cell derived clones of such lines are genetically heterogeneous. Therefore, some differences in transcript levels among such clones would not be surprising. There are almost no reports in the literature, however, on comparative gene expression studies in clones of primary fibroblasts. One of the reasons may be that it is difficult to generate such clones. In 1981, Griffin et al [29] reported variation in the activity of steroid 5alpha reductase, the enzyme that converts testosterone to dihydrotestosterone, in cloned human skin fibroblasts. Activity of this enzyme is low in non-genital skin but high in genital skin cells that grow in response to androgen. Foreskin-derived fibroblast clones varied greatly (up to 200 fold) in enzyme activity. The range of activity for other microsomal enzymes was much lower in the same fibroblast clones. These authors did not study mRNA levels but reductase activity, therefore, variation at the level of protein stability could also play a role.
Our data could be interpreted to suggest that microarray experiments should be performed on greater numbers of samples and/or samples from mutant and wild-type brain to generate meaningful data. It is instructive, however, that Tudor and colleagues compared global gene expression in a large number of wild-type and Mecp2-null male mice and failed to identify striking changes in gene expression in the brain [30].
Conclusions
Both fibroblast and lymphoblast data together agree in the most important positive result of our studies: MeCP2 deficiency does not result in global deregulation of gene expression. This result is novel and unexpected in light of the purported function of MeCP2 as a global transcriptional regulator that was based on in vitro biochemical studies. There are several possibilities that might explain our findings. First, redundancy in methyl-CpG binding proteins that recruit transcriptional silencing complexes may mask the effects of MeCP2 deficiency; for example, MBD1, MBD2 or Kaiso, may complement the defect in the cell types we studied. Second, if the inter-clonal variation we detected by the quantitative RT-PCR study of 5 genes in 22 clones from two different Rett individuals were representative of a general feature of diploid fibroblast clones, it could have masked any consistent biologically meaningful expression changes. If our limited findings were confirmed by larger-scale studies, we would need to conclude that clonal fibroblast strains are not a suitable resource for genome-wide expression profiling studies. Third, deregulation of very few genes expressed only in parts of the brain, or only under certain conditions, may be responsible for the neuronal abnormalities in Rett brains, and this would not be detected by microarray studies. Fourth, MeCP2 action on chromatin, as suggested by observed changes in histone acetylation [31,32], may not involve protein coding genes that are represented on microarrays, but instead, may affect centromeric heterochromatin or the expression of small non-coding RNAs that are abundant in normal brain [33]. The results presented in this paper contradict the established model of MeCP2 function and necessitate a rethinking of strategies to unravel the pathologic process that leads to neuronal dysfunction in Rett syndrome.
List of abbreviations
cDNA complementary deoxyribonucleic acid
DNA deoxyribonucleic acid
EST expressed sequence tag
mRNA messenger ribonucleic acid
PCR polymerase chain reaction
μl microliter
μm micrometer
ml milliliter
mm millimeter
mM millimolar
ng nanogram
nM nanomolar
RNA ribonucleic acid
RT reverse transcription
RT-PCR reverse transcription – polymerase chain reaction
U unit
IVT in-vitro transcription
Competing interests
None declared.
Authors' contributions
JT carried out the cell culture, single cell cloning, nucleic acid extraction, X chromosome inactivation studies, mutation analysis, sample preparation for array hybridization, microarray analysis, real-time quantitative PCR and some of the statistical analysis, and drafted the manuscript. PA performed the immunoblot analysis. LL performed the most of the statistical analysis. UF conceived the study design, supervised and coordinated its progress and prepared the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
The authors acknowledge helpful advice from Mimi Wan, Stephen S.J. Lee and other members of the Francke lab. The work was supported by the Howard Hughes Medical Institute, research grants from the NINDS and the March of Dimes Birth Defects Foundation (UF), from NIGMS (LL) and the institutional postdoctoral training grant T32 GM08748 (JT).
