

Abstract
Background
Recently, Almeida and Vinga offered a new approach for the representation of arbitrary discrete sequences, referred to as Universal Sequence Maps (USM), and discussed its applicability to genomic sequence analysis. Their work generalizes and extends Chaos Game Representation (CGR) of DNA for arbitrary discrete sequences.
Results
We have considered issues associated with the practical implementation of USMs and offer a variation on the algorithm that: 1) eliminates the overestimation of similar segment lengths, 2) permits the identification of arbitrarily long similar segments in the context of finite word length coordinate representations, 3) uses more computationally efficient operations, and 4) provides a simple conversion for recovering the USM coordinates. Computational performance comparisons and examples are provided.
Conclusions
We have shown that the desirable properties of the USM encoding of nucleotide sequences can be retained in a practical implementation of the algorithm. In addition, the proposed implementation enables determination of local sequence identity at increased speed.
Background
Attempts to develop new representations of biological sequences that facilitate analysis and comparison continue today. Representations that preserve the statistical properties and contextual information of the sequence would offer considerable value in the analysis of the enormous volume of genomic data being accumulated. In 1990, Jeffery [1] published a representation known as the Chaos Game Representation (CGR) that exploited iterative function systems to map nucleotide sequences into a continuous two dimensional space on the unit square. Properties of the CGR representation have been generalized and studied extensively [2,3].
Recently Almeida and Vinga [4] proposed an extension to this method, termed Universal Sequence Maps (USM), that provides a scale-independent method for representing and comparing any sequence of discrete units, which encompasses genomic, proteomic, and even linguistic information. As discussed in that report, scale independency in the context of sequence analysis corresponds to the ability to recognize the length of a re-occurring segment while comparing the representation of any of its analogous unit components. This property enables scale-free (e.g. order free) word statistics, the critical first step to recognize sequence conservation when overall sequence identity is too low for alignment. The application of USM to the representation of a sequence can be summarized in the following steps.
Step 1
Identify the unique symbols in the analyzed sequences. For a nucleotide sequence the unique symbols would be A, G, C, and T for the four nucleotides found in DNA sequences.
Step 2
Map each symbol to a unique corner in the unit hypercube. The dimension of the unit hypercube, n, is chosen as the upper integer of log2(uu) where uu is the number of unique symbols. Therefore n has the value of 2 for DNA and 5 for proteins. For each symbol mapped in such a way, let usj be the jth coordinate of the corner of the hypercube to which symbol s is mapped. For DNA one possible mapping is uA = [0,0], uC = [0,1], uG = [1,0], and uT = [1,1]. There are sparser implementations of USM that may use values of n up to the number of unique units, uu, as detailed in the original proposition [4]. However, those solutions are not essentially different and the implementation presented here can be straightforwardly ported to sparser USM representations.
Step 3
Iteratively generate the forward USM coordinates for each of the k symbols and each of the n coordinates as follows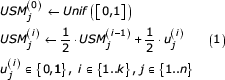
Step 4
Iteratively generate the backward USM coordinates for each symbol and each coordinate as follows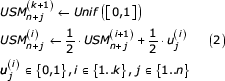
The given procedure results in the 2n USM coordinates for each of the k symbols in the transformed sequence. The similarity of two sequences at any pair of symbols can be measured using the distance measure defined by Almeida and Vinga [4]. The measure is defined by
D = df (aibj) + db (aibj) (3)
where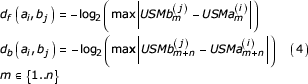
Almeida and Vinga present the distance measure as an estimator of the length of the similar segment containing the compared symbols. They also note that the measure, as defined, necessarily overestimates the length of these segments.
