

Abstract
We describe here a method for measuring DNA replication and, thus, cell proliferation in slow turnover cells that is suitable for use in humans. The technique is based on the incorporation of 2H2O into the deoxyribose (dR) moiety of purine deoxyribonucleotides in dividing cells. For initial validation, rodents were administered 4% 2H2O in drinking water. The proliferation rate of mammary epithelial cells in mice was 2.9% per day and increased 5-fold during pregnancy. Administration of estradiol pellets (0–200 μg) to ovariectomized rats increased mammary epithelial cell proliferation, according to a dose–response relationship up to the 100 μg dose. Similarly, proliferation of colon epithelial cells was stimulated in a dose–response manner by dietary cholic acid in rats. Bromodeoxyuridine labeling correlated with the 2H2O results. Proliferation of slow turnover cells was then measured. Vascular smooth muscle cells isolated from mouse aorta divided with a half-life in the range of 270–400 days and die-away values after 2H2O wash-out confirmed these slow turnover rates. The proliferation rate of an adipocyte-enriched fraction from mouse adipose tissue depots was 1–1.5% new cells per day, whereas obese ad libitum-fed ob/ob mice exhibited markedly higher fractional and absolute proliferation rates. In humans, stable long-term 2H2O enrichments in body water were achieved by daily 2H2O intake, without toxicities. Labeled dR from fully turned-over blood cells (monocytes or granulocytes) exhibited a consistent amplification factor relative to body 2H2O enrichment (≈3.5-fold). The fraction of newly divided naive-phenotype T cells after 9 weeks of labeling with 2H2O was 0.056 (CD4+) and 0.043 (CD8+) (replacement rate <0.1% per day). In summary, 2H2O labeling of dR in DNA allows safe, convenient, reproducible, and inexpensive measurement of cell proliferation in humans and experimental animals and is well suited for slow turnover cells.
Keywords: deuterated water, cell proliferation, DNA synthesis, adipogenesis, vascular smooth muscle cell proliferation
Cell division and death play central roles in normal tissue homeostasis and in numerous pathophysiologic processes. The most characteristic biochemical feature of cell division is DNA synthesis, occurring essentially only during S-phase of the cell cycle (1). Accordingly, cell division rates have usually been measured by DNA synthesis, based on the incorporation of labeled biosynthetic precursors into replicating polydeoxyribonucleotide chains (2, 3). The true precursors for DNA synthesis, the four deoxyribonucleotide-triphosphates (dNTPs; Fig. 1) in the cell's nucleus typically have been labeled by administration of tagged pyrimidine deoxyribonucleosides, e.g., [3H]thymidine or 5-bromodeoxyuridine (3HdT and BrdUrd, Fig. 1; refs. 2 and 3). This approach has several important limitations, however. In particular, these reagents cannot be used safely in humans because of toxicities (4, 5), are difficult to interpret during the die-away or de-labeling phase for assessment of cell death (6) and are particularly problematic for measurement of kinetics of cell populations that turn over slowly in vivo.
Fig. 1.

(A) Labeling pathways for measuring DNA synthesis and thus cell proliferation. (B) Sites of 2H incorporation from 2H2O into C–H bonds of dR in replicating DNA. GNG, gluconeogenesis/glycolysis; PPP, pentose–phosphatase pathway; RR, ribonucleotide reductase; DNPS, de novo purine/pyrimidine synthesis pathway; DNNS, de novo nucleotide synthesis pathway.
Toxicities of 3HdT and BrdUrd include mutagenicity, impairment of cell division (e.g., myelosuppressive effects), and killing of susceptible cells (4, 5). Biochemical complications, such as reutilization of labeled pyrimidine nucleosides released from dying cells (7, 8) and unpredictable uptake efficiency of administered label by dividing cells (5–8), constrain measurement of slow turnover (long-lived) tissues.
Therefore, it should be no surprise that controversies remain concerning the rate (or even the occurrence) of cell division for many cells believed to be long-lived. Cells in this class include mature adipocytes (9, 10), vascular smooth muscle cells (VSMC), neurons, epithelial stem cells (11), naive-phenotype T lymphocytes (12), and others.
