

Abstract
On the basis of the crystallographic snapshots of Rayment and his collaborators [Fisher, A. J., Smith, C. A., Thoden, J. B., Smith, R., Sutoh, K., Holden, H. M., & Rayment, I. (1995) Biochemistry 34, 8960–8972], we have understood some basic principles about the early stages of myosin catalysis, namely, ATP is drawn into the active site, over which the cleft closes. Catalyzed hydrolysis occurs, and the first product (orthophosphate) is released from the backdoor of the cleft. In the cleft-closing process, the active site incidentally signals its movement to a particular remote tryptophan residue, Trp-512. In this work, we expand on some of these ideas to rationalize the behavior of a mutated system in action. From the behavior of recombinant myosin systems in which Arg-247 and Glu-470 were substituted in several ways, we draw the conclusions that (i) the force between Arg-247 and γ-phosphate of ATP may assist in closing the cleft, and incidentally in signaling to the remote Trp, and (ii) in catalysis, Glu-470 is involved in holding the lytic H2O (w1). We also propose that w1 and also a second water, w2, enter into a structure that bridges Glu-470 and the γ-phosphate of bound ATP, and at the same time positions w1 for its in-line hydrolytic attack.
Research of the last half century established that the free energy of ATP+H2O, under the influence of the acto-myosin system, degrades in stages while accomplishing external work. The coupled gross movements that we observe are what we call muscle contraction. Underlying such changes are transition: namely, bindings and unbindings of actin or nucleotides, nucleotide hydrolyses, “influence” transmissions, and domain movements, etc. These transitions could not have been probed more deeply without ascertaining the structure of myosin at atomic resolution. This progress was pioneered by Rayment and his collaborators beginning in 1993 (1). Here, we are interested in the events that begin with the binding of ATP to the active site of myosin. In the Trentham–Bagshaw (2, 3) sequence of states, this is the formation of M⋅ATP.¶ We are also concerned with the transition to the prehydrolysis state, M*⋅ATP. Some inferences about the structural changes during these events have already been made by Smith and Rayment (5) based on their “before” and “after” crystallographic snapshots. Specifically, they note that, in the transition state of hydrolysis, Glu-470 pairs with Arg-247 to form a salt-bridge, as a loop (switch II) rotates about a hinge composed of Ile-466, Ala-467, and Gly-468. We have sought to check and enhance their ideas, using the logic that, if these ideas are correct, they should predict the functional behavior of M as it is altered in various site-directed mutations. Our results lead us to suggest that each of the two pairing residues has an important role in early ATP-myosin interaction, and that Glu-470 is probably a key residue in myosin ATP hydrolysis.
Materials and Methods
Protein Preparations.
F-actin was prepared from rabbit skeletal muscle as in ref. 6.
Preparation of Recombinant Heavy Meromyosins (HMM).
cDNA constructs for wild-type and five mutant (R247A, R247E, E470A, E470R, and R247E/E470R) HMM heavy chains linked to a His tag (at the N terminus) and a myc tag (at the C terminus) were prepared as described in Kojima et al. (7). Site-directed mutations were performed by Kunkel's method (8) using various primers (9, 10). Transformation of Escherichia coli and production of recombinant baculoviruses were carried out according to the manufacturer's protocol (Life Technologies, Rockville, MD). AcNPV/ELC/RLC viruses (for essential and regulatory light chains) were prepared (11). Expression and purification of recombinant HMMs were carried out (7, 12), except that affinity-purified His-tagged HMMs were further purified by chromatography using a Pharmacia MonoQ column. To remove bound nucleotides from mutant HMMs, the preparation was dialyzed for 2 days against 500 ml of buffer, 0.45 M KCl, 1 mM EDTA, 20 mM Tris⋅HCl (pH 7.5), and 0.5 mM DTT, on ice with four-time buffer changes.
Fluorimetry and Stopped-Flow Experiments.
Fluorimetry was performed with an F-4500 fluorescence spectrophotometer (Hitachi, Tokyo). Stopped-flow experiments were performed with a Hi-Tech Scientific (Salisbury, U.K.) SF61-DX2 stopped-flow spectrophotometer using a 75-W Xe/Hg lamp and a monochromator for excitation wavelength selection. The binding of mant fluorophores to the active site of HMMs was detected by measuring the fluorescence energy transferred from Trp residues to fluorophores. Trp residues were excited at 290 nm, and the emitted light from fluorophores was monitored at 455 nm with a bandwidth of 5 nm (for fluorimetry), or after passing through an LWPF 410-nm filter (Asahi Techno Glass, Tokyo) (for stopped-flow experiments). For Trp fluorescence, excitation was performed at 290 nm, and emission was recorded after passing through a WG 320-nm filter (Schott, Mainz, Germany). Software provided by Hi-Tech was used for curve fitting of the data.
ATPase Assays.