As can be seen in the USM procedure, each symbol in the sequence is encoded as a set of USM coordinates. These coordinates are constructed in such a way as to encode the symbol itself as well as the preceding symbols (forward coordinates) and following symbols (backward coordinates). Scale independence is a property of this representation that allows the complete recovery of the encompassing sequence (preceding and following symbols) from the USM coordinates of any symbol in the sequence to any resolution (any scale) up to the complete length of the sequence. In practical implementations we are faced with the limitations of finite word length representations of USM coordinates. In these implementations our ability to recover the encompassing sequence is bounded by the word length of the coordinate representation. For this reason, we refer to USM and bUSM implementations with finite precision coordinates as bounded scale independent representations. In this paper we consider the implementation of the USM algorithm and propose a modification to Almeida and Vinga's approach [4] that eliminates the overestimation and allows determination of similar segment lengths of bounded length and offer an algorithm for overcoming the bounded length restriction.
Results
Source of the USM distance metric over-estimation
The application of USM as a tool for measuring scale-independent discrete sequence similarity and its particular application to genomics and proteomics exploits a distance metric providing an estimate of the length of similar regions surrounding a pair of symbols. In the approach presented by Almeida and Vinga, this distance metric is shown to overestimate the true length of the similar segment. We propose a variation on their approach that retains the distance property, eliminates the over-estimation, and uses more computationally efficient operations. We begin this discussion by first providing a more complete proof of the distribution of overestimation in the unidirectional USM. This proof aids in the illumination of the source of the over-estimation.
The USM distance metric estimates the length of ungapped identical segments in the region surrounding the symbols being compared. As such, we now consider two sequences, V and W with k symbols in agreement starting at symbol indices mv and mw respectively.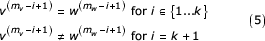
From the definition of the USM recursion (Equation 1) we can see that the jth coordinate at the kth step can also be written as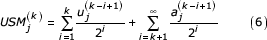
Where the 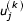 is the jth coordinate of the kth symbol and the
is the jth coordinate of the kth symbol and the 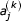 are determined by the initial values of the coordinates. These values are assigned in the initialization step of the USM encoding process. We write a coordinate of the sequence at the mvth and mwth step in the recursion as:
are determined by the initial values of the coordinates. These values are assigned in the initialization step of the USM encoding process. We write a coordinate of the sequence at the mvth and mwth step in the recursion as: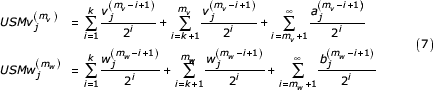
These representations are given as three summations corresponding to the k symbols in agreement, the symbols preceding the similar segment back to the beginning of each sequence, and the initial value of the coordinate. From our definition of these sequences (k most recent symbols in agreement) we see that that the first terms (first summation) in each of the expressions are equal. We can factor a common term from each of the remaining two summations giving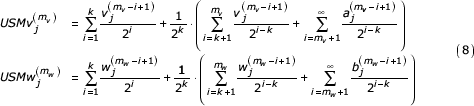
By change of index we get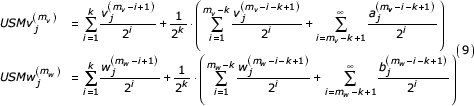
We let 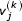 ,
, 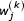 , and
, and 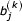 for the non-similar segment be independent Bernoulli random variables with p = 1/2. The term in brackets is recognized as a uniformly distributed random value on [0,1). We can rewrite USMv and USMw as follows
for the non-similar segment be independent Bernoulli random variables with p = 1/2. The term in brackets is recognized as a uniformly distributed random value on [0,1). We can rewrite USMv and USMw as follows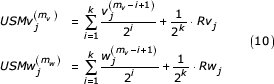
Where Rvj and Rwj are uniformly distributed on [0,1). Next consider the differences between the USM coordinates for each of these sequences.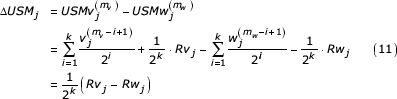
The terms in the similar segment are eliminated and the difference becomes a scaled difference between two uniformly distributed random variables. The scale factor of this difference gives the length of the similar segment.
