

Abstract
The recent observation [Wentworth, P., Jones, L. H., Wentworth, A. D., Zhu, X. Y., Larsen, N. A., Wilson, I. A., Xu, X., Goddard, W. A., Janda, K. D., Eschenmoser, A. & Lerner, R. A. (2001) Science 293, 1806–1811] that antibodies form H2O2 from 1O2 plus H2O was explained in terms of the formation of the H2O3 species that in the antibody reacts with a second H2O3 to form H2O2. There have been few reports of the chemistry for forming H2O3, but recently Engdahl and Nelander [Engdahl, A. & Nelander, B. (2002) Science 295, 482–483] reported that photolysis of the ozone–hydrogen peroxide complex in argon matrices leads to significant concentrations of H2O3. We report here the chemical mechanism for this process, determined by using first-principles quantum mechanics. We show that in an argon matrix it is favorable (3.5 kcal/mol barrier) for H2O2 and O3 to form a [(HO2)(HO3)] hydrogen-bonded complex [head-to-tail seven-membered ring (7r)]. In this complex, the barrier for forming H2O3 plus 3O2 is only 4.8 kcal/mol, which should be observable by means of thermal processes (not yet reported). Irradiation of the [(HO2)(HO3)-7r] complex should break the HO–OO bond of the HO3 moiety, eliminating 3O2 and leading to [(HO2)(HO)]. This [(HO2)(HO)] confined in the matrix cage is expected to rearrange to also form H2O3 (observed experimentally). We show that these two processes can be distinguished isotopically. These results (including the predicted vibrational frequencies) suggest strategies for synthesizing H2O3 and characterizing its chemistry. We suggest that the [(HO2)(HO3)-7r] complex and H2O3 are involved in biological, atmospheric, and environmental oxidative processes.
Peroxone (the combination of ozone and hydrogen peroxide) is used to treat soil, groundwater, and wastewater contaminated with volatile organic compounds, polycyclic aromatic hydrocarbons, petroleum hydrocarbons, chlorinated solvents, metals, munitions, diesel fuel, methyl tert-butyl ether (MTBE), BTEX (benzene, toluene, ethylbenzene, and xylene), trinitrotoluene (TNT), and other waste constituents (1). This mixture of ozone with hydrogen peroxide is far more reactive than either alone (1), but no mechanistic studies have been reported to explain why.
A recent paper (2) showed that mixing O3 and H2O2 in an argon matrix leads to a complex that when photolyzed produces significant concentrations of H2O3, indicating that complex intermediates might be involved in the peroxone process.
Another recent paper (3) showed the surprising result that H2O3 is apparently involved in the formation of H2O2 from 1O2 plus H2O by antibodies. Here a H2O3 species reacts with a second H2O3 to form [(HOO)(HOOO)-7r] (a head-to-tail seven-member ring complex), which subsequently leads to H2O2 (4, 5).
These studies suggest that other poorly understood but important oxidative processes involved in biological, atmospheric, and aqueous systems may involve H2O3, [(HOO)(HOOO)-7r], and related unknown or poorly characterized intermediates. Consequently we have been studying the chemistry of some of these possibly important species.
This report presents a detailed mechanistic study of the formation of H2O3 from H2O2 + O3. Our results show that a complex of H2O2 and O3 rearranges first to form the [(HOO)(HOOO)-7r] complex claimed in ref. 4 to be involved in the antibody-catalyzed process. Under irradiation, the HO–OO bond should break, leading to 3O2 plus [(HO2)(HO)] confined in the matrix cage, which is expected to rearrange to form H2O3, which has been observed in ref. 2. In addition, the same cyclic intermediate complex can form H2O3 by a thermal unimolecular rearrangement process. We show that these two processes can be distinguished isotopically.
The computational details are given in the next section, followed by a presentation of the results, a discussion of the implications, and finally a presentation of some conclusions.