We recently developed a stable isotope-mass spectrometric technique for measuring DNA synthesis that resolved some of these problems (12–14). The deoxyribose (dR) moiety of dNTPs in replicating DNA can be labeled endogenously, through the de novo nucleotide synthesis pathway by using stable isotope 2H- or 13C-labeled glucose (Fig. 1A). The isotopic enrichment of the purine deoxyribonucleosides [deoxyadenosine (dA), or deoxoguanosine (dG)] are then determined by gas chromatographic/mass spectrometric (GC/MS) analysis after isolation and hydrolysis of genomic DNA. Because no radioactivity or genotoxic agents are involved, this technique is safe for use in humans. Also, several technical advantages simplify interpretation: the de novo synthesis pathway contribution to purine dNTPs predominates and is relatively constant (7, 8, 13), is unaffected by extracellular nucleoside concentrations (13), and derives almost entirely from extracellular glucose (Fig. 1A; ref. 13), so that the precursor–product relationship can be used in a predictable way across cell types (13). Also, reutilization of free dR from degraded DNA does not occur, and reutilization of purine deoxyribonucleotides is minor (7, 8, 13); enzymes of the de novo synthesis pathway, particularly ribonucleotide reductase, are up-regulated during S-phase of the cell cycle (in contrast to enzymes of the nucleoside salvage pathway, which are suppressed during S-phase; refs. 7 and 8). The [2H]glucose technique has been applied to the proliferation of T lymphocytes (12, 14), granulocytes (13), hepatocytes (13), monocytes,¶ thymocytes (13), colonocytes,∥ and other cells.**
Use of stable isotope-labeled glucose is not ideal for the measurement of slowly dividing cell populations, however, because of the rapid metabolic removal of glucose and the relatively high cost of maintaining a constant level of isotopic enrichment in the dNTP precursor pool for prolonged periods of time. Accordingly, our primary goal here was to develop a stable isotope-mass spectrometric technique for measuring DNA synthesis that would be suitable for measurement of slow-turnover cells in whole animals, including humans. The approach described here is based on the incorporation of deuterated water (2H2O) into dR of newly synthesized DNA (Fig. 1). We apply this method to a number of cell types for which the replacement rate remains uncertain. Portions of this work have been reported previously in abstract form.††,‡‡,§§
Methods
General Procedures.
Biochemistry of 2H2O incorporation into dR of replicating DNA.
Hydrogen atoms from cellular water enter into the seven carbon–hydrogen (C–H) bonds of dR in dNTPs through several well characterized enzymatic steps (Fig. 1B), including the pentose-phosphate pathway (contributing labeled hydrogen atoms to positions C-2 and C-4 of dR), ribonucleotide reductase (position C-2 of dR), and gluconeogenesis/glycolysis (all C–H bonds in dR; ref. 15). Entry of 2H from 2H2O into the base-moiety of purine and pyrimidine dNTPs also occurs in a variable manner (depending upon the activity of the free base salvage pathway; Fig. 1A). To avoid this latter complication, the derivative that we analyzed contains only the dR moiety, not the base portion, of purine deoxyribonucleosides.
Another important analytic point is that amplification of isotope enrichment occurs in dR relative to 2H2O in cellular water. There are seven positions on the dR carbon skeleton that potentially can be labeled from H2O (Fig. 1B). Because the likelihood of double labeling of dR is low in the body 2H2O enrichment range of 1–4% (e.g., <0.3% double-labeled species if 2H2O is 2.0% enriched; ref. 16), the fraction of single-labeled (M1)-dR species represents a nearly linear sum of the 2H-enrichments at each C–H position (see below).
Measurement of stable isotope enrichment in dR from DNA.
After isolation of specific cell types, genomic DNA was prepared as described (12, 14). The DNA was enzymatically hydrolyzed to free deoxyribonucleosides, then the dA in the hydrolysate was purified with an LC18 SPE column (Supelco; ref. 12). Purified dA was derivatized in one of two ways. The deoxyribose-aldonitrile-triacetyl (dRATA) derivative was synthesized as described (12). In addition, a pentane-tetraacetate (PTA) derivative of the sodium borohydride-reduced dR was used (17).