The unidirectional distance metric given by Almeida and Vinga is defined as
d = -log2 (max|ΔUSMj|) for j = 1..n (12)
where n is the number of coordinates in the USM vector. Exploiting the fact that log is monotone increasing we substitute our expression for the USM difference and write d as: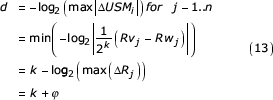
where ΔRj = |Rvj - Rwj|. The overestimation is given by φ. Since Rvj and Rwj are uniformly distributed on [0,1), the distribution of ΔRj is given by: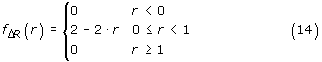
And the associated cumulative distribution is given by: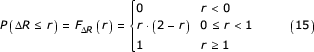
Therefore for n independent coordinates distributed as defined, the distribution for the maximum is
P(Rmax ≤ r) = P(ΔR1 ≤ r, ΔR2 ≤ r,...,ΔRn ≤ r) = (P(ΔR ≤ r))n = (r·(2 - r))n (16)
Under the transformation
φ(r) = -log2 (r) (17)
the distribution of φ can be determined as follows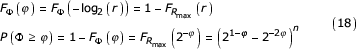
This confirms the result originally reported by Almeida and Vinga [4]. We see from this derivation that the exact length of the similar segment, given by k, is determined by the exponent of the common factor of 1/2 factored from the non-similar segment. The remaining factor in that term constitutes the overestimation. Overestimation is, therefore, determined by the difference between terms in the series representation of the maximum coordinate difference beyond the similar segment. The symbol sequence encoded in this portion of the coordinate provides no information as to the length of the similar segment and so we wish to eliminate its effect on the estimation of the similar length.
Boolean USMs
It is clear from the discussion above that overestimation of the length of similar segments by USM results from contributions to the coordinate difference from terms in the coordinate summation preceding the similar segment. This effect is due to the use of an arithmetic difference in the computation of Almeida and Vinga's distance metric. Consider the following example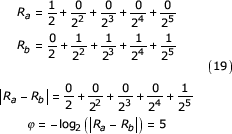
where Ra and Rb are the uniformly distributed random values on [0,1) described in the previous derivation. Finite length coordinates are used here for illustration purposes. These two quantities must differ in the most significant position but are not constrained in the remaining terms. In this example we see that for the least significant subtraction to occur, the difference must borrow from the next more significant term. The borrow propagates the length of the sum and the length is overestimated, in this case, by 5. The overestimation is therefore, due to the interaction of the symbols prior to the first symbol at which the sequences differ.
We propose a variation on the USM encoding and difference metric in which arithmetic operations (subtraction, maximum, and base 2 logarithm) are replaced with equivalent Boolean operations in which the values of individual terms in the above expression do not interact. We now present the proposed change of arithmetic.
Consider the following representation of a bUSM (Boolean USM) coordinate
c = 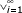 Riaiwhereai ∈ {0,1} (20)
Riaiwhereai ∈ {0,1} (20)
Where represents the bit-wise logical OR of a series of terms and Riis the right shift operator repeated i times. The value c is then an infinite bit representation that encodes one bit from each symbol in the encoded sequence. The bUSM recursion is then written as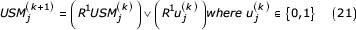
The representation of a coordinate of the USM after the kth step of the recursion is given by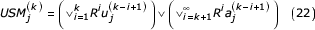
Again, consider two sequences, V and W, as defined previously. We write the coordinates of these sequences at the mvth and mwth step in the recursion as: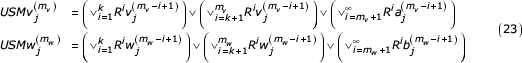
As before, we break the representation into three terms; one representing the similar segment, one representing terms prior to the similar segment, and one representing the initial value of the coordinate. The proposed recursion replaces division by 2 with a right shift operation and addition with a bit-wise logical OR. Resulting coordinates are histories of one bit of the symbols preceding the encoded symbol with the most recent symbol's bit stored in the left-most bit position (most significant position).
Given the proposed coordinate representation and recursion, we now examine the bit-wise binary equivalent to the computation of the distance metric. Consider the exclusive OR of the coordinates of two symbols being compared.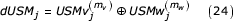
The exclusive OR operation yields true (bit is set) if the bits differ and false (bit is not set) otherwise. Based on our definition of sequences V and W none of the bits in the similar segment of the newly defined bUSM coordinate difference are set and the first bit beyond the similar segment must be set. The exact length of the similar segment is given by one less than the position of the left-most set bit in the set of coordinate differences.