Computational Details
All quantum mechanical (QM) calculations used the B3LYP flavor of density functional theory (DFT) (6–10), which includes a generalized gradient approximation (GGA) and some exact exchange (hybrid method). The 6-31G** basis set (11, 12) was used on all atoms, and a full geometry optimization was carried out for all stable complexes and saddle points. Vibrational frequencies (from the analytic Hessian) were calculated to ensure that each minimum is a true local minimum (containing only positive frequencies) and that each transition state has only a single imaginary frequency (one negative eigenvalue of the Hessian). The Pulay modified scaled quantum mechanical (SQM) method was used to predict the vibrational frequencies (13). Scale factors for O–O stretching, O–H stretching, O–O–O bending, O–O–H bending, and O–O–O–H torsion are 0.917, 0.905, 0.920, 0.939, and 0.855, respectively. All QM calculations were carried out with jaguar (14).
To obtain more accurate energetics, we also carried out calculations with the cc-pVTZ basis set (15), using the optimized geometries from the 6-31G** basis. It was reported that calculations with B3LYP/6-311+G (3df,2p) lead to a mean absolute error of 3.11 kcal/mol for a collection of 148 simple inorganic/organic molecules (16).
For molecules such as 1O2 and O3, which have significant open-shell character, standard density functional theory methods often lead to much larger errors. Thus with B3LYP the singlet and triplet gap for O2 is ΔE (1Δg − 3Σ ) = 10.4 kcal/mol, in poor agreement with the experimental value of 22.5 kcal/mol (17). Consequently, we used spin-projection techniques (18) to ensure a proper description of the complexes involving 1O2. This procedure leads to ΔE (1Δg − 3Σ
) = 10.4 kcal/mol, in poor agreement with the experimental value of 22.5 kcal/mol (17). Consequently, we used spin-projection techniques (18) to ensure a proper description of the complexes involving 1O2. This procedure leads to ΔE (1Δg − 3Σ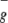 ) = 20.5 kcal/mol, in reasonable agreement with experiment.
) = 20.5 kcal/mol, in reasonable agreement with experiment.
Fig. 1 summarizes the whole reaction profile from H2O2 + O3 to H2O3 + O2, where LM stands for “local minimum” and TS stands for “transition state.” All energetics are reported for ΔH (298 K), in kcal/mol, where the calculated vibrational frequencies were used with standard quantum statistical formulas to obtain the entropy and enthalpy as a function of temperature.
Fig 1.

Reaction profile from H2O2 + O3 to H2O3 + 3O2. TS, transition state.
Results
Formation of [(HO2)(HO3)-7r] Cyclic Complex.
Starting with the free reactants (LM0: H2O2 + O3), we find two molecular complexes, LM1-endo and LM1-exo: [(H2O2)(O3)], leading to Eqs. 1a and 1b.
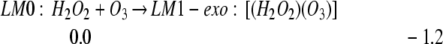 |
 |
where LM1-endo has a pseudoring configuration and LM1-exo has a pseudolinear configuration (see Fig. 1). LM1 can be formally considered as σ−complexes with H of H2O2 pointing to the Pσ lone pair orbital of a terminal O of O3. This hydrogen bond is weak (2.0 kcal/mol, compared with 5.0 kcal/mol for water dimer) because the Mulliken charge on the terminal oxygen is only −0.2 (compared with −0.6 in H2O).
We find LM1-exo leads to the formation of LM2-linear: [(HO2)(HO3)-L], whereas LM1-endo leads to the formation of LM2-ring: [(HO2)(HO3)-7r] (seven-member ring), as in Eqs. 2a and 2b. In addition, LM2-linear can rearrange to LM2-ring as in Eq. 2c.