The steady-state ATPase activity was measured at 20°C in 0.24 mg/ml HMM, 0.1 mg/ml BSA, 0.45 M KCl, 3 mM MgCl2, 20 mM Tris⋅HCl (pH 7.5), 0.5 mM DTT, 0.5 mM ATP, and 1 mM EDTA. Actin-activated ATPase activity was measured as a function of actin concentration in 0.054 mg/ml (wild-type) or 0.24 mg/ml (mutants) HMM, 0.04 M KCl, 2 mM MgCl2, 20 mM Tris⋅HCl (pH 7.5), 0.5 mM DTT, 1 mM ATP, 4 μg/ml chicken gizzard myosin light chain kinase, 1 μg/ml bovine testis calmodulin, and 0.05 mM CaCl2. Released inorganic phosphate was estimated by the malachite green method (13). The activity is per HMM head. Initial phosphate burst was measured as in ref. 9.
ATP Detection by HPLC.
The amount of ATP in HMM preparations was measured by HPLC (14).
Results and Discussion
Kinetic Scheme for Myosin Mg-ATPase.
It has been established (2) that myosin Mg-ATPase can be described by the following minimal reaction scheme:
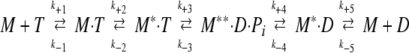 |
where M is myosin, and T, D, and Pi are ATP, ADP, and inorganic phosphate, respectively. k+i and k−i are forward and backward rate constants, respectively. The single and double asterisks (* and **) refer to different conformations as detected by intrinsic protein fluorescence. Step 1 corresponds to the formation of a collision complex of myosin and ATP. Binding of ATP is followed by a conformational change (step 2), and, subsequently, ATP is hydrolyzed (step 3). The produced Pi and ADP are released from the ternary complex in steps 4 and 5, respectively. In the wild-type system, steps 1, 2, and 3 are fast, and step 4 rate limits the overall reaction. Here, in analyzing the Mg-ATPase of myosin mutants, we used the foregoing reaction scheme with selected rate constants. An assumption is that differences between the behavior of wild-type and mutant systems are expressible solely as differences in rate constants, keeping the reaction connectivities the same.
Binding of mantATP to HMMs.
To detect binding of nucleotides by wild-type and mutant HMMs, we used a fluorescent nucleotide [2′(3′)-O-(N-methylanthranilloyl) adenosine 5′-triphosphate (mantATP)]. Because the excitation spectrum of mant fluorophores closely overlaps the emission spectrum of Trp residues, the observed fluorescence is energy transferred from Trp residues to fluorophores bound at the active site of HMMs. Addition of excess mantATP produced increases in fluorescence of 82%, 93%, 87%, 58%, and 91% for R247A, R247E, E470A, E470R, and R247E/ E470R mutants, respectively, compared with 100% for wild-type (denoted as “fast” in Table 1). In each of these HMMs, the fluorescence-time transient was well fitted by a single exponential curve. The observed rate constants (kobs) were linearly dependent on the mantATP concentration, in the range from 5 to 60 μM (Fig. 1). The slope of the plots of kobs against [mantATP] depends on the occupants of positions 247 and 470. The second-order rate constant (K1k+2) of nucleotide binding was greatly decreased for R247A, and decreased even more for R247E, whereas it was slightly increased for E470A and increased more for E470R (Table 1). We explain these results by assuming that γ-phosphate occupies a position closer to position 247 than to position 470. For R247E/E470R, K1k+2 was close to that of wild type (Table 1). Therefore, we think that, in the inverted arrangement, γ-phosphate moves to interact with R at 470.
Table 1.
Summary of the rate constants and extents for the interaction of nucleotides with wild-type and salt-bridge mutant HMMs
| Measurements | Parameters | Wild-type | R247A | R247E | E470A | E470R | R247E/E470R |
|---|---|---|---|---|---|---|---|
| MantATP binding | Fast, % | 100 | 82 | 93 | 87 | 58 | 91 |
| K1k+2 (s−1⋅M−1) | 3.5 × 105 | 0.49 × 105 | 0.28 × 105 | 4.5 × 105 | 5.7 × 105 | 2.2 × 105 | |
| Slow, % | 33 | ||||||
| kslow, s−1 | 0.0001 | ||||||
| Trp fluorescence | Increase, % | 19 | 1.8 | 1.1 | 16 | 4 (6.7) | 15 |
| K1k+2 (s−1⋅M−1) | 2.5 × 105 | 0.33 × 105 | 0.23 × 105 | 3.1 × 105 | 4.5 × 105 | 1.6 × 105 | |
| kmax, s−1 | 166 | 92 | 172 | ||||
| K0.5, μM | 650 | 330 | 980 | ||||
| Phosphate burst | Mole per mole of HMM head | 0.68 | 0 | 0 | 0 | 0 | 0 |
| MantATP release | k+3 or k+3k+4/(k+3 + k−3), s−1 | 0.009 | 0.003 | 0.0011 | 0.00014 | 0.000036 | 0.001 |
| MantADP release | k+5, s−1 | 1.3 | 0.027 | 0.0036 | 0.28 | 0.34 | 0.031 |
| Steady-state ATPase | kcat, s−1 | 0.005 | 0.002 | 0.0005 | 0.00008 | 0.00003 | 0.0004 |
| Actin-activated ATPase | kmax, s−1 | 1.9 | 0 | 0 | 0 | 0 | 0 |
| Kactin, μM | 120 |
Conditions: 0.45 M KCl, 3 mM MgCl2, 1 mM EDTA, 20 mM Tris⋅HCl (pH 7.5) at 20°C.
