

Abstract
In a recent article [Olah, G. A., Prakash, G. K. S. & Rasul, G. (1999) Proc. Natl. Acad. Sci. USA 96, 3494–3495] the authors found that the helionitronium trication, HeNO , is an unusually stable helium-containing polyatomic ion. This result was based on second-order many-body perturbation (MP2) calculations that showed that strong binding should occur between the oxygen and helium atoms in the assumed singlet ground state. The dissociation energy with respect to NO+ and HeO2+ was predicted to be 7.95 eV. We show here by thorough multireference configuration interaction (MRCI) studies that the ground state for the helionitronium trication is a triplet 3B1 state with He binding to the N atom (C2v). The He—O bound structure of Cs symmetry is not stable. Dissociation of the helionitronium trication occurs toward NO
, is an unusually stable helium-containing polyatomic ion. This result was based on second-order many-body perturbation (MP2) calculations that showed that strong binding should occur between the oxygen and helium atoms in the assumed singlet ground state. The dissociation energy with respect to NO+ and HeO2+ was predicted to be 7.95 eV. We show here by thorough multireference configuration interaction (MRCI) studies that the ground state for the helionitronium trication is a triplet 3B1 state with He binding to the N atom (C2v). The He—O bound structure of Cs symmetry is not stable. Dissociation of the helionitronium trication occurs toward NO and He+, and the trication is bound by at most 0.25 eV. These results indicate that the helionitronium trication is unstable under ambient conditions. The discrepancies between our results and the previous study are explained by the strong multireference character of the wave function of the trication.
and He+, and the trication is bound by at most 0.25 eV. These results indicate that the helionitronium trication is unstable under ambient conditions. The discrepancies between our results and the previous study are explained by the strong multireference character of the wave function of the trication.
Structural and energetic properties of helium-containing polyatomic cations are of great interest because they offer new possibilities and sources of superacids. There have been a number of studies (1–7) that have examined the stability of such cations. The studies of Koch and Frenking (4), Schleyer (5), and Wong et al. (6) all have shown that polyatomic cations are capable of forming strong bonds with helium. Olah et al. (7) reported a remarkably stable helionitronium trication, HeNO , that is bound with respect to dissociation into NO+ and OHe2+ by 7.95 eV. The unusual stability of the helionitronium trication, HeNO
, that is bound with respect to dissociation into NO+ and OHe2+ by 7.95 eV. The unusual stability of the helionitronium trication, HeNO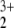 catalyzed our interest in this class of novel species. In the present work we report results of more sophisticated calculations on the structure and stability of the helionitronium trication.
catalyzed our interest in this class of novel species. In the present work we report results of more sophisticated calculations on the structure and stability of the helionitronium trication.
Methodology
To compute reliable energies and geometries for the multiple cationic species in question, it turned out that multireference ab initio techniques need to be applied as will be shown in the course of this article. All results for the complexes were obtained by the internally contracted multireference singles–doubles configuration interaction method (8) including the multireference version of Davidson correction (denoted by “+Q”; ref. 9). The basis of reference orbitals was generated by the complete active space self-consistent field (CASSCF) or multiconfiguration self-consistent field (MCSCF) method. The reference wave function was state-averaged in all cases where multiple roots of the Hamiltonian were needed. For the asymptotes some calculations were performed by the restricted open-shell coupled-cluster singles–doubles method, including perturbative triples [RCCSD(T)); ref. 10]. In all calculations the augmented double-ζ ANO basis of Widmark et al. (11) was used.
The multireference calculations [CASSCF/complete active space configuration interaction (CASCI) and MCSCF/multireference configuration interaction (MRCI)] were carried out in two different schemes regarding active space and reference configurations. For the specification of the active space, it is the easiest to treat the fragments NO2 and He separately. In all calculations the 1s orbitals of N and O were treated as core orbitals and were excluded from the correlation procedure. The space of valence orbitals is spanned by the He 1s orbital and three σ , three σ
, three σ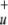 , two πu, and one πg orbital of the linear NO2 fragment (NO
, two πu, and one πg orbital of the linear NO2 fragment (NO , NO
, NO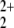 , and NO
, and NO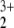 all have linear equilibrium structures). This results in a full valence space of 13 active orbitals, containing 16 electrons. Careful test calculations by CASSCF showed that the highest valence σ
all have linear equilibrium structures). This results in a full valence space of 13 active orbitals, containing 16 electrons. Careful test calculations by CASSCF showed that the highest valence σ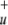 orbital is not needed to describe the static correlation in any of the treated processes and thus was excluded, resulting in an active space of 16 electrons in 12 orbitals (16e/12o).
orbital is not needed to describe the static correlation in any of the treated processes and thus was excluded, resulting in an active space of 16 electrons in 12 orbitals (16e/12o).