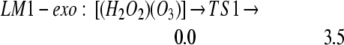 |
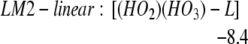 |
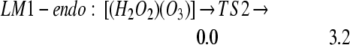 |
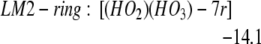 |
 |
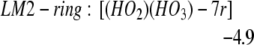 |
The barrier is 3.5 kcal/mol from LM1-exo to LM2-linear, whereas it is 3.2 kcal/mol from LM1-endo to LM2-ring. Both reactions are noteworthy in that they convert the closed-shell singlet molecules H2O2 and O3 to a biradical complex [(HO2)(HO3)], which is significantly more stable (10.4 kcal/mol for linear and 15.3 for cyclic) than the free reactants H2O2 + O3. Both products LM2-linear and LM2-ring are planar, with the radical (singly occupied) orbitals perpendicular to the molecular plane. In the HOO moiety, ≈70% of the spin density is localized at the terminal oxygen, whereas in the HOOO moiety, ≈90% of the spin density is on the two terminal oxygens. In this biradical the energy surface for the spin singlet initial state is degenerate with the triplet surface (we calculate that the singlet state is 0.01 kcal/mol lower), leading to rapid interconversion to the triplet. (The rate is determined by the spin orbit matrix elements and the energy spacings between the various populated vibrational—rotational levels, which we guess might be microseconds.) Neglecting decomposition and reactions from this state, the equilibrium concentration ratio of triplet to singlet would be 3:1.
It was reported that codepositing O3 and H2O2 in argon matrices leads to new IR bands indicating complex formation, but no details were reported (2). We predict that this complex is most likely to be the cyclic [(HO2)(HO3)-7r] complex (LM2-ring). To help test this prediction, we report the predicted vibrational frequencies in Table 1 for various isotopic cases.
Table 1.
Predicted fundamentals for the [(HO2)(HO3)-7r] complex, in wavenumbers, based on the scaled quantum mechanical analysis of the Hessian from B3LYP/6-31G** calculations
| Mode
|
Character
|
Wavenumber, cm−1 | ||
|---|---|---|---|---|
| (HO2)(HO3) | (HO2)(H18O3) | (DO2)(DO3) | ||
| ν1 | Antisymmetric OH stretch | 3,190.3 (737.3) | 3,188.8 | 2,331.8 |
| ν2 | Symmetric OH stretch | 3,030.1 (260.9) | 3,020.6 | 2,220.2 |
| ν3 | HOO bend of HO3 | 1,511.6 (64.7) | 1,506.1 | 1,216.9 |
| ν4 | HOO bend of HO2 | 1,466.9 (65.4) | 1,460.1 | 1,180.5 |
| ν5 | OO stretch of HO2 | 1,173.8 (9.1) | 1,173.7 | 1,096.3 |
| ν6 | Symmetric OO stretch of HO3 | 1,167.2 (29.7) | 1,102.7 | 1,074.7 |
| ν7 | Antisymmetric OO stretch of HO3 | 956.8 (72.1) | 956.3 | 780.5 |
| ν8 | HOOO-oop | 787.9 (100.6) | 743.6 | 690.8 |
| ν9 | HO2–HO3-slide | 588.5 (161.4) | 588.1 | 512.0 |
| ν10 | O–O–O bend in HO3 | 533.2 (5.6) | 505.0 | 432.3 |
| ν11 | Antisymmetric hydrogen-bond stretch | 273.2 (49.9) | 271.4 | 261.8 |
| ν12 | Ring torsion-twist | 219.6 (3.2) | 210.2 | 216.8 |
| ν13 | Symmetric hydrogen-bond stretch | 175.1 (1.9) | 171.1 | 170.4 |
| ν14 | Ring torsion-chair | 116.3 (0.5) | 110.3 | 116.0 |
| ν15 | Ring torsion-boat | 65.3 (0.8) | 64.7 | 64.2 |
The calculated infrared intensities for [(HO2)(HO3)-7r] are listed in parentheses.
Formation of H2O3 by Thermal Processes.