The estimated increase for E470R of which sites are vacant.
Conditions: 200 μM F-actin, 0.04 mM KCl, 2 mM MgCl2, 20 mM Tris⋅HCl (pH 7.5) at 25°C.
Fig 1.

Rates of binding of mantATP to wild-type and mutant HMMs. Various concentrations of mantATP were mixed with 0.56 μM HMMs in a rapid mixing stopped flow fluorometer, and the increases in the energy transfer from Trp residues to bound mantATP were recorded. Observed rate constants kobs were plotted as a function of the mantATP concentration. The slope of the plots of kobs vs. [mantATP] are the second-order rate constants of mantATP binding (K1k+2) and are listed in Table 1. HMMs: Wild-type (○), R247A (▵), R247E (□), E470A (▴), E470R (▪), and R247E/E470R (•). Conditions: 0.45 M KCl, 3 mM MgCl2, 1 mM EDTA, 20 mM Tris⋅HCl (pH 7.5) at 20°C.
It should be noted that E470R shows a slow transient of fluorescence increase (denoted as “slow” in Table 1). In that way, the fluorescence increase of this mutant produced by adding mantATP finally reached 91% of the value obtained with wild type. In contrast to the faster one, the rate of the slower component was independent of the mantATP concentration (data not shown). This behavior was also very similar to that in a single turnover of mant fluorophore recorded on chasing with excess ATP. Moreover, analysis by reverse-phase HPLC revealed that E470R contains about one-third mole of ATP per mole of head (data not shown). Therefore, we can assume that some of the enzyme sites of E470R are occupied with ATP. This result suggests that the slow rate of mantATP binding is due to the slow release of nonfluorescent occupants from enzyme sites.
Increase in Trp Fluorescence of HMMs by Binding of ATP.
Myosins respond to binding of ATP with an increase in intrinsic protein fluorescence. Several studies (12, 15–17) have suggested that Trp-512 (of smooth muscle myosin) is the responsible Trp residue. Crystallographic studies demonstrated that the ATP-induced rotation of the lower piece of 50 kDa, which bears Trp-512 at its tip, can perturb this fluorophore (5). Here, we took the increase in Trp fluorescence as an indicator of the rotation of the 50-kDa lower piece, the rotation being presumably equivalent to the transition from the “open” to the “closed” state identified in crystallography. The maximum increase in Trp fluorescence depends strongly on the occupants of positions 247 and 470. The addition of excess ATP increased the Trp fluorescence by 1.8%, 1.1%, 18%, 4%, and 15% in the case of R247A, R247E, E470A, E470R, and R247E/E470R, respectively, compared with 19% for wild type (Table 1). These results suggest that R at 247 is the most important residue for the open → closed transition occurring in step 2, but that in the inverted arrangement, R at 247 is functionally replaceable by R at 470. As described above, the E470R used had one-third of its enzyme sites occupied with ATP. Therefore, the real increase in Trp fluorescence might have been as large as 6.7%, still smaller than the value for wild type. We explain this result by assuming that a repulsive strain between two R residues at 247 and 470 partially interferes with the open-closed transition.
Trp fluorescence-time transients were well fitted by a single exponential. The observed rate constants were linearly dependent on the concentration of ATP at low ATP concentrations (Fig. 2). K1k+2 can be obtained for the rate of developing Trp fluorescence as well as the rate of developing mantATP fluorescence (Table 1). The numbers are very similar for both. These results support our assumption that the position of γ-phosphate of bound ATP is closer to position 247 than to position 470. At higher ATP concentrations, kobs was no longer linearly dependent on the ATP concentration. The plots of kobs against [ATP] were well described by hyperbolas, kobs = kmax [ATP]/([ATP] + K0.5). The maximum rates (kmax) of 166 s−1, 92 s−1, and 172 s−1 were obtained for wild type, E470A, and R247E/E470R, respectively (Table 1). In wild type, the transition from M*⋅T to M**⋅D⋅Pi (step 3) is also fast. Although the maximum rate of this transition could not be obtained from Pi measurements, we assigned to kmax of the fluorescence signal the rate constant of step 2, (k+2 + k−2), because the fluorescence signal showed no deviation from a single exponential curve. Because E470A and R247E/E470R have very slow transitions from M*⋅T to M**⋅D⋅Pi (see Fig. 3), we suggest that kmax definitely corresponds to k+2 + k−2.
Fig 2.