The optimization of HeNO and the asymptotic fragments was carried out by the CASSCF/CASCI approach, which required us to reduce further the active orbital space. For calculations of the complex this was accomplished by treating the 3σ
and the asymptotic fragments was carried out by the CASSCF/CASCI approach, which required us to reduce further the active orbital space. For calculations of the complex this was accomplished by treating the 3σ and 2σ
and 2σ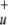 orbitals of NO2 as well as the 1s orbital of He as closed-shell orbitals that were kept doubly occupied in all reference configurations. This results in an active space of 10 electrons in 8 orbitals (10e/8o). However, in the CASCI calculations the singles and doubles excitations were performed on all occupied orbitals except the three 1s core orbitals of N and O. Preliminary CASSCF calculations with the above-mentioned 16e/12o active space justified the exclusion of these three orbitals, because in those calculations the occupation of these orbitals was always very close to 2.0. However, at the [He + NO2]3+ asymptote instead of the He 1s orbital the 3σ
orbitals of NO2 as well as the 1s orbital of He as closed-shell orbitals that were kept doubly occupied in all reference configurations. This results in an active space of 10 electrons in 8 orbitals (10e/8o). However, in the CASCI calculations the singles and doubles excitations were performed on all occupied orbitals except the three 1s core orbitals of N and O. Preliminary CASSCF calculations with the above-mentioned 16e/12o active space justified the exclusion of these three orbitals, because in those calculations the occupation of these orbitals was always very close to 2.0. However, at the [He + NO2]3+ asymptote instead of the He 1s orbital the 3σ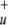 orbital of the NO2 fragment had to be excluded from the CAS space to obtain a reasonable 10e/8o active space. This is due to the fact that at this asymptote the electronic ground state refers to a charge distribution of He+ and NO
orbital of the NO2 fragment had to be excluded from the CAS space to obtain a reasonable 10e/8o active space. This is due to the fact that at this asymptote the electronic ground state refers to a charge distribution of He+ and NO , and thus the He 1s orbital needs to be part of the active space. In the C2v point group, in which most of the calculations were carried out, the asymptotic active space thus is composed of [5a1 6a1 7a1 8a1 1b1 2b1 4b2 1a2], and for the complex the active space consists of [6a1 7a1 8a1 1b1 2b1 3b2 4b2 1a2].
, and thus the He 1s orbital needs to be part of the active space. In the C2v point group, in which most of the calculations were carried out, the asymptotic active space thus is composed of [5a1 6a1 7a1 8a1 1b1 2b1 4b2 1a2], and for the complex the active space consists of [6a1 7a1 8a1 1b1 2b1 3b2 4b2 1a2].
Because the lowest energy asymptote corresponds to NO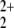 + He+, the He 1s orbital has to be included in the active space when the dissociation process is treated. Thus, for the treatment of the decomposition coordinate the active space was increased to 16 electrons in 12 active orbitals, and only the three 1s core orbitals of NO2 were kept closed. To keep the calculations tractable, a limited set of reference configurations had to be selected for both the MCSCF reference wave function and the MRCI calculations. Careful analysis of the CASSCF wave functions at equilibrium geometries and asymptotes showed that in addition to a charge transfer from He to NO2, the latter fragment also undergoes an electronic excitation from the X̃2Πg to the Ã2Πu state after bond rupture. To accommodate this effect, the most significant electron configurations at equilibrium geometry and asymptote were selected on which all singles and doubles excitations within the active space were applied. Only configurations with CI coefficients in the CASSCF wave function larger than 0.5 for any of the treated states were selected as reference for the singles and doubles excitations. Four primary references were chosen for the b1 state symmetry from which 537 configurations were created, resulting in 1,856 configuration state functions. In this way a well defined and smooth transition between the equilibrium region and the asymptote is ensured.