Starting with the LM2-ring complex, a simple H atom transfer process allows the hydrogen of HO2 to transfer to the terminal oxygen of HO3, leading directly to the formation of HOOOH, as indicated in Eq. 3 (see Fig. 1). Because the LM2-ring is expected to have nearly degenerate singlet and triplet spin states, we considered both spin states.
 |
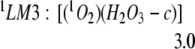 |
 |
 |
For the triplet case, the net reaction is 17.0 kcal/mol exothermic (because of formation of the ground state triplet molecule, 3O2), leading to a barrier of only 4.8 kcal/mol. Forming 1O2 is endothermic by 3.0 kcal/mol with a barrier of 11.5 kcal/mol. Thus at low temperatures the triplet process is expected to dominate. Because of the rapid singlet–triplet conversion in [(HO2)(HO3)-7r], it is likely to produce H2O3 even at low temperature.
The H2O3 formed in Eqs. 3a and 3b is in the cis form, with both O–H groups pointing toward the same side of the O–O–O plane of H2O3 (See Fig. 1). The O2 and H2O3 complexes (LM3) formed are very weakly bound (binding energy = 0.2 kcal/mol), so that they are likely to dissociate even in the matrix.
The cis H2O3 can rotate its OH bond (either inward or outward) to convert to the trans global minimum of H2O3 with a low barrier as in Eq. 4 (refs. 4 and 19; ref. 19 reported detailed theoretical studies of H2O3).
Starting with 18O3 + H2O2, the thermal process would lead to the formation of H18O18O18OH. Table 2 gives the predicted vibrational frequencies, which should be useful for experimental validation of these results.
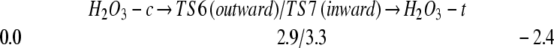 |
Table 2.
Predicted fundamentals of cis HOOOH and trans HOOOH, H18OOOH, H18O18O18OH, HOOOD, and DOOOD, in wavenumbers, based on the scaled quantum mechanical analysis of the Hessian from B3LYP/6-31G** calculations
| Molecule
|
Wavenumber, cm−1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ν1 | ν2 | ν3 | ν4 | ν5 | ν6 | ν7 | ν8 | ν9 | |
| H2O3-c | 3,522.0 | 3,518.7 | 1,359.9 | 1,329.5 | 900.7 | 768.0 | 485.5 | 418.4 | 190.4 |
| H2O3-t | 3,533.1 | 3,528.4 | 1,353.6 | 1,349.6 | 899.5 | 770.2 | 506.5 | 410.4 | 346.9 |
| (3,529.6) | (3,529.6) | (1,359.1) | (1,347.4) | (821.0) | (776.3) | (509.1) | (387.0) | (346.4) | |
| H18OOOH-t | 3,531.2 | 3,518.3 | 1,352.4 | 1,347.0 | 887.6 | 759.9 | 495.1 | 408.9 | 346.3 |
| (3,529.6) | (3,520.3) | (1,357.0) | (1,344.3) | (NR) | (768.0) | (NR) | (386.6) | (346.0) | |
| HO18OOH-t | 3,528.4 | 3,533.1 | 1,348.7 | 1,347.0 | 874.8 | 745.6 | 502.9 | 408.4 | 346.1 |
| H218O3-t | 3,521.1 | 3,516.4 | 1,344.8 | 1,343.8 | 848.3 | 726.1 | 480.3 | 405.6 | 344.8 |
| HO3D-t | 3,530.7 | 2,580.5 | 1,351.4 | 1,009.2 | 896.3 | 770.0 | 499.5 | 387.1 | 276.6 |
| (3,529.6) | (2,610.4) | (1,349.9) | (NR) | (814.6) | (772.0) | (NR) | (369.2) | (NR) | |
| D2O3-t | 2,582.2 | 2,579.0 | 1,013.6 | 1,004.2 | 892.8 | 769.8 | 492.6 | 317.1 | 252.8 |
| (2,610.4) | (2,610.4) | (1,007.3) | (NR) | (NR) | (762.6) | (NR) | (301.6) | (273.5) | |
The experimental data (2) are listed in parentheses for comparison (NR, not reported). The modes are as follows: ν1, symmetric OH stretch; ν2, antisymmetric OH stretch; ν3, antisymmetric HOO bend; ν4, symmetric HOOO bend; ν5, symmetric OO stretch; ν6, antisymmetric OO stretch; ν7, OOO bend; ν8, antisymmetric torsion; and ν9, symmetric torsion.