Under the original approach, the maximum of the differences across all coordinates is taken prior to computing the base 2 logarithm. Under the proposed approach we replace this operation with the bit-wise OR of the differences across all coordinates. The left-most bit set in the result corresponds to the bUSM coordinate that determines the length of the similar segment (equivalent to the coordinate winning the max operation in the standard USM). The computation of the distance metric in the original approach employs a base-2 logarithm. Under the proposed approach we substitute the logarithm with a scan for the position of the most significant bit set in the bit-wise OR of the coordinate differences. By forming both the forward and reverse bUSM coordinates and adding the forward and backward distances, the exact length of the similar segment can be determined for all pairs within the segment.
For the standard USM, initial values for coordinates are taken as random draws on [0,1). This allows the statistical properties of the overestimation to remain consistent at the beginning and end of the sequences. The Boolean USM does not overestimate the similar length and we must, therefore, reexamine the initialization approach so as to preserve the determination of exact lengths at the beginning and end of the sequences. This can be accomplished through the addition of two unique symbols, a tail symbol for sequence V and a tail symbol for sequence W. The use of two special symbols to mark the ends of the sequences results in no change to the recursion or similar segment length determination. It does impact the initialization and possibly the computational costs due to a potential increase in the number of coordinates required to represent the alphabet and the tail symbols. The addition of two extra symbols will, in some cases, increase the number of coordinates required. If, for example, an alphabet of 4 is required, the addition of two symbols increases the number of coordinates from 2 to 3. If, however, an alphabet of 20 is required, the addition of two symbols results in no increase in the number of coordinates. Naturally, this would not ever happen for a sparse implementation of USM, where n = uu[4], and, consequently, the number of coordinates increases with every new symbol. Nevertheless, this would not, in any way, change the bUSM proposition as the sole difference between sparse and compact representations concerns the binary representation of each symbol, uj = 1,...,uu. It is noteworthy, however, that the incentive to use sparser USM representations, which is the smaller extent of over-determination, does not exist for bUSM, where determination of length unit identity is exact, as shown below.
The initial value of the Boolean USM coordinates for sequence V are set to indicate that an occurrence of the sequence V tail symbol precedes the first symbol in V. An instance of the tail symbol is also added to the end of the sequence (follows the last symbol in V). The initial value for sequence W's coordinates are set similarly using the sequence W tail symbol. Since tail symbols differ from each other and from all non-tail symbols, similar regions will be terminated at the beginning and end of the sequence and exact distances will be determined as required.
Both forms of the USM coordinates can be considered a form of embedding and reorientation of the sequence data as illustrated in Figure 1. Instead of coding the information as a sequence of symbol codes, we code it as a collection of coordinates containing one code bit for each symbol in the sequence. Each USM coordinate stores one bit for each symbol preceding (forward coordinates) or following (backward coordinates) the symbol associated with the coordinate. The sequence of coordinates redundantly embeds the symbols surrounding the current symbol.
Figure 1.

Comparison of encodings for the original sequence, an embedded representation and USM coordinates. Sample encodings for a nucleotide sequence illustrating the equivalence between an embedded encoding and a finite word length, block floating point representation of a standard USM encoding. The values indicated as ii,j represent the initial values of the USM coordinates and the subscripted A, G, T, and C indicate the 0th and 1st bits of the 2-bit USM representation of the associated coordinates.
The standard USM coordinates can be directly obtained from the bUSM form by interpreting the bUSM coordinates as block floating point representations of the USM coordinates with the binary decimal point set to the left of the most significant bit. Dividing the unsigned word representation of the bUSM coordinate by 2W, where W is the word length, yields the equivalent standard USM coordinate with W symbols of precision. We also recognize that for both the standard and Boolean USM coordinates, the determination of similar segment lengths is limited by the length (or precision) of the word used to represent the coordinate. The original implementation of the standard USM was created in Matlab and used 64-bit floating point coordinates. As such, lengths for similar segments longer than 53 symbols (IEEE 754 format provides 53 bits of precision [5]) cannot be determined. The bUSM coordinates are similarly limited. The comparable implementation encodes bUSM coordinates in 64-bit fixed point representations and so exact similar segment lengths up to a maximum length of 63 can be determined.