+ He+, the He 1s orbital has to be included in the active space when the dissociation process is treated. Thus, for the treatment of the decomposition coordinate the active space was increased to 16 electrons in 12 active orbitals, and only the three 1s core orbitals of NO2 were kept closed. To keep the calculations tractable, a limited set of reference configurations had to be selected for both the MCSCF reference wave function and the MRCI calculations. Careful analysis of the CASSCF wave functions at equilibrium geometries and asymptotes showed that in addition to a charge transfer from He to NO2, the latter fragment also undergoes an electronic excitation from the X̃2Πg to the Ã2Πu state after bond rupture. To accommodate this effect, the most significant electron configurations at equilibrium geometry and asymptote were selected on which all singles and doubles excitations within the active space were applied. Only configurations with CI coefficients in the CASSCF wave function larger than 0.5 for any of the treated states were selected as reference for the singles and doubles excitations. Four primary references were chosen for the b1 state symmetry from which 537 configurations were created, resulting in 1,856 configuration state functions. In this way a well defined and smooth transition between the equilibrium region and the asymptote is ensured.
For all multireference ab initio calculations the MOLPRO package was used (12). The geometry optimizations were performed with an external rational function optimizer (13) in symmetrized valence coordinates. The use of this optimizer allowed for efficient parallelization of the numerical gradient determination and perfect step-length control for optimization of the weak complexes.
Because we observed discrepancies between our multireference calculations and the results reported by Olah et al. (7), we carefully repeated and verified their computations. For those second-order many-body perturbation (MP2) calculations the GAUSSIAN program (14) and the 6-31G** basis set were used to render the results exactly comparable.
Results and Discussion
The two ionization potentials of He are well known to be 24.59 and 54.42 eV, respectively, whereas the first ionization potential of NO2 is known to be rather low (9.75 eV) (15). We calculated the first ionization potential of NO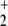 at the RCCSD(T) level of theory to be 24.25 eV, thus showing that the lowest asymptote for He-bond cleavage has to be NO
at the RCCSD(T) level of theory to be 24.25 eV, thus showing that the lowest asymptote for He-bond cleavage has to be NO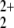 (2Πg) + He+ (2S). This is a quadruply degenerate asymptote, which after interaction of the fragments can split into two singlet and two triplet states. Thus the singlet results of Olah et al. for this energy reference are too high by 5.58 eV. The asymptotic energies of all considered states are depicted in Fig. 1.
(2Πg) + He+ (2S). This is a quadruply degenerate asymptote, which after interaction of the fragments can split into two singlet and two triplet states. Thus the singlet results of Olah et al. for this energy reference are too high by 5.58 eV. The asymptotic energies of all considered states are depicted in Fig. 1.
Fig 1.

Asymptotic states and energies of decomposition of the HeNO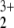 cation as computed by RCCSD(T).
cation as computed by RCCSD(T).
If we consider the asymptote NO+ + HeO2+, which have ground states of 1Σ+ and 3Σ− symmetry, respectively, our RCCSD(T) calculations show that this charge distribution is indeed 2.97 eV lower in energy than the alternative NO2+ (2Π) + HeO+ (2Π). It also turns out that NO+ + HeO2+ is 2.42 eV lower in energy than the fragmentation into NO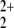 + He+. This is in disagreement with the finding of Olah et al. that the latter asymptote would be ≈29 eV higher in energy even if the 5.58 eV for the wrong charge distribution are subtracted. However, the lowest asymptote is a triplet of nondegenerate spatial symmetry that does not connect adiabatically with the singlet species. The 1Σ+ state of HeO2+ is 2.81 eV above the ground state, and thus NO
+ He+. This is in disagreement with the finding of Olah et al. that the latter asymptote would be ≈29 eV higher in energy even if the 5.58 eV for the wrong charge distribution are subtracted. However, the lowest asymptote is a triplet of nondegenerate spatial symmetry that does not connect adiabatically with the singlet species. The 1Σ+ state of HeO2+ is 2.81 eV above the ground state, and thus NO + He+ is the lowest possible singlet asymptote.
+ He+ is the lowest possible singlet asymptote.