Contributor Information
Jeff Traynor, Email: jtraynor@stanford.edu.
Priyanka Agarwal, Email: pagarwal@stanford.edu.
Laura Lazzeroni, Email: lazzeroni@stanford.edu.
Uta Francke, Email: francke@cmgm.stanford.edu.
References
- Hagberg B, Hagberg G. Rett syndrome: epidemiology and geographical variability. Eur Child Adolesc Psychiatry. 1997;6:5–7. [PubMed] [Google Scholar]
- Rett A. Uber ein eigenartiges hirnatrophisches Syndrom bei Hyperammonamie im Kindesalter. Wien Med Wochenschr. 1966;116:723–738. [PubMed] [Google Scholar]
- Trevathan E, Moser HW, Opitz JM, Percy AK, Naidu S, Holm VA, Boring CC, RS J, Yeargrin-Allsopp M, Adams MJ. Diagnostic criteria for Rett syndrome. Ann Neurol. 1988;23:425–428. [Google Scholar]
- Schanen NC, Dahle EJ, Capozzoli F, Holm VA, Zoghbi HY, Francke U. A new Rett syndrome family consistent with X-linked inheritance expands the X chromosome exclusion map. Am J Hum Genet. 1997;61:634–641. doi: 10.1086/515525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schanen NC, Francke U. A severely affected male born into a Rett syndrome kindred supports X-linked inheritance and allows extension of the exclusion map. Am J Hum Genet. 1998;63:267–269. doi: 10.1086/301932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirianni N, Naidu S, Pereira J, Pillotto RF, Hoffman EP. Rett syndrome: confirmation of X-linked dominant inheritance, and localization of the gene to Xq28. Am J Hum Genet. 1998;63:1552–1558. doi: 10.1086/302105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, Carrie A, de Roux N, Vinet MC, Jonveaux P, Couvert P, Villard L, Arzimanoglou A, Beldjord C, Fontes M, Tardieu M, Chelly J. MECP2 mutations account for most cases of typical forms of Rett syndrome. Hum Mol Genet. 2000;9:1377–1384. doi: 10.1093/hmg/9.9.1377. [DOI] [PubMed] [Google Scholar]
- Huppke P, Laccone F, Kramer N, Engel W, Hanefeld F. Rett syndrome: analysis of MECP2 and clinical characterization of 31 patients. Hum Mol Genet. 2000;9:1369–1375. doi: 10.1093/hmg/9.9.1369. [DOI] [PubMed] [Google Scholar]
- Lee SS, Wan M, Francke U. Spectrum of MECP2 mutations in Rett syndrome. Brain Dev. 2001;23:S138–143. doi: 10.1016/s0387-7604(01)00339-4. [DOI] [PubMed] [Google Scholar]
- Nan X, Meehan RR, Bird A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993;21:4886–4892. doi: 10.1093/nar/21.21.4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nature Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- Coy JF, Sedlacek Z, Bachner D, Delius H, Poustka A. A complex pattern of evolutionary conservation and alternative polyadenylation within the long 3'-untranslated region of the methyl-CpG-binding protein 2 gene (MeCP2) suggests a regulatory role in gene expression. Hum Mol Genet. 1999;8:1253–1262. doi: 10.1093/hmg/8.7.1253. [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. 2002;11:115–124. doi: 10.1093/hmg/11.2.115. [DOI] [PubMed] [Google Scholar]
- Holm VA. Physical growth and development in patients with Rett syndrome. Am J Med Genet Suppl. 1986;1:119–126. doi: 10.1002/ajmg.1320250513. [DOI] [PubMed] [Google Scholar]
- Ellis KJ, Shypailo RJ, Hardin DS, Perez MD, Motil KJ, Wong WW, Abrams SA. Z score prediction model for assessment of bone mineral content in pediatric diseases. J Bone Miner Res. 2001;16:1658–1664. doi: 10.1359/jbmr.2001.16.9.1658. [DOI] [PubMed] [Google Scholar]