Formation of H2O3 by Photolysis.
We find that free HOOO radical has a planar cis configuration. It is bound by only 3 kcal/mol with respect to HO and 3O2, with a barrier to decomposition of only 4 kcal/mol.
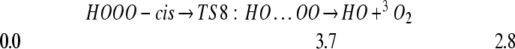 |
This configuration is consistent with the experimental observation that HOOO can be formed when hydroxyl radicals are added to molecular oxygen in an argon matrix (20). The terminal OO bond in HOOO is 1.26 Å, indicating a partial double bond resulting from π resonance (just as in HOO), whereas the other OO bond is 1.50 Å, slightly longer than a single bond (1.46 Å) in H2O2. Thus photolyzing HOOO is expected to break the weak bond between HO and OO, producing HO + 3O2 by a spin-allowed process.
[(HO2)(HO3)-7r] is stabilized by 8.1 kcal/mol (Eq. 6), which should retard the decomposition of HO3.
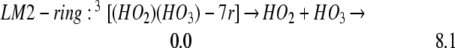 |
 |
Because the barrier for the thermal process to produce H2O3 + 3O2 is only 4.8 kcal/mol, we expect that reaction 6 would not be so important in a low-temperature process.
Photolysis of [(HO2)(HO3)-7r] is expected to produce 3O2, leaving behind a [(HOO)(HO)] complex in the matrix cage. Because [(HO2)(HO3)-7r] is ≈75% triplet, we expect the [(HOO)(HO)] complex formed in this decomposition to be predominantly singlet as in Eq. 7, which can close rapidly to form HOOOH as in Eq. 8
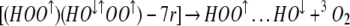 |
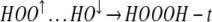 |
Assuming no bonding in the HOO↑…HO↓ on the left of Eq. 8, reaction 8 is 28.2 kcal/mol exothermic. In this case the product is expected to be dominantly in the trans form.
In contrast, reacting gas phase HO with HOO (in an overall triplet state) leads to H2O + 3O2 with no effective barrier (21§).
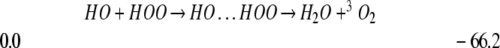 |
Thus the gas-phase reaction of HO with HOO would lead mostly to water with very little formation of HOOOH.
Experimentally, photolysis at 266 nm converts the initial complex [which we predict to be ring-(HO2)(HO3)] completely to water plus a species identified as H2O3 by the presence of two torsion bands and two OO stretches (2). Starting with 18O3 and H2O2, the photolysis experiment leads to formation of 100% H18OOOH (2). This result is consistent with our mechanism (from Eqs. 1 and 2 to 7 and 8), which also leads exclusively to the formation of H18OOOH. Table 2 summarizes the calculated frequencies of various isotopes of HOOOH, which are in good agreement with experiment (2).
Ref. 2 presented an alternative mechanism for the authors' observation of 100% H18OOOH. They speculated that in their experiments irradiation at 266 nm decomposed O3 to O(1D) and O2(1Δ) and that O(1D) inserted into the O–H of HOOH to make H18OOOH. However, we believe that O(1D) might also insert into the OO bond of HOOH, leading to HO18OOH. Thus we believe that this mechanism would give a mixture of H18OOOH and HO18OOH, in disagreement with their experiments.
Decomposition of H2O3.