Beyond this finding, attempts to reproduce the MP2 results of Olah et al. (7) faced unexpected difficulties. There are two isomers of HeNO . In the first isomer (structure 1) the helium forms a bond with the oxygen, whereas in the second isomer (structure 2) the binding is through the nitrogen. The structures are shown in Fig. 2. At the MP2/6-31G** level of theory, these authors found that the binding through oxygen yields a lower energy than through the nitrogen by 2.7 eV. We repeated the calculations of Olah et al. (7) and were able to reproduce their results as shown in Table 1. However, we found a further structure of C2v symmetry lower in energy than either structure 1 or 2. The geometrical parameters and energy of the new structure are given in Table 1 as structure 3. This stationary point is characterized by a long NO bond of 1.365 Å and a He—N bond of 1.399 Å. The new structure is found to be 1.08 eV lower in energy than the lowest energy structure of Cs symmetry and 3.79 eV lower than the C2v symmetric structure 2. This suggests that neither the C2v structure with the short He—N distance (structure 2) nor the Cs structure, where binding through the oxygen occurs, is the lowest energy structure at the MP2 level of theory. When we performed a vibrational frequency calculation to examine whether the new structure is a minimum, we found that the MP2/6-31G** calculation showed all real and no imaginary frequencies, but there were unusually large vibrational frequencies. This is often an indication of intrinsic problems of the applied methodology. It might point to symmetry breaking of the electronic wave function, or vibronic coupling phenomena, or, as will be shown to be the case here, strong multireference effects that cannot be handled appropriately by single-reference methods such as MP2.
. In the first isomer (structure 1) the helium forms a bond with the oxygen, whereas in the second isomer (structure 2) the binding is through the nitrogen. The structures are shown in Fig. 2. At the MP2/6-31G** level of theory, these authors found that the binding through oxygen yields a lower energy than through the nitrogen by 2.7 eV. We repeated the calculations of Olah et al. (7) and were able to reproduce their results as shown in Table 1. However, we found a further structure of C2v symmetry lower in energy than either structure 1 or 2. The geometrical parameters and energy of the new structure are given in Table 1 as structure 3. This stationary point is characterized by a long NO bond of 1.365 Å and a He—N bond of 1.399 Å. The new structure is found to be 1.08 eV lower in energy than the lowest energy structure of Cs symmetry and 3.79 eV lower than the C2v symmetric structure 2. This suggests that neither the C2v structure with the short He—N distance (structure 2) nor the Cs structure, where binding through the oxygen occurs, is the lowest energy structure at the MP2 level of theory. When we performed a vibrational frequency calculation to examine whether the new structure is a minimum, we found that the MP2/6-31G** calculation showed all real and no imaginary frequencies, but there were unusually large vibrational frequencies. This is often an indication of intrinsic problems of the applied methodology. It might point to symmetry breaking of the electronic wave function, or vibronic coupling phenomena, or, as will be shown to be the case here, strong multireference effects that cannot be handled appropriately by single-reference methods such as MP2.
Fig 2.

Structures of the two assumed isomers of the HeNO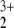 cation. (A) Structure 1. (B) Structure 2 (note that structure 3 is represented by the same conformation).
cation. (A) Structure 1. (B) Structure 2 (note that structure 3 is represented by the same conformation).
Table 1.
Calculated geometries and energetics for the helionitronium trication
| Structure, state, symmetry | Method | NO, Å | NO′, Å | HeO, Å | HeN, Å | ONO′, ° | HeON, ° | Energy, hartree |
|---|---|---|---|---|---|---|---|---|
| 1 |1A′〉 (Cs) | MP2 | 1.146 | 1.586 | 1.163 | 171.0 | 102.4 | −204.98651 | |
| 2 |1A1〉 (C2v) | MP2 | 1.318 | 1.080 | 117.8 | −204.88682 | |||
| 3 |1A1〉 (C2v) | MP2 | 1.365 | 1.399 | 162.8 | −205.02607 | |||
| |3B1〉 (C2v) | MP2 | 1.268 | 1.819 | 147.4 | −204.93968 | |||
| |11A1〉 (C2v) | MRCI | 1.246 | 1.973 | 164.6 | −205.09742 | |||
| |21A1〉 (C2v) | MRCI | 1.243 | 1.925 | 162.2 | −205.06413 | |||
| |3B1〉 (C2v) | MRCI | 1.261 | 2.059 | 169.3 | −205.16319 |
CASSCF/CASCI (10e/8o).