Rates of increases in Trp fluorescence of wild-type and mutant HMMs on addition of ATP. HMMs (0.56 μM) were mixed with various concentrations of ATP in a rapid mixing stopped-flow fluorometer, and the increases in the Trp fluorescence were recorded. Observed rate constants kobs were plotted as a function of the ATP concentration. The slopes of the regressions to the linear portions at lower ATP concentrations are the second-order rate constants of ATP binding (K1k+2). The plot was well described by a hyperbola [kobs = kmax[ATP]/([ATP] + K0.5)]. kmax is the predicted value for kobs at infinite [ATP] and K0.5 is the ATP concentration in which kobs = kmax/2. K1k+2, kmax, and K0.5 are listed in Table 1. HMMs: Wild-type (○), R247A (▵), R247E (□), E470A (▴), E470R (▪), and R247E/E470R (•). Conditions: 0.45 M KCl, 3 mM MgCl2, 1 mM EDTA, and 20 mM Tris⋅HCl (pH 7.5) at 20°C.
Fig 3.

Initial Pi bursts in the hydrolysis of ATP by wild-type and mutant HMMs. Assay conditions were 0.20 mg/ml of each of wild-type (○), R247A (▵), R247E (□), E470A (▴), E470R (▪), and R247E/E470R (•) HMMs, 0.45 M KCl, 3 mM KCl, 1 mM EDTA, 20 mM Tris⋅HCl, pH 7.5, 0.5 mM DTT, and 4 μM [γ-32P]ATP. The data of wild-type HMM were cited from ref. 10. The burst size, determined by extrapolating the steady-state Pi release to zero time, was 0.68 moles of Pi per mole of HMM head for the wild-type HMM, but no burst was obtained for any of the mutant HMMs.
Initial Pi Burst in ATP Hydrolysis.
Hydrolysis of ATP by wild type occurs rapidly, and results in a metastable ternary complex between myosin, ADP, and Pi. When the ATPase reaction is quickly terminated by acid, a stoichiometric Pi burst is obtained (18). Previously, we have found that the burst size in wild type was 0.68 moles per mole of HMM head, whereas four single mutants (R247A, R247E, E470A, and E470R) exhibited no Pi burst (9, 10). In the same paper, we also reported that the burst size of R247E/E470R was quite similar to that of wild type. Later, however, we discovered that, due to a labeling mistake, the mutant that we described as the double mutant was actually P548G (19). When we reexamined, using the authentic double mutant, we found that no burst at all was generated by the true double mutant. In the inverted arrangement, the ability to form the salt-bridge did not per se restore normal ATP hydrolysis. These results clearly show that hydrolysis does not always accompany the open → closed transition measured by the ATP-induced increase in Trp fluorescence. Earlier work (9, 10) and also the present work show that R247A, R247E, E470A, E470R, and R247E/E470R exhibit no Pi burst, but do exhibit the ATP-induced increase in Trp fluorescence, so we must assume that, in such cases, [M*⋅T] is greater than [M**⋅D⋅Pi], i.e., the equilibrium constant for step 3 (K3) is much smaller than 1. For rabbit skeletal subfragment 1, it was found that K3 = ≈10 (100 mM KCl, pH 7.0, 20°C; ref. 2).
Turnover of Fluorescent Intermediates in the mantATP Hydrolysis.
Fig. 4A shows time-dependent changes in mant fluorescence on sequential additions of mantATP and ATP to wild-type HMM. We can assume that the high, constant phase represents saturation of the enzyme sites with fluorophores. When 1 μM mantATP was added, this phase was followed by a phase in which fluorescence was decreasing. This decrease can be taken to measure the diminishing fraction of sites that are occupied by fluorophore. When 4 μM mantATP was added, the high, constant phase became longer. After maximal mantATP binding, the HMM–mantATP complex was then mixed with 100 μM nonfluorescent ATP, which took over all vacant sites. Because, according to the kinetic scheme (2, 3), the Pi release step (step 4) is rate-limiting, the decay of mant fluorescence on adding excess ATP is an indirect measure of the single turnover rate of ATP hydrolysis. A rate constant of 0.009 s−1 for k+4 was obtained by this measure. The time course of the decay in fluorescence after adding excess ATP was rather similar to that of the decrease in fluorescence after adding 1 μM mantATP.
Fig 4.

Turnover rates of fluorescent intermediates measured by the decay of fluorescence from wild-type HMM– and mutant HMM–mantATP complexes on addition of excess ATP. (A) MantATP (1 and 4 μM) was added to 0.56 μM wild-type HMM at the arrows labeled with 60 and 530 s, respectively. Increasing fluorescence was used to monitor nucleotide binding to the enzyme sites. At 600 s, 100 μM nonfluorescent ATP was added to the reaction mixture. Because there is no longer any “new” mantATP that can bind to enzyme sites, the rate of decrease in fluorescence is expressed by the turnover rate of the fluorescent intermediate. (B and C) Decays of the fluorescences from mant fluorophores bound to mutants on adding excess ATP. MantATP (1 μM) was added to 0.56 μM HMMs (▵, R247A, □, R247E, and •, R246E/E470R, in B; and ▴, E470A, and ▪, E470R, in C), bringing the system to a high, maintained fluorescence level. While in the steady state, 100 μM ATP was added (at time 0). This addition causes the fluorescence to start decaying. The continuous lines are the best fits to single exponential decays. Numerical values of kobs are listed in Table 1. Conditions: 0.45 M KCl, 3 mM MgCl2, 1 mM EDTA, 20 mM Tris⋅HCl (pH 7.5), and 0.5 mM DTT, at 20°C.