Experimentally, prolonged irradiation eventually eliminates the bands assigned to HOOOH (2). Indeed, we find
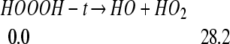 |
The HO and HO2 formed here can go through reaction 9, leading to the formation of H2O + 3O2.
We also find that H2O can catalyze the decomposition of HOOOH to 1O2 (4, 22). Thus
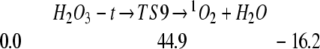 |
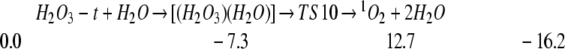 |
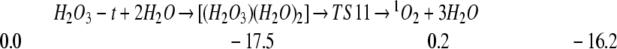 |
Thus complexes of one or two H2O with H2O3-t lead to decomposition barriers of 20.0 and 17.7 kcal/mol.
Discussion
Hydrogen peroxide, H2O2, is a widely known oxidant, but little is known of the stability and chemistry of higher hydrogen polyoxides. H2O3 has been postulated repeatedly (since Bertholet in 1880) to rationalize processes in combustion, explosions, atmospheric chemistry, water chemistry, biochemistry, and the radiation chemistry in aqueous systems (3–5, 22–32), but until recently it has not been known how or whether H2O3 is formed, when H2O3 can exist, and under which circumstances H2O3 will decompose. Thus H2O3 was proposed as a transient intermediate in the oxygenation of alkanes with ozone in superacid media (30) and in the pulse radiolysis of air-saturated perchloric acid solution (23). By means of 17O NMR spectroscopy, Plesnicar and coworkers (32) concluded that the low-temperature ozonation of isopropyl alcohol and isopropyl methyl ether yields hydrogen trioxide H2O3 (in addition to the hydrotrioxides of these compounds). Recently the antibody-catalyzed oxidation of water to hydrogen peroxide by 1O2 has been proposed to proceed through H2O3 as the critical intermediate (3–5).
This situation has changed with the recent synthesis of H2O3 in argon matrices by the photolysis of the ozone–hydrogen peroxide complex (2), and we have now clarified the mechanism by using first-principles quantum mechanics. We find that H2O3 is formed from the [(HO2)(HO3)-7r] precursor by both thermal and photolytic processes. Interestingly, use of labeled ozone (18O3) is predicted to yield H18O18O18OH by the thermal process and H18OOOH by the photochemical process. We report the predicted vibrational frequencies to aid in experimentally characterizing [(HO2)(HO3)-7r] and its chemistry and for developing strategies for synthesizing it and H2O3.
Our calculations reveal that formation of H2O3 from H2O2 + O3 is favorable both thermodynamically and kinetically. Irradiation of H2O3 with 266-nm radiation produces radicals (HO, HO2) that can form water, which would in turn induces H2O3 decomposition by reactions 10 and 11b and 11c. Thus we anticipate that systems in which the H2O3 is formed by thermal processes should exhibit a much longer lifetime for the H2O3 than those in which the H2O3 is formed photolytically (because this would also form H2O).
We suggest that [(HO2)(HO3)-7r] and H2O3 may both play roles in oxidative processes ranging from the peroxone environmental cleanup process to the antibody conversion of 1O2 and H2O to H2O2. Free OH is often postulated to be the reactive intermediate in such systems, despite the short lifetime. [(HO2)(HO3)-7r] is essentially a capped OH radical, giving it a much longer lifetime than free OH. Thus the chemistry of [(HO2)(HO3)-7r] would resemble that of free OH. For example, attack on an aromatic ring might proceed as
 |
where the HO2 in the complex can extract the H from the ring after attack by the OH.
It has long been recognized that hydrogen peroxide is an efficient catalyst for the decomposition of ozone (33) but that peroxone (O3 + H2O2) is effective at destroying organic contaminants in water chemistry (1). We believe that the spectacular effectiveness of the peroxone process results from a variety of reactive intermediates (H2O3, [(HO2)(HO3)-7r]) and their products (HO3, HO2, OH). Knowledge of the mechanistic details connecting these species should help to understand and improve the peroxone processes for environmental applications and should help establish the role of these intermediates in a variety of biological and environmental oxidations.