A closer look at the MP2 wave functions revealed that a single-reference approach is inappropriate for the HeNO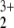 system. We computed natural orbitals of the correlated wave functions that have fractional occupation numbers. If the single determinant reference function gives a fair representation of the true wave function, the deviations of the fractional with respect to integer occupation numbers are small. This is not the case for the MP2 calculations of HeNO
system. We computed natural orbitals of the correlated wave functions that have fractional occupation numbers. If the single determinant reference function gives a fair representation of the true wave function, the deviations of the fractional with respect to integer occupation numbers are small. This is not the case for the MP2 calculations of HeNO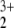 . We give the most significant natural orbital occupation numbers (NOONs) in Table 2. The orbitals are characterized by their symmetry label in the actual C2v framework as well as by their corresponding labels in linear NO2, because the latter dominates the character of the wave function.
. We give the most significant natural orbital occupation numbers (NOONs) in Table 2. The orbitals are characterized by their symmetry label in the actual C2v framework as well as by their corresponding labels in linear NO2, because the latter dominates the character of the wave function.
Table 2.
NOONs of the 1A1 state of HeNO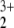 obtained from MRCI, MP2 with true density (|Ψ|2), and MP2 with effective density. Calculations were performed with the 6-31G** basis set at the geometry of structure 2
obtained from MRCI, MP2 with true density (|Ψ|2), and MP2 with effective density. Calculations were performed with the 6-31G** basis set at the geometry of structure 2
| Orbital sym.
|
Orbital type (c.f. lin. NO2)
|
Occupation number | ||
|---|---|---|---|---|
| MP2 (eff. density) | MP2 (|Ψ|2) | MRCI | ||
| b1 | πu | 2.34188 | 1.94267 | 1.94032 |
| a1 | σ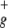
|
2.23610 | 1.95859 | 1.95554 |
| b2 | σ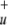
|
2.00099 | 1.93335 | 1.94313 |
| a2 | πg | 1.65996 | 1.87207 | 1.61548 |
| b2 | πg | 1.20377 | 0.24098 | 0.74020 |
| a1 | πu | 0.89307 | 1.73636 | 1.36244 |
| b1 | π*u | −0.07418 | 0.16349 | 0.29461 |
| b2 | σ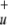 * *
|
−0.43958 | 0.03782 | 0.00530 |
At first glance, it is apparent that a number of natural orbitals show occupations that strongly deviate from integers. The deviations are particularly strong for the NOONs that are obtained by diagonalizing the effective MP2 density, which is usually done in standard quantum chemistry codes such as GAUSSIAN. Although a closed-shell 1A1 state is calculated, the NOONs of 0.89 and 1.20 for the a1 (πu) and b2 (πg) orbitals, respectively, look more like a triplet or open-shell singlet wave function. The a2 (πg) orbital also shows a rather low occupation of 1.66. Such occupation numbers clearly indicate that static correlation plays a substantial role and that the wave function cannot be expressed by a single-reference function corrected by small perturbations. Thus the corresponding orbitals have to be included into the active space of a multireference treatment. Somewhat bewildering is the finding that natural orbitals with unphysical occupation numbers of >2 and <0 are obtained. This effect was already discovered by Gordon et al. (16), who found that such unphysical NOONs, obtained from the effective MP2 density, are excellent indicators for the multireference character of a wave function.
For comparison we also calculated the natural orbitals by diagonalizing the true MP2 density |Ψ|2. In this way no unphysical NOONs are obtained. Again we find four orbitals with occupations between 1.87 and 0.16, indicating that those orbitals are strongly involved in static correlation effects. Analysis of the orbitals reveals that those orbitals with unphysically high occupation in the effective MP2 density are noticeably depopulated in the |Ψ|2 analysis. On the other hand the b1 (π*u) orbital shows negative occupation in the effective but significantly positive occupation in the true MP2 density. However, the b2 (σ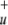 *) orbital has a strongly negative eigenvalue for the effective MP2 density but no unusually high occupation in |Ψ|2.
*) orbital has a strongly negative eigenvalue for the effective MP2 density but no unusually high occupation in |Ψ|2.