- Hendrich B, Guy J, Ramsahoye B, Wilson VA, Bird A. Closely related proteins MBD2 and MBD3 play distinctive but interacting roles in mouse development. Genes Dev. 2001;15:710–723. doi: 10.1101/gad.194101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokhortchouk A, Hendrich B, Jorgensen H, Ruzov A, Wilm M, Georgiev G, Bird A, Prokhortchouk E. The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. 2001;15:1613–1618. doi: 10.1101/gad.198501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet. 1992;51:1229–1239. [PMC free article] [PubMed] [Google Scholar]
- Pegoraro E, Schimke RN, Arahata K, Hayashi Y, Stern H, Marks H, Glasberg MR, Carroll JE, Taber JW, Wessel HB, et al. Detection of new paternal dystrophin gene mutations in isolated cases of dystrophinopathy in females. Am J Hum Genet. 1994;54:989–1003. [PMC free article] [PubMed] [Google Scholar]
- Wan M, Lee SS, Zhang X, Houwink-Manville I, Song HR, Amir RE, Budden S, Naidu S, Pereira JL, Lo IF, Zoghbi HY, Schanen NC, Francke U. Rett syndrome and beyond: recurrent spontaneous and familial MECP2 mutations at CpG hotpots. Am J Hum Genet. 1999;65:1520–1529. doi: 10.1086/302690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- Colantuoni C, Jeon OH, Hyder K, Chenchik A, Khimani AH, Narayanan V, Hoffman EP, Kaufmann WE, Naidu S, Pevsner J. Gene expression profiling in postmortem Rett Syndrome brain: differential gene expression and patient classification. 2001;8:847–865. doi: 10.1006/nbdi.2001.0428. [DOI] [PubMed] [Google Scholar]
- Maeda T, Honda Y, Hanawa M, Yamada E, Ono Y, Shikata T, Shimizu YK. Production of antibodies directed against microtubular aggregates in hepatocytes of chimpanzees with non-A, non-B hepatitis. J Gen Virol. 1989;70:1401–1407. doi: 10.1099/0022-1317-70-6-1401. [DOI] [PubMed] [Google Scholar]
- Kitamura A, Takahashi K, Okajima A, Kitamura N. Induction of the human gene for p44, a hepatitis-C-associated microtubular aggregate protein, by interferon-alpha/beta. Eur J Biochem. 1994;224:877–883. doi: 10.1111/j.1432-1033.1994.00877.x. [DOI] [PubMed] [Google Scholar]
- Novak JP, Sladek R, Hudson TJ. Characterization of variability in large-scale gene expression data: implications for study design. Genomics. 2002;79:104–113. doi: 10.1006/geno.2001.6675. [DOI] [PubMed] [Google Scholar]
- Griffin JE, Allman DR, Durrant JL, Wilson JD. Variation in steroid 5 alpha-reductase activity in cloned human skin fibroblasts. Shift in phenotypic expression from high to low activity upon subcloning. J Biol Chem. 1981;256:3662–3666. [PubMed] [Google Scholar]
- Tudor M, Chen RZH, Akbarian S, Jaenisch R. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci (USA) 2002. [DOI] [PMC free article] [PubMed]
- Wan M, Zhao K, Lee SS, Song HR, Francke U. MECP2 truncating mutations cause histone H4 hyperacetylation in Rett syndrome. Hum Mol Genet. 2001;10:1085–1092. doi: 10.1093/hmg/10.10.1085. [DOI] [PubMed] [Google Scholar]
- Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- Kiss T. Small nucleolar RNAs: an abundant group of noncoding RNAs with diverse cellular functions. Cell. 2002;109:145–148. doi: 10.1016/s0092-8674(02)00718-3. [DOI] [PubMed] [Google Scholar]