In mutants, because step 3 is very slow, the rate constant of decay of mant fluorescence on addition of 100 μM ATP is not as simple as it is in wild type. Its value is k+3 if k+3 < k+4 and k+3k+4/(k+3 + k−3) if k+3 > k+4. Fig. 4 B and C shows that this rate constant depends on occupants at positions 247 and 470, but the dependency is very different from either the K1k+2 of nucleotide binding (Figs. 1 and 2, and Table 1), or the ATP-induced increase in Trp fluorescence (Table 1). The rates for R247A and R247E were reduced 3- and 9-fold, respectively, compared with wild type, but those for E470A and E470R were reduced by 65- and 225-fold, respectively. The especially slow rate for mutants at position 470 suggests that the charge change (− to 0, or − to +), or perhaps the replacement of a carbonyl group at 470, is very significant in inhibiting hydrolysis. The rate of decay in mant fluorescence in R247E/E470R (Fig. 4B) was 25-fold faster than that in E470R, but its rate was 9-fold slower than in wild type. We can assume that either the second charge change (R247E) or the ability to form the inverted salt-bridge partially restores the steady-state ATP hydrolysis, but never as well as in the wild-type arrangement.
MantADP Removal from HMM-mantADP Complexes.
The rate of ADP removal from M*⋅D (step 5) was measured by displacing mantADP from HMM–mantADP complexes with excess ATP. The rate of the decrease in fluorescence expresses the rate of removal of mantADP. If the rate of mantADP removal is faster than the rate of removal of earlier steps, we conclude that removal of bound ADP cannot be the overall rate-limiting step in the sequence. When 100 μM ATP was added to a prepared mixture of 0.56 μM HMMs and 2 μM (for mutants) or of 10 μM (for wild type) mantADP, the plots of transient decreases against time of fluorescence could be fitted by single exponentials. The observed rate constants of mantADP removal (k+5) were 1.3 s−1 for wild type, 0.027 s−1 for R247A, 0.0011 s−1 for R247E, 0.28 s−1 for E470A, 0.34 s−1 for E470R, and 0.031 s−1 for R247E/E470R, respectively (Table 1). In wild type, the rate of removal of mantADP by adding excess ATP was much faster than the rate of mant fluorescence decrease when the initial substrate was mantATP (see Fig. 4), supporting the established conclusion that the rate-limiting step in the steady state is the transition from M**⋅D⋅Pi to M*⋅D (step 4), rather than the removal of ADP from M*⋅D (step 5). In two of the single mutants, R247A and R247E, the rates at which mant fluorescence decreases, when the initial substrates were mantADP and mantATP, were still different, but much nearer to one another (the ratio of the former vs. the latter was 9 for R247A and 3 for R247E, compared with 140 for wild type). Interestingly, two other single mutants showed opposite effects, being the ratio of 2,000 for E470A and 8,500 for E470R. Again, this observation supports our idea that Arg at 247 and Glu at 470 have different roles in the ATP hydrolysis.
Steady-State ATP Hydrolysis.
From experiments consisting of chasing bound mant fluorophores by excess ATP, we can identify any one of several intermediates that can be only slowly removed. To decide whether the step determined by chase experiments is rate-limiting in the cycle of ATP hydrolysis, we performed steady-state measurements of ATPase at 0.5 mM ATP. The steady-state ATPase activity (kcat) was 0.005 s−1 for wild type, 0.002 s−1 for R247A, 0.0005 s−1 for R247E, 0.00008 s−1 for E470A, 0.00003 s−1 for E470R, and 0.0004 s−1 for R247E/E470R (Table 1). Because the dissociation constant of HMMs for ATP is much less than 0.5 mM, the observed rates are maximal at saturating ATP concentration. When compared at the corresponding HMMs, the kcat values and the rates of decay of mant fluorescence on adding excess ATP to HMM-mantATP complexes were very similar. Therefore, we can conclude that the decay of mant fluorescence in chase experiments is limited by whatever is the slowest step in the ATPase cycle.