These findings led us to investigate the character of the HeNO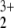 singlet species by a rigorous MCSCF/MRCI (16e/12o) treatment. Again we obtained natural orbitals and the respective occupations. All natural orbitals of Table 2 are included in the active reference space. The MRCI results confirm the conclusion drawn from the MP2 |Ψ|2 data that four orbitals have considerable noninteger occupations and play the most significant role in the static correlation.
singlet species by a rigorous MCSCF/MRCI (16e/12o) treatment. Again we obtained natural orbitals and the respective occupations. All natural orbitals of Table 2 are included in the active reference space. The MRCI results confirm the conclusion drawn from the MP2 |Ψ|2 data that four orbitals have considerable noninteger occupations and play the most significant role in the static correlation.
It further turns out that besides the singlet ground state of 1A1 symmetry there exists a second 1A1 state with an excitation energy of ≈0.91 eV. For both states equilibrium geometries were optimized by the CASSCF/CASCI approach using the (10e/8o) active space. The optimized structures show rather long He—N bonds of 1.973 and 1.925 Å for |11A1〉 and |21A1〉, respectively, which is markedly different from the results of Olah et al. and would indicate a rather weak bond. The N—O bonds are 1.246/1.243 Å and the He—N—O bond angles are 97.7/98.9° for the two singlet states.
An analysis of the state character shows that both states are determined by two electron configurations that correspond to a double excitation between the 4b2 and 1a2 orbitals (Table 3). The CI coefficients clearly show the extreme multireference character of the respective states, which readily explains why single-reference methods such as MP2 are unable to yield reliable results for such a species. We also computed excited states of other symmetries by CASSCF, but the two 1A1 states are the most stable ones by far, and thus we refrained from looking at the higher states in more detail.
Table 3.
Leading configurations and their CI coefficients of HeNO electronic states
electronic states
| 5a1 | 6a1 | 7a1 | 8a1 | 1b1 | 2b1 | 4b2 | 1a2 | c1|11A1〉 | c2|21A1〉 | c3|13B1〉 |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 2 | 0 | 0 | 2 | 0 | 2 | 0 | −0.572 | 0.630 | 0.000 |
| 2 | 2 | 0 | 0 | 2 | 0 | 0 | 2 | 0.624 | 0.566 | 0.000 |
| 2 | 2 | 0 | 0 | 2 | 0 | 1 | 1 | 0.000 | 0.000 | 0.818 |
Instead we notice that a double excitation such as the one between the 4b2 and 1a2 orbitals of the HeNO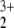 complex points toward the existence of a low-lying triplet state. Indeed we found that the ground state of this system is the X̃3B1 state. This state is characterized mainly by an electron configuration with the two unpaired electrons in the orbitals 4b2 and 1a2 and is 1.79 eV lower in energy than the lowest singlet state. However, the dominating but low CI coefficient of 0.8 (compare Table 3) indicates that again fairly strong multireference effects are present. The optimized geometry at CASSCF/CASCI (10e/8o) level of theory (compare Table 1) shows an even longer He—N bond of 2.059 Å and slightly longer N—O bonds of 1.261 Å, which go along with reduced He—N—O angles of 95.4°.
complex points toward the existence of a low-lying triplet state. Indeed we found that the ground state of this system is the X̃3B1 state. This state is characterized mainly by an electron configuration with the two unpaired electrons in the orbitals 4b2 and 1a2 and is 1.79 eV lower in energy than the lowest singlet state. However, the dominating but low CI coefficient of 0.8 (compare Table 3) indicates that again fairly strong multireference effects are present. The optimized geometry at CASSCF/CASCI (10e/8o) level of theory (compare Table 1) shows an even longer He—N bond of 2.059 Å and slightly longer N—O bonds of 1.261 Å, which go along with reduced He—N—O angles of 95.4°.
Although this triplet state might be treated, in principle, by single-reference methods such as MP2, it took some effort to locate any stationary point on the MP2 surface for a stable triplet complex. The finally obtained structure is given in Table 1. However, the noticeable multireference character of the MRCI wave function and a thorough analysis of the orbital occupation of the MP2 results fostered doubts about the reliability of the MP2 calculations. A very strong spin contamination is noticed with 〈S2〉 = 3.37 and atomic spin densities of 1.73 at the O and −0.98 at the N atoms, pointing to a mixture of triplet and quintet spin state. Accordingly there is a rather large difference between unprojected and projected MP2 energy. The natural orbitals show a similarly strong deviation from integer occupation numbers as for the singlet states, which again indicates that the MP2 method is not suitable for treatment of the HeNO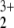 triplet state either.
triplet state either.