For some samples, nucleosides in the DNA hydrolysate were separated by HPLC (13). The dA, dG, dC, and dT peaks were collected and converted to the PTA derivative of dR for mass spectrometric analysis.
Measurement of Body 2H2O Enrichments.
Enrichment of 2H2O in body water (blood or urine) was measured by a recently developed GC/MS technique (17). H2O enrichments are calculated by comparison to standard curves generated by mixing 100% 2H2O with natural abundance H2O in known proportions.
Mass Spectrometric Analyses.
A model 5973 mass spectrometer was used with a model 6890 gas chromatograph and autosampler (all from Hewlett–Packard). Isotopic enrichments of either derivative were calculated by comparison to abundance-corrected standards prepared from unlabeled dA. Both PTA and dRATA were analyzed by using a 20-m DB-225 column (0.18 mm i.d., 0.2 μm film thickness, J & W Scientific, Folsom, CA). The PTA derivative was analyzed by methane chemical ionization mass spectrometry at m/z 245 (M0) and 246 (M1). The dRATA was also analyzed by methane chemical ionization mass spectrometry at m/z 198 (M0) and 199 (M1). In both cases, unlabeled standards were analyzed during the same run to provide a profile of the dependence of the ratio of labeled to unlabeled ions on amount of sample injected. This dependence can be characterized by plotting the abundance of the unlabeled mass (M0, i.e., m/z 198 or 245) vs. the ratio of labeled to unlabeled ions [i.e., 199/(198 + 199) or 246/(245 + 246)]. At low abundances of material injected, a cut-off was determined below which no samples would be included. Above that abundance, a linear regression of the ratio vs. M0 abundance was calculated. This regression line was then used to calculate the baseline ratio at any particular M0 abundance (18).
Chemicals and Reagents.
2H2O (70% or 99% enriched) was purchased from Cambridge Isotope Laboratories (Cambridge, MA). Chemicals were purchased from Sigma, except where indicated otherwise.
Animal Studies: Housing and Care.
Sprague–Dawley rats (200–250 g) and C57BL/6J mice (10–20 g, The Jackson Laboratory) were used. All procedures were approved by the University of California Berkeley Office of Laboratory Animal Care. Housing was in individual cages for rats and fewer than five per cage for mice. Diet was Purina Chow, provided ad libitum. A 12-h light/12-h dark cycle was maintained.
2H2O Administration Protocol.
The 2H2O labeling protocol was the same in all animals and consisted of an initial i.p. priming bolus of 99% 2H2O to 2.0–2.5% body water enrichment (based on estimated 60% body weight as water), followed by administration of 4% 2H2O in the drinking water for the duration of the study. Dry chow (Purina) and water intake were provided ad libitum. Mice were killed by cervical dislocation; rats were killed by CO2 asphyxiation. The duration of 2H2O administration and timing of tissue collection are described separately for each of the protocols (see below).
Measurement of Proliferation of Different Tissues.
Proliferation of mammary epithelial cells (MEC).
Both rats and mice were studied: an estradiol dose-response study in ovariectomized rats and the effects of pregnancy in mice. Rats were ovariectomized, then implanted s.c. 14 days before killing with estrogen-containing miniosmotic pellets containing 0–200 μg of estradiol per pellet, as described (19). 2H2O was administered concurrently during the 14-day treatment period. Mice were mated by housing one male with two female mice per cage; female mice were checked daily for vaginal plugs. 2H2O was provided from 1 day before mating until full-term delivery (25 days total), at which time the mice were killed. Of 14 females, 13 became pregnant and were studied. Control, virgin female mice were given 2H2O for the same time period (total of 25 days) for comparison. The mammary glands of rats and mice were dissected postmortem and MEC were isolated by the method of Nandi et al. (19), by enzymatic cell dissociation and Percoll-gradient (Amersham Pharmacia) centrifugation.
Proliferation of colon epithelial cells.
Male rats were given 2H2O for 3 days before killing while on ad libitum chow diets. Cholic acid (CHA) was provided in the diet at 0%, 0.2%, and 0.5%, by weight. The rats were killed by carbon dioxide asphyxiation and colon was excised immediately. The cecum was discarded and colonocytes were isolated, with the removal of intraepithelial lymphocytes as described (20).