Actin activated the ATPase activity of wild-type HMM, yielding a maximum rate of 1.9 s−1 and an apparent dissociation constant of 120 μM for actin. Previous kinetic studies on rabbit skeletal acto-subfragment 1 ATPase showed that actin primarily accelerates the step of releasing inorganic phosphate from M**⋅D⋅Pi. If any one of earlier steps is limited by mutations, we would not observe actin activation. To examine whether salt-bridge mutants are such a case, we measured the steady-state ATPase activity in the presence of 200 μM [actin]. Practically no actin activation was observed in any of the mutant cases (Table 1). These results are consistent with our observation that the rate of hydrolysis of bound ATP (step 3) is strongly limited in these mutations (Fig. 3).
Possible Roles of R247, E470, and the Salt-Bridge in ATP Hydrolysis.
In this work, we take a rapid increase in fluorescence emission from HMM as evidence that, on binding at the active site, an influence quickly travels from the site to a particular residue, Trp-512 (12, 15–17). In the case in which a mant fluorophore has access to the active site, under illumination at 290 nm, we take a rapid increase in fluorescence at 455 nm as evidence that the fluorophore is more or less statically bound to the active site (assuming that such a fluorophore is tuned only to accept energy from Trp and then re-emit it at 455 nm), but we do not claim to know the identity of the originating Trp.
From the successive crystallographic snapshots by Rayment and his colleagues (5, 20), we have gained an insight into the events after the binding of ATP to the active site of myosin: The cleft closes, and Arg-247 and Glu-470, once long separated, come together into a salt-bridge. In this process, Arg-247 comes to lie close to the γ-phosphate of the bound ATP, as does Glu-470. Simultaneously, these effects are transmitted toward Trp-512. Also, in some hitherto unexplained way, a water molecule at a particular location is prepared to attack the P-O-P bridge of ATP. After the bridge severance, the cleft reopens and orthophosphate is released from the back door of the cleft.
In the present work, we find that, even after making substantial structural substitutions by site-directed mutation, we can continue to track in time various functional parameters. Specifically, these are the rate and extent at which mant-ATP occupies the active site, the rate and extent at which signals from the active site reach Trp-512, and the rate at which bound ATP is hydrolyzed. When we measure these parameters in various mutants, we find a very interesting pattern of results (Table 1). We denote paired mutations at the salt-bridge positions as 247 and 470, and identify RE as wild-type; we have also studied AE, EE, RA, RR, and ER. A simple and very good correlation is found between the presence of R and the rate of occupation of the active site by a mant analog. Similarly there is a good correlation between influences reaching Trp-512 and the presence of R. On the other hand, there is a strong correlation between catalyzed hydrolysis of ATP and the presence of E. Lastly, it is evident that occupation of the active site, and perturbation of Trp-512, can both occur without hydrolysis (e.g., RA). Translating these results into tentative conclusions, we suggest that, as surmised by Smith and Rayment (5), Arg-247 and Glu-470 certainly have important functions in forming a salt-bridge. In our view, the electrical force between Arg-247 and the γ-phosphate of the ATP bound to the active site, although attenuated in solution, is probably significant for cleft closure; this closure is required to form the salt-bridge. If some other force, e.g., interactions between the P-loop and the triphosphate moiety of the ATP, is involved in the cleft closure, the long-range force between R and the γ-phosphate would at least stabilize the closed interaction. In anomalous circumstances, closure can occur without hydrolysis. Also, in the present work, the tight correlation between hydrolysis and the presence of E leads us to revive our old hypothesis that Glu-470 per se is involved in catalyzing the hydrolysis (9). Formation of the salt-bridge would move Glu-470 to obligatorily participate in the catalysis. In the cases of AE and RA, the reasons for failing to catalyze properly would be different. In the case of AE, closure is poor and E does not move to the proper position, whereas in the case of RA, closure occurs, but E is lost.
Our results, reported above, raise the question as to how the two residues, Arg-247 and Glu-470, can interact specifically with a bound γ-phosphate separated by >5 Å, and why crystallography reveals no amino acid side chain in the γ-phosphate pocket that is near enough to the phosphate to function as the catalytic base in hydrolysis. Building on the reports of Fisher et al. (20) and of Smith and Rayment (ref. 5, discussion about plasticity conferred on the γ-phosphate pocket by hydrogen bonding), and on our present results, we speculate about the mechanism of myosin catalysis. We suppose that two water molecules are involved, the lytic water, w1, and also a second water, w2. In the structure of the MgADP⋅BeFx complex that mimics the prehydrolysis state, M⋅T, w1 is hydrogen-bonded to a fluoride of BeFx and to the main-chain carbonyl oxygen of Ser-246 (Fig. 5A). w1 also interacts electrostatically with the guanydyl group of Arg-247. We propose that Arg-247 thus indirectly interacts with γ-phosphate via this water, and so, for this reason, Arg-247 is important in nucleotide binding to the active site, and in inducing cleft closure. On the other hand, in the same motor domain, but now with MgADP⋅vanadate (which mimics the transition state to M**⋅D⋅Pi), w2 appears in the phosphate pocket (see Fig. 5B). w2 is hydrogen-bonded to the main-chain carbonyl oxygen of Gly-468 and to one of the two oxygen atoms in the carboxyl group of Glu-470. This finding suggests that, after cleft closure, movement of Glu-470 results, first, in bridging it to Arg-247, then in releasing w1 from Arg-247, and, finally, in bonding of the two hydrogen atoms of w1 with the oxygen atom of w2 and with the main-chain carbonyl oxygen of Ser-246. Then, the conformation becomes optimal, orienting w1 for a nucleophilic attack on the γ-phosphorus. The two hydrogen atoms of w2 are oriented toward the carboxyl group of Glu-470 and to the main-chain carboxyl oxygen of Gly-468. These oxygen atoms seem to function as proton acceptors, so we can speculate that w2 accepts a proton from w1, after w1 has made its attack on the γ-phosphorus (see Fig. 5B). Alternatively, the hydrogen bond network around w1 may stabilize a trigonal-bipyramidal arrangement of oxygen atoms around the γ-phosphorus in the transition state.