The MCSCF/MRCI (16e/12o) calculations of the triplet complex show that it is thermodynamically unstable with respect to the NO + He+ ground state asymptote by 9.06 eV. However, this asymptote is not reachable adiabatically. The energy diagram for the expected products and their state designations accessible by the helionitronium trication is shown in Fig. 3. The Mulliken charges of the complex reveal that the three positive charges are more or less equally distributed over the N and O atoms and that He is uncharged. This means that to reach the lowest asymptote a charge transfer has to occur, because there is no covalent bond between He and NO2, which could be split homolytically. Thus the complex connects diabatically to a highly unfavorable He + NO
+ He+ ground state asymptote by 9.06 eV. However, this asymptote is not reachable adiabatically. The energy diagram for the expected products and their state designations accessible by the helionitronium trication is shown in Fig. 3. The Mulliken charges of the complex reveal that the three positive charges are more or less equally distributed over the N and O atoms and that He is uncharged. This means that to reach the lowest asymptote a charge transfer has to occur, because there is no covalent bond between He and NO2, which could be split homolytically. Thus the complex connects diabatically to a highly unfavorable He + NO asymptote. Even the homolytic splitting of a hypothetical covalent He—N bond would yield a highly excited NO
asymptote. Even the homolytic splitting of a hypothetical covalent He—N bond would yield a highly excited NO state with three unpaired electrons. However, the lowest adiabatically connected asymptote, at least within C2v symmetry, is the Ã2Πu state of NO
state with three unpaired electrons. However, the lowest adiabatically connected asymptote, at least within C2v symmetry, is the Ã2Πu state of NO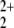 and He+, which is found 5.19 eV lower in energy than the equilibrium structure. The |X̃2Πg〉 ← |Ã2Πu〉 excitation energy is 3.87 eV, and thus the total dissociation energy is calculated to be 9.06 eV. In fact, the reason why the He—NO
and He+, which is found 5.19 eV lower in energy than the equilibrium structure. The |X̃2Πg〉 ← |Ã2Πu〉 excitation energy is 3.87 eV, and thus the total dissociation energy is calculated to be 9.06 eV. In fact, the reason why the He—NO ion is bound at all is that the crossing of the repulsive charge separated state with the bound NO
ion is bound at all is that the crossing of the repulsive charge separated state with the bound NO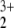 —He state results in an avoided crossing, which establishes a barrier to dissociation. This result is in perfect agreement with earlier works of Frenking et al. (17), who found such curve crossings in general for rare gas–ion complexes. An upper bound for the barrier height is calculated from fitting quasidiabatic potential curves for the bond rupture for both states and determining the intersection point. A value of 0.25 eV (5.87 kcal/mol) is found, indicating that the complex will decompose easily at all but extremely low temperatures.
—He state results in an avoided crossing, which establishes a barrier to dissociation. This result is in perfect agreement with earlier works of Frenking et al. (17), who found such curve crossings in general for rare gas–ion complexes. An upper bound for the barrier height is calculated from fitting quasidiabatic potential curves for the bond rupture for both states and determining the intersection point. A value of 0.25 eV (5.87 kcal/mol) is found, indicating that the complex will decompose easily at all but extremely low temperatures.
Fig 3.

Correlation diagram for dissociation of HeNO along an arbitrary C2v pathway χ.
along an arbitrary C2v pathway χ.
Although in C2v symmetry only the first excited asymptote can be reached adiabatically, this is not the case for distortion to Cs geometries within the molecular plane. However, our calculations show that the optimized C2v structure is a true minimum and that distortion of the He atom out of the symmetry axis requires substantial energy. Finally we also investigated the stability of He—O bound complex. However, all attempts failed to optimize any Cs structure similar to that found by Olah et al. (7). The complexes decomposed either to NO2+ + HeO+ or NO + He+, and this finding was independent of the spin state. At least for the triplet state this is not surprising, because the NO
+ He+, and this finding was independent of the spin state. At least for the triplet state this is not surprising, because the NO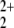 (2πg) is preformed already and the closed-shell He atom forms a repulsive interaction with the doubly occupied πg orbital of NO
(2πg) is preformed already and the closed-shell He atom forms a repulsive interaction with the doubly occupied πg orbital of NO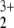 . In contrast, in the C2v complex the He atom interacts with the empty component of the πu orbital, which explains the attractive potential.