These same reactive intermediates (H2O3, [(HO2)(HO3)-7r]) and their products (HO3, HO2, OH) might also be formed in the atmosphere, where conditions of low temperature and low H2O concentrations might lead to modest concentrations of such intermediates. It is hoped that the mechanistic analysts and vibrational frequencies for the various intermediates provided here might help identify these species in atmospheric or laboratory experiments.
Conclusions
Detailed mechanisms for the formation of H2O3 from H2O2 + O3 are explored in this report. We predict that the codepositing of O3 and H2O2 in an argon matrix leads to formation of the [(HO2)(HO3)-7r] head-to-tail cyclic complex. We have predicted the vibrational frequencies of this complex to aid experimental validation.
We predict that thermal decomposition of [(HO2)(HO3)-7r] (by means of hydrogen transfer from the HO2 moiety to the HO3 moiety) will lead directly to H2O3 (with all 3 O atoms arising from the O3), whereas H2O will decompose this species to form 1O2 plus H2O. These predictions should help to define optimal experimental conditions for making H2O3.
On the other hand, UV irradiation of the [(HO2)(HO3)-7r] complex will break the HO–OO bond of the HO3 moiety, eliminating 3O2 and forming a [(HO2)(HO)] complex. We expect that this [(HO2)(HO)] confined in the matrix cage will rearrange to form H2O3. In this case just one of the terminal O atoms comes from O3. This formation of only H18OOOH from 18O3 has been observed experimentally.
We suggest that [(HO2)(HO3)-7r] and H2O3 are involved in biological, atmospheric, and environmental oxidative processes.
Acknowledgments
We thank Prof. Richard Lerner for pointing out the peroxone process and some references to us. This research was funded by National Institutes of Health Grant HD 36385-02 and the National Science Foundation. The facilities of the Materials and Process Simulation Center used in these studies were funded by a Shared University Research Grant from IBM and grants from the Defense University Research Instrumentation Program (Army Research Office and Office of Naval Research) and the National Science Foundation Major Research Instrumentation. In addition, the Materials and Process Simulation Center is funded by grants from the Department of Energy–Accelerated Strategic Computing Initiative–Academic Strategic Alliances Program, Army Research Office Multidisciplinary University Research Initiative, National Institutes of Health, National Science Foundation, ChevronTexaco, Seiko-Epson, Kellogg's, 3M, Asahi Kasei, Nippon Steel, and Toray.
Abbreviations
LM, local minimum
As pointed out in ref. 21, we find that the planar 3A" HO⋅⋅⋅HO2 has one imaginary frequency for the H–O⋅⋅⋅H bending, thus is not a true local minimum. Nonplanar HO⋅⋅⋅HO2 structure leads automatically to H2O + 3O2.