Fig 5.

Schematic representation of the γ-phosphate pocket of the prehydrolytic state (A) and of the transition state for hydrolysis (B). In the former, the γ-phosphate of ATP is in a tetrahedral arrangement with its four oxygen atoms, whereas, in the latter, five oxygen atoms bound to the γ-phosphate adopt a trigonal bipyramidal arrangement. Two water molecules involved in hydrogen bonding are denoted as w1 and w2. We assume that w1 in A is HOH1181 in the Fisher et al. structure (ref. 20; Protein Data Bank ID code ), and that w1 and w2 in B take the place of the terminal oxygen of the vanadate moiety and that of HOH697, respectively, in the Smith and Rayment structure (ref. 5; Protein Data Bank ID code ). Only those stereochemical interactions that participate in positioning w1, w2, and three oxygen atoms (labeled 1, 2, and 3) of the γ-phosphate are depicted. Covalent bonds are shown as solid lines and hydrogen bonds and ionic interactions in dashed lines. Phosphorus, oxygen, hydrogen, and magnesium are depicted in yellow, red, violet, and green, respectively.
The structures found by Rayment harmonize well with our reported correlation between the presence of E in the salt-bridge and the ability of the system to catalyze hydrolysis. The inverted mutant, R247E/E470R, supports a fluorescence enhancement of ≈80% of that in a wild-type system, but not a phosphate burst. Presumably this result is because the influence of R on the γ-phosphate is long-ranged, whereas the positioning of w2 in the catalytic apparatus has to be precise. In other hydrolyzing systems, e.g., G-proteins, the lytic water is required to move to an in-line position (21–24). Perhaps, in myosin catalysis, the Glu-470-w2-Gly-468 network plays the role that the catalytic Gln plays in G-proteins. In the interesting simulations of Okimoto et al. (25) about events in ATPase, obligatory participation is assigned to Glu-470. However, the simulations do not take explicit account of w2; perhaps for that reason, they deduce an unusually large energy of activation.
In wild-type myosin, formation of M**⋅D⋅Pi is very fast compared with release of the produced Pi, so this intermediate accumulates when the system is in the steady state. A reasonable speculation for the slowness with which Pi is released is that the release corresponds to the cleft reopening, and that such a process takes time. Neither AE nor EE exhibits a Pi burst. This result suggests that [M**⋅D⋅Pi] is very small, even though the mutants achieve an ATPase activity comparable to that of the wild-type system. In our view, this result would mean that, in these mutants, either the cleft reopens faster or it never closes well. Recently, Manstein and colleagues (26) found that, in Dictylstelium, with the mutation corresponding to EE, the cleft remains open, even in the presence of the tightly binding nucleotide analog, MgADP⋅BeFx. Our speculation seems to be consistent with their finding.
We close with a technological note. Mutants such as RR, in which M*⋅T is highly stabilized (presumably by overattraction to γ-phosphate), are very difficult to purify by dialysis, and for that reason they exhibit properties that are easy to misinterpret.
Acknowledgments
We are grateful to Professor H. M. Martinez for his important counsel and Professors T. Burghardt, I. Rayment, and J. T. Pearson for significant improvements in our manuscript. This work was supported by Grant-in-Aids for Scientific Research and by Special Coordination Funds for Promoting Science and Technology from the Ministry of Education, Culture, Sports, Science, and Technology (to H.O. and N.M.) and by Grant MCB 9603670 from the National Science Foundation (to M.F.M.).
Abbreviations
HMM, heavy meromyosin
mantATP, 2′(3′)-O-(N-methylanthranilloyl) adenosine 5′-triphosphate
w1, lytic water
w2, second water
Whereas M is to be read as myosin, the protein used in our work has the sequence of the truncated, doubly headed heavy meromyosin (HMM) extracted from chicken smooth muscle. Myosin exhibits many species differences, but the residues of interest here are highly conserved. The sequence numeration that we use is given in ref. 4. We note that Ser-245, Ser-246, Arg-247, Gly-468, Glu-470, and Trp-512 correspond to Ser-236, Ser-237, Arg-238, Gly-457, Glu-459, and Trp-501 in Dictyostelium myosin.