. In contrast, in the C2v complex the He atom interacts with the empty component of the πu orbital, which explains the attractive potential.
Conclusions
The present multireference configuration interaction study shows that the helionitronium trication, HeNO , is at best a weakly bound charge-transfer complex. It is not unusually stable, and the Cs structure, reported by Olah et al. to be the minimum structure, is found to be unbound. The singlet C2v structure (|11A1〉), in which the helium bonds to the nitrogen, is an excited state. We find that the true ground state is a triplet state (|3B1〉). The lesson to be learned from this investigation is that many-body perturbation methods and other single-determinant methods may lead to artifactual results for the structure and energetics for polyatomic polycations, which is because these systems may have wave functions of extreme multireference character that cannot be handled properly by approximation methods based on a single determinant. A careful look on the natural orbital populations can help reveal the necessity to treat such a system by multireference techniques.
, is at best a weakly bound charge-transfer complex. It is not unusually stable, and the Cs structure, reported by Olah et al. to be the minimum structure, is found to be unbound. The singlet C2v structure (|11A1〉), in which the helium bonds to the nitrogen, is an excited state. We find that the true ground state is a triplet state (|3B1〉). The lesson to be learned from this investigation is that many-body perturbation methods and other single-determinant methods may lead to artifactual results for the structure and energetics for polyatomic polycations, which is because these systems may have wave functions of extreme multireference character that cannot be handled properly by approximation methods based on a single determinant. A careful look on the natural orbital populations can help reveal the necessity to treat such a system by multireference techniques.
Acknowledgments
We thank the Leibniz-Rechenzentrum for generous allocation of computer time.
Abbreviations
CASSCF, complete active space self-consistent field
MCSCF, multiconfiguration self-consistent field
RCCSD(T), restricted open-shell coupled-cluster singles–doubles method, including perturbative triples
CASCI, complete active space configuration interaction
MRCI, multireference configuration interaction
MP2, second-order many-body perturbation
NOON, natural orbital occupation number
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Frenking G. & Cremer, D. (1990) Struct. Bonding (Berlin) 73 17-95. [Google Scholar]
- 2.Wilson S. & Green, S. (1980) J. Chem. Phys. 73 419-424. [Google Scholar]
- 3.Cooper D. L. & Wilson, S. (1981) Mol. Phys. 41 161-163. [Google Scholar]
- 4.Koch, W. & Frenking, G. (1986) J. Chem. Soc. Chem. Commun., 1095–1096.
- 5.Schleyer, P. v. R. (1985) Adv. Mass. Spectrom., 287–301.
- 6.Wong, M. W., Nobes, R. H. & Radom, L. (1987) J. Chem. Soc. Chem. Commun., 233–234.
- 7.Olah G. A., Prakash, G. K. S. & Rasul, G. (1999) Proc. Natl. Acad. Sci. USA 96 3494-3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Werner H.-J. & Knowles, P. J. (1988) J. Chem. Phys. 89 5803-5814. [Google Scholar]
- 9.Davidson E. R. (1975) J. Comput. Phys. 17 87-97. [Google Scholar]
- 10.Knowles P. J., Hampel, C. & Werner, H.-J. (1993) J. Chem. Phys. 99 5219-5227. [Google Scholar]
- 11.Widmark P. O., Malmqvist, P. A. & Roos, B. (1990) Theor. Chim. Acta 77 291. [Google Scholar]
- 12.Werner, H.-J. & Knowles, P. J. (1996) MOLPRO (Univ. Stuttgart, Stuttgart).
- 13.Eisfeld, W. (1999) WEGO (Garching, Germany), Version 1.6.
- 14.Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Zakrzewski, V. G., Montgomery, J. A., Stratmann, R. E., Burant, J. C., et al. (1998) GAUSSIAN 98 (Gaussian, Pittsburgh), Rev. A.3.
- 15.Lide D. R., (1996) Handbook of Chemistry and Physics (CRC, Boca Raton, FL).
- 16.Gordon M. S., Schmidt, M. W., Chaban, G. M., Glaesemann, K. R., Stevens, W. J. & Gonzalez, C. (1999) J. Chem. Phys. 110 4199-4207. [Google Scholar]
- 17.Frenking G., Koch, W., Reichel, F. & Cremer, D. (1990) J. Am. Chem. Soc. 112 4240-4256. [Google Scholar]