Overcoming finite word length limitations
In theory, USM encoded sequences could be used to detect arbitrarily long similar segments. Previously we discuss the constraint imposed by finite-length binary representations of USM and bUSM coordinates. An algorithm for overcoming this limitation will now be presented. First we define the following two functions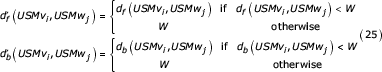
Under the finite word length limitation we note that df and db, the true forward and backward distances, cannot be determined from a single symbol pair comparison. We define two new functions  and
and 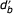 which can be determined from W-bit coordinate representations. These functions provide the length of similar segments up to the length of the underlying representation W. The true length of the forward and backward similar segments are returned for lengths less than W and the word length is returned otherwise. Next we present the following recursive functions
which can be determined from W-bit coordinate representations. These functions provide the length of similar segments up to the length of the underlying representation W. The true length of the forward and backward similar segments are returned for lengths less than W and the word length is returned otherwise. Next we present the following recursive functions
The functions Df and Db recursively locate the end of the similar segment by stepping backward (Df) and forward (Db) through the similar segment until the end of the region is detected. The exact forward and backward lengths of similar segments of arbitrary length can be determined from these recursions. If the similar segment extends to the end of the sequence, the recursion will terminate on the last step due to the tail symbols added to the beginning and end of the sequence. The exact length of the similar segment containing symbols vi and wj is given by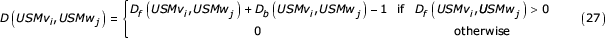
The proposed recursive definition of similar segment length overcomes the finite word length limitation and provides a practical method for recovering exact distances for arbitrarily long similar segments.
Performance comparisons
Computational comparisons of the two approaches were performed using the C-code implementations developed as described in Methods. This comparison examines the performance gains achieved through the use of binary operations (shift, exclusive OR, OR, bit scan) as replacements for the equivalent functions in the standard USM (division by 2, subtraction, max, and log). Since the Boolean USM requires two additional symbols to represent the tail symbols for each of the sequences, three coordinates are required for each of the forward and reverse directions (six total). Four coordinates were required for the standard USM. Standard USM coordinates were stored as 64 bit floating point numbers and bUSM coordinates as 64 bit unsigned fixed point numbers. The standard and binary USM implementations were, therefore, limited to detecting similar segments up to lengths 53 and 63 respectively in a single comparison (not using the recursive distance algorithm).
Elapsed execution time measurements were made for each of the 10 test cases and are presented in Table 1. For small sequence lengths (less than 10,000 symbols) the symbol-pair distance calculations for the binary USM achieved approximately 8.3 M comparisons per second on our test platform while the standard USM achieved rates of approximately 2.3 M comparisons per second. The binary USM approach was approximately 3.7 times the speed of the standard USM. For these small sequence test cases, the test application and the USM representations of the small sequences fit within the processor's on-chip cache (512 kilobytes). For cases where the USM encoded sequences exceed the cache size and require reads from main memory, the performance decreases as indicated for the larger cases (10,000 symbols and above). For these cases, the binary USM executes at 3.3 M comparisons per second and the standard USM at 1.7 M comparisons per second for a performance ratio of approximately 1.9.
Table 1.