References
- 1.Rayment I., Rypniewski, W. R., Schmidt-Bäse, K., Smith, R., Tomchick, D. R., Benning, M. M., Winkelmann, D. A., Wesenberg, G. & Holden, H. M. (1993) Science 261 50-58. [DOI] [PubMed] [Google Scholar]
- 2.Bagshaw C. R. & Trentham, D. R. (1974) Biochem. J. 141 331-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bagshaw C. R., Eccleston, J. F., Eckstein, F., Goody, R. S., Gutfreund, H. & Trentham, D. R. (1974) Biochem. J. 141 351-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yanagisawa M., Hamada, Y., Katsuragawa, Y., Imamura, M., Mikawa, T. & Masaki, T. (1987) J. Mol. Biol. 198 143-157. [DOI] [PubMed] [Google Scholar]
- 5.Smith C. A. & Rayment, I. (1996) Biochemistry 35 5404-5417. [DOI] [PubMed] [Google Scholar]
- 6.Spudich J. A. & Watt, S. (1971) J. Biol. Chem. 246 4866-4871. [PubMed] [Google Scholar]
- 7.Kojima S., Fujiwara, K. & Onishi, H. (1999) Biochemistry 38 11670-11676. [DOI] [PubMed] [Google Scholar]
- 8.Kunkel T. A., Roberts, J. D. & Zakour, R. A. (1987) Methods Enzymol. 154 367-382. [DOI] [PubMed] [Google Scholar]
- 9.Onishi H., Morales, M. F., Kojima, S., Katoh, K. & Fujiwara, K. (1997) Biochemistry 36 3767-3772. [DOI] [PubMed] [Google Scholar]
- 10.Onishi H., Kojima, S., Katoh, K., Fujiwara, K., Martinez, H. M. & Morales, M. F. (1998) Proc. Natl. Acad. Sci. USA 95 6653-6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Onishi H., Maéda, K., Maéda, Y., Inoue, A. & Fujiwara, K. (1995) Proc. Natl. Acad. Sci. USA 92 704-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Onishi H., Konishi, K., Fujiwara, K., Hayakawa, K., Tanokura, M., Martinez, H. M. & Morales, M. F. (2000) Proc. Natl. Acad. Sci. USA 97 11203-11208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kodama T., Fukui, K. & Kometani, K. (1986) J. Biochem. (Tokyo) 99 1465-1472. [DOI] [PubMed] [Google Scholar]
- 14.Samizo K., Ishikawa, R., Nakamura, A. & Kohama, K. (2001) Anal. Biochem. 293 212-215. [DOI] [PubMed] [Google Scholar]
- 15.Johnson W. C., Bivin, D. B., Ue, K. & Morales, M. F. (1991) Proc. Natl. Acad. Sci. USA 88 9748-9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hiratsuka T. (1992) J. Biol. Chem. 267 14949-14954. [PubMed] [Google Scholar]
- 17.Batra R. & Manstein, D. J. (1999) Biol. Chem. 380 1017-1023. [DOI] [PubMed] [Google Scholar]
- 18.Kanazawa T. & Tonomura, Y. (1965) J. Biochem. (Tokyo) 57 604-615. [PubMed] [Google Scholar]
- 19.Onishi H., Kojima, S., Katoh, K., Fujiwara, K., Martinez, H. M. & Morales, M. F. (2001) Proc. Natl. Acad. Sci. USA 98 5369a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fisher A. J., Smith, C. A., Thoden, J. B., Smith, R., Sutoh, K., Holden, H. M. & Rayment, I. (1995) Biochemistry 34 8960-8972. [DOI] [PubMed] [Google Scholar]
- 21.Pai E. F., Krengel, U., Petsko, G. A., Goody, R. S., Kabsch, W. & Wittinghofer, A. (1990) EMBO J. 9 2351-2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coleman D. E., Berghuis, A. M., Lee, E., Linder, M. E., Gilman, A. G. & Sprang, S. R. (1994) Science 265 1405-1412. [DOI] [PubMed] [Google Scholar]
- 23.Sondek J., Lambright, D. G., Noel, J. P., Hamm, H. E. & Sigler, P. B. (1994) Nature 372 276-279. [DOI] [PubMed] [Google Scholar]
- 24.Schweins T., Geyer, M., Scheffzek, K., Warshel, A., Kalbitzer, H. R. & Wittinghofer, A. (1995) Nat. Struct. Biol. 2 36-44. [DOI] [PubMed] [Google Scholar]
- 25.Okimoto N., Yamanaka, K., Ueno, J., Hata, M., Hoshino, T. & Tsuda, M. (2001) Biophys. J. 81 2786-2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kliche W., Fujita-Becker, S., Kollmar, M., Manstein, D. J. & Kull, F. J. (2001) EMBO J. 20 40-46. [DOI] [PMC free article] [PubMed] [Google Scholar]