References
- 1.Prousek J. (1996) Chem. Listy 90 229-237. [Google Scholar]
- 2.Engdahl A. & Nelander, B. (2002) Science 295 482-483. [DOI] [PubMed] [Google Scholar]
- 3.Wentworth P., Jones, L. H., Wentworth, A. D., Zhu, X. Y., Larsen, N. A., Wilson, I. A., Xu, X., Goddard, W. A., Janda, K. D., Eschenmoser, A. & Lerner, R. A. (2001) Science 293 1806-1811. [DOI] [PubMed] [Google Scholar]
- 4.Xu X., Muller, R. P. & Goddard, W. A., III. (2002) Proc. Natl. Acad. Sci. USA 99 3376-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Datta D., Vaidehi, N., Xu, X. & Goddard, W. A., III (2002) Proc. Natl. Acad. Sci. USA 99 2636-2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slater J. C. (1951) Phys. Rev. 81 385. [Google Scholar]
- 7.Vosko S. H., Wilk, L. & Nusair, M. (1988) Can. J. Phys. 58 1200-1211. [Google Scholar]
- 8.Becke A. D. (1980) Phys. Rev. A 38 3098-3100. [DOI] [PubMed] [Google Scholar]
- 9.Becke A. D. (1993) J. Chem. Phys. 98 5648-5652. [Google Scholar]
- 10.Lee C. T., Yang, W. T. & Parr, R. G. (1988) Phys. Rev. B 37 785-789. [DOI] [PubMed] [Google Scholar]
- 11.Hehre W. J., Ditchfield, R. & Pople, J. A. (1972) J. Chem. Phys. 56 2257-2261. [Google Scholar]
- 12.Hariharan P. C. & Pople, J. A. (1973) Theor. Chim. Acta 28 213-222. [Google Scholar]
- 13.Baker J., Jarzecki, A. A. & Pulay, P. (1998) J. Phys. Chem. A 102 1412-1424. [Google Scholar]
- 14.Schrödinger & Inc., , (1998) jaguar (Schrödinger, Portland, OR), Version 3.5.
- 15.Dunning T. H. (1989) J. Chem. Phys. 90 1007-1023. [Google Scholar]
- 16.Curtiss L. A., Raghavachari, K., Redfern, P. C. & Pople, J. A. (1997) J. Chem. Phys. 106 1063-1079. [Google Scholar]
- 17.Herzberg G., (1965) Spectra of Diatomic Molecules, Molecular Spectra, and Molecular Structure (Van Nostrand, New York), Vol. 1, pp. 560. [Google Scholar]
- 18.Wittbrodt J. M. & Schlegel, H. B. (1996) J. Chem. Phys. 105 6574-6577. [Google Scholar]
- 19.Cremer D. (1978) J. Chem. Phys. 69 4456-4471. [Google Scholar]
- 20.Nelander B., Engdahl, A. & Svensson, T. (2000) Chem. Phys. Lett. 332 403-408. [Google Scholar]
- 21.Gonzalez C., Theisen, J., Schlegel, H. B., Hase, W. L. & Kaiser, E. W. (1992) J. Phys. Chem. 96 1767-1774. [Google Scholar]
- 22.Koller J. & Plesnicar, B. (1996) J. Am. Chem. Soc. 118 2470-2472. [Google Scholar]
- 23.Czapski G. & Bielski, B. H. J. (1963) J. Phys. Chem. 67 2180-2184. [Google Scholar]
- 24.Giguere P. A. & Herman, K. (1970) Can. J. Chem. 48 3473-3482. [Google Scholar]
- 25.Nangia P. S. & Benson, S. W. (1979) J. Phys. Chem. 83 1138-1142. [Google Scholar]
- 26.Diem M., Tso, T.-L. & Lee, E. K. C. (1982) J. Chem. Phys. 76 6452-6454. [Google Scholar]
- 27.Sugimoto H. & Sawyer, D. T. (1988) J. Am. Chem. Soc. 110 8707-8708. [Google Scholar]
- 28.Plesnicar B., Kovac, F. & Schara, M. (1988) J. Am. Chem. Soc. 110 214-222. [Google Scholar]
- 29.Plesnicar B., Cerkovnik, J., Koller, J. & Kovac, F. (1991) J. Am. Chem. Soc. 113 4946-4953. [Google Scholar]
- 30.Cerkovnik J. & Plesnicar, B. (1993) J. Am. Chem. Soc. 115 12169-12170. [Google Scholar]
- 31.Speranza M. (1996) Inorg. Chem. 35 6140-6151. [Google Scholar]
- 32.Plesnicar B., Cerkovnik, J., Tekavec, T. & Koller, J. (2000) Chem. Eur. J. 6 809-819. [DOI] [PubMed] [Google Scholar]
- 33.Bray W. C. (1938) J. Am. Chem. Soc. 60 82-87. [Google Scholar]