Execution time performance for standard and Boolean USM implementations.
| Length of Sequence A |
Length of Sequence B |
Total Time (Boolean USM) |
Distance Compute Time (Boolean USM) |
Total Time (standard USM) |
Distance Compute Time (standard USM) |
Rate (Boolean USM distance calculations per second) |
Rate (standard USM distance calculations per second) |
Speed Ratio (Boolean to standard) |
Memory (kB) |
|---|---|---|---|---|---|---|---|---|---|
| 1,000 | 1,000 | 0.12 | 0.12 | 0.44 | 0.44 | 8,333,333 | 2,272,727 | 3.67 | 94 |
| 2,000 | 2,000 | 0.48 | 0.48 | 1.78 | 1.77 | 8,333,333 | 2,244,669 | 3.71 | 188 |
| 3,000 | 3,000 | 1.09 | 1.09 | 3.98 | 3.98 | 8,249,313 | 2,264,151 | 3.64 | 281 |
| 4,000 | 4,000 | 1.92 | 1.92 | 7.08 | 7.08 | 8,324,662 | 2,259,887 | 3.68 | 375 |
| 5,000 | 5,000 | 3.04 | 3.03 | 11.07 | 11.07 | 8,212,878 | 2,259,376 | 3.64 | 469 |
| 10,000 | 10,000 | 30.53 | 30.52 | 58.17 | 58.16 | 3,275,145 | 1,719,011 | 1.91 | 938 |
| 15,000 | 15,000 | 68.68 | 68.67 | 131.01 | 131.00 | 3,276,158 | 1,717,452 | 1.91 | 1,406 |
| 17,000 | 17,000 | 88.23 | 88.21 | 168.29 | 168.28 | 3,275,678 | 1,717,253 | 1.91 | 1,594 |
| 20,000 | 20,000 | 122.09 | 122.07 | 233.00 | 232.99 | 3,276,406 | 1,716,775 | 1.91 | 1,875 |
| 40,000 | 40,000 | 488.46 | 488.43 | 931.74 | 931.71 | 3,275,587 | 1,717,219 | 1.91 | 3,750 |
Results of performance comparisons of standard USM and Boolean USM implementations in C (gcc 2.95.3, cygwin, Windows 2000, PIII 1 GHz). Sequence lengths are given in nucleotides. Times measure elapsed execution time in seconds. Total times include both USM sequence preparation time and distance calculations for all symbol pairs. Memory is measured in kilobytes and represents the space required to store the USM coordinates for both sequences.
Two examples applying both the standard and binary USM approaches were prepared to illustrate the difference in results obtained from overestimated distance and exact similar segment length determination. The first case duplicates the example given by Almeida and Vinga. Phrases from the Wendy Cope poem were converted to standard and binary USM for each of the sequences. The pair-wise comparisons were performed using both approaches and the results are shown as pixel maps (Figure 2). In these pixel maps, brighter regions indicate higher distance values and should correspond to symbol pairs found in similar segments. It is clear from the images in this figure that the major similar segments (lengths 7, 9, and 11) are clearly visible in both images. However, the exact distances in the Boolean USM image clearly show the shorter segments (lengths 3, 4, and 5) that are somewhat hidden by the standard USM overestimation error (Figure 2A). A similar illustration is provided using a sample nucleotide sequence. The sequence coding the human insulin receptor was acquired through NCBI (XM_048346, INSR) and used in a BLAST search for similar sequences. The second sequence (M69243, CTK-1) was taken from that list. A 100 nucleotide segment of the of the human insulin receptor (XM_048346, 3056–3155) associated with the predicted tyrosine kinase domain and a 100 nucleotide segment from the chicken tyrosine kinase (M69243, 51–150) were converted to standard USM and bUSM coordinates and pair-wise compared using the associated distance metrics. Pixel maps of the distance metrics were prepared (Figure 3). Again, the long similar segments are clearly visible in both images. The overestimation noise in the standard USM (image A) again masks many of the shorter similar segments seen in the Boolean USM (image B).
Figure 2.

Comparison of standard and Boolean USM similar segment length measurements. Pixel images of bi-directional distance determination for standard (A) and Boolean (B) USM implementations. Brighter pixels indicate longer similar segments.
Figure 3.

Comparison of standard and Boolean USM length measurements for sample nucleotide sequences. Pixel images of bi-directional distance determination for standard (A) and Boolean (B) USM implementations. The sequences are 100 nucleotide segments from the human insulin receptor (INSR) and a chicken tyrosine kinase (CTK-1). Brighter pixels correspond to longer similar segments. The dominant segment is an exact match that is 17 nucleotides long.
Discussion
Almeida and Vinga presented a fundamentally interesting and practically useful extension of the Chaos Game Representation iterative function (CGR) referred to as Universal Sequence Maps (USM) and demonstrated the application of this representation and an associated distance metric in the identification of similar segments of discrete sequences. In this report we have presented considerations for the practical implementation of these methods and offer an implementation of USM that 1) eliminates the overestimation of the length of similar segments, 2) eliminates the inability to recognize similar segments longer than the word length of the coordinate representation, 3) can be implemented with more efficient operations, and 4) provides a simple conversion that recovers the standard USM coordinates. As currently defined, the USM distance metric (and associated bUSM implementation) is limited to the estimation of lengths of local identity about the pair of symbols being compared.
The nature of the overestimation by the unidirectional distance metric was revealed in a proof of the distribution of the overestimation from the standard method. The algebraic difference taken in computing the distance results in the overestimation of length. This observation leads to a modification of the algorithm that eliminates the interaction of symbols when computing distance. We also recognize, in this derivation, that to achieve this result we must assume that the ith coordinates for the symbol representations are equally likely (p = 1/2). The symbol coding selections for standard USM sequences must be balanced so as to make the occurrence of 1 and 0 in a standard USM coordinate equally likely for the given symbol set (and frequency of occurrence). This indicates that instead of choosing the first n binary representations of symbols as suggested in [4], the symbols should be chosen from the 2n possible values in such a way as to balance the occurrences of 1 and 0 for each coordinate. The Boolean USM approach places no constraints other than uniqueness on the symbol representation.
The Boolean USM approach eliminates the overestimation problem noted in the standard USM and can recover more symbols than the standard approach for a given amount of storage. The binary approach is faster than the standard approach (based on a straightforward implementation) and offers the potential for further enhancement through, for example, the use of processor instructions designed specifically to find the first or last bit set in a word (e.g. Pentium Bit Scan Reverse (BSR) instruction [6]). In our test cases the binary approach performed 1.9 to 3.7 times that of the standard approach even though it processed 6 (3 forward, 3 backward) rather than 4 (2 forward, 2 backward) coordinates. These measurements are specific to the test platform (processor, OS, compiler, etc.) and with optimizations these ratios will change considerably.
The computational cost of preparing the coordinates is insignificant when compared to the cost of the distance calculations in cases we examined. This may, in part, be due to the fact that we performed N × M comparisons for sequences of length N and M and with a minimum value for N or M of 1000. Since the storage requirements for a USM representation of a sequence are considerably larger than that of the sequence itself, it may be more efficient to store the sequence in its native form (a sequence of symbols) and compute the USM coordinates just prior to sequence comparison when a large number of comparisons are being performed. We also observed that the storage size of the coordinates had an impact on computational performance. The performance of the algorithm decreases sharply when the USM coordinate representation exceeds the on-chip cache of the processor. This may indicate that we should be considering tradeoffs between word length and the size of the similar segments we wish to detect without using the recursive length function. If, for example, the probability of sequences longer than 16 symbols is sufficiently small, then a 16 bit coordinate might be used.
A single pairwise comparison grows as the upper integer of the base 2 logarithm of the number of unique symbols. Almeida and Vinga note that for a given length of interest w, we need to sample the distance metric at no more than NA·NB/w indices to locate all sequences of length w or greater. In an application focused on locating all identical segments of length w or greater, the computational cost therefore grows as [log2(nn)]·(NA·NB)/w. The exact distance given in the binary approach allows us to locate, from this sampling, the beginning and end of the similar segment from the sampled symbol indices and the forward and reverse distances. Subtracting one less than the exact forward distance from the indices of the symbols being compared and adding one less than the exact backward distance from the indices we can identify the start and end of the similar segment containing the given symbol pair. The Boolean USM, offers this advantage due to its unique ability to determine the exact length of the similar segment.
Conclusions
USM representations of discrete sequences in genomics and proteomics offer the possibility of scale-independent representations of sequence information surrounding points of comparison in those sequences. In order to maximize the potential for application of these methods we must consider both their theoretical properties and the computational methods for efficient implementation that retain these properties. We have offered one further step in that direction by identifying a Boolean implementation of USM (bUSM) that not only preserves the theoretical properties of numerical USM but actually uses the binary environment of the computational implementation to achieve a more exact logical solution. The proposed implementation leads to a distance metric that exactly determines similarity length between sequences. Ultimately, this achievement can be described as one that replaces the determination of logarithmic, numerical distance, with computationally more efficient logic operations. It could then be argued that, given the discrete nature of biological sequences, new scale-independent numerical representations, such as USM, are all but the first step in the identification of more accurate Boolean equivalents of fundamental relevance.
Methods
Software implementation
Test versions of both the standard USM and the Boolean USM algorithms were developed in order to facilitate performance comparisons of practical implementations. A single "generic" implementation of the framework for both methods was first developed and then used as the starting point for developing method-specific implementations. Care was taken so as to minimize differences in any code other than that implementing the recursion and the distance metric calculations. Programs were written in C and compiled and tested in the same environment (compiler, linker, libraries, and host) and executed against the same test cases. No attempt was made to optimize either implementation (beyond that performed by the compiler). Both applications were compiled using the GNU compiler (gcc version 2.95.3) under the cygwin environment http://www.cygwin.com/ on a 1 GHz Pentium III-based system running Windows 2000. The source code, Makefiles, and test data are available from http://bioinformatics.musc.edu/resources.html.
Software performance testing
Test cases were produced by replicating two different 1 kbp segments of the e-coli genome to create 2 kbp, 3 kbp, 4 kbp, 5 kbp, 10 kbp, 15 kbp, 17 kbp, 20 kbp, and 40 kbp sequences. Each implementation is run as a separate program. The programs load the two sequences to be compared from disk, compute the USM (or bUSM) coordinates for all symbols in each sequence, and compute the local distance metric ( or ) for all pairs of symbols. Compute time was measured from the point following the load of the sequence from disk to the point at which all of the distance calculations were completed. Times were measured using the unix clock() function. Compute time was also measured from the point at which the USM coordinate calculations were completed to the end of the distance calculations.
Acknowledgements
The authors thankfully acknowledge support by the training grant 1-T15-LM07438-01 "training of toolmakers for Biomedical Informatics" by the National Library of Medicine of the National Institutes of Health, USA (NLM/NIH, http://www.nlm.nih.gov/ep/T15Training.html).
Authors' contributions
First author developed bolean implementation of Universal Sequence Map (bUSM). Second author, the original proponent of USM [4] identified theoretical context.
Contributor Information
John Schwacke, Email: schwacke@musc.edu.
Jonas S Almeida, Email: almeidaj@musc.edu.
References
- Jeffrey HJ. Chaos game representation of gene structure. Nucleic Acids Res. 1990;18:2163–2170. doi: 10.1093/nar/18.8.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida JS, Carrico JA, Maretzek A, Noble PA, Fletcher M. Analysis of genomic sequences by Chaos Game Representation. Bioinformatics. 2001;17:429–437. doi: 10.1093/bioinformatics/17.5.429. [DOI] [PubMed] [Google Scholar]
- Tino P. Spatial representation of symbolic sequences through iterative function systems. IEEE Transactions on Systems, Man, and Cybernetics – Part A: Systems and Humans. 1999;29:386–393. doi: 10.1109/3468.769757. [DOI] [Google Scholar]
- Almeida JS, Vinga S. Universal sequence map (USM) of arbitrary discrete sequences. BMC Bioinformatics. 2002;3:6. doi: 10.1186/1471-2105-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IEEE. 754–1985 IEEE Standard for Binary Floating-Point Arithmetic 1985. Book 754–1985 IEEE Standard for Binary Floating-Point Arithmetic 1985 (Editor ed.^eds.). City. 1985.
- Intel. IA-32 Intel Architecture Software Developer's Manual Volume 2: Instruction Set Reference. Book IA-32 Intel Architecture Software Developer's Manual Volume 2: Instruction Set Reference (Editor ed.^eds.). City: Intel Corporation. 2001.