Colon epithelial cells were isolated by mechanical agitation from three different proliferative zones of the crypt by using a modification of methods published (21). The three zones are, from the bottom to the top of the crypt, proliferative, transit, and mature cell zones. Data are shown here for the proliferative zone only.
Rats received an i.p. dose of BrdUrd (160 μg/kg body weight in DMSO) 3 h before killing. Tissue for immunohistochemistry was taken from the distal end of the colon (1 cm) and fixed in 10% (vol/vol) PBS-buffered formalin overnight, followed by BrdUrd staining, as described (22). Labeling index [(no. of cells incorporating BrdUrd)/total no. of cells counted] was determined by using a Zeiss confocal microscope with video camera and monitor. At least 10–15 well defined crypts (1,000–1,500 total cells) were counted.
Proliferation of vascular smooth muscle cells (VSMC) in mouse aorta.
Female mice received 2H2O for 3 weeks (from 3–6, 15–18, or 18–21 weeks of age; n = 4 per group) and then were killed. VSMC were isolated from aorta according to the protocol of Travo et al. (23). Briefly, the aortas (from the aortic arch to the iliac bifurcation) were incubated for 30 min with HBBS (GIBCO) containing 1 mg/ml collagenase type 1 (Sigma), then the adventitial layer was stripped off with forceps. The endothelial layer was scraped off mechanically. The resulting sleeve-like medial layer contains VSMC (23), from which DNA was isolated and isotopic enrichment was measured as detailed above. The purity of VSMC in this preparation was confirmed histologically, by staining for smooth muscle cell actin and the macrophage surface marker CD19 (Histo-Tec, Hayward, CA). Histologic evaluation also confirmed the absence of macrophages in the vessel wall, including the medial layer (courtesy of N. R. Raju, Covance Laboratories, Berkeley, CA).
Proliferation of mature adipocyte-enriched fraction of mouse adipose tissue.
Female ob/ob mice (The Jackson Laboratory; n = 4) and lean C57/Bl6 ungenotyped (+/?) littermates (n = 7) were studied. Starting at age 5 weeks, 4% 2H2O was administered for 21 days, and then animals were killed. Mice were fed a purified mouse diet ad libitum (3.7 kcal/g, 4.9% fat calories, Bioserv, Frenchtown, NJ) and were housed individually in hanging wire cages.
Cell fractions enriched for mature adipocytes and for stromal-vascular cells were isolated from fat tissue by the method of Rodbell (24). In brief, fat was dissected quantitatively from several anatomic depots (epidydimal, retroperitoneal, mesenteric, and inguinal). Collagenase digestion was performed to release cells from the fibrous connective tissue, followed by centrifugation (24). The stromal-vascular compartment was taken from the infranatant cell fraction. The supernatant fraction was filtered through 350-μm nylon mesh filters (Spectrum Laboratories, Houston) to remove debris for recovery of the mature adipocyte-enriched fraction. Microscopic evaluation after toluene blue and oil red O staining revealed scattered areas containing strands of incompletely digested, stromal-type cells among the predominant lipid-rich cells; consequently, we termed this fraction adipocyte-enriched (or stromal-vascular-depleted) rather than pure mature adipocytes.
Collection of bone marrow cells.
Bone marrow cells were used as a potential measure of a fully turned-over tissue (i.e., completely replaced DNA) for calculation of fractional replacement rates of less rapidly turned-over cells (ref. 13, and see Calculations).
Human Studies.
2H2O administration.
Healthy subjects (n = 21, age 38 ± 5 years) were recruited by advertisement. Because transient vertigo or dizziness has been reported as a rare adverse effect of rapid changes in body 2H2O enrichment (25), we administered 2H2O to human subjects for the first 24 h under observation in a metabolic ward setting at the General Clinical Research Center of the San Francisco General Hospital. The 2H2O administration protocol consisted of 50-ml doses given every 3 h for 18–24 h in the metabolic ward. No subject experienced significant adverse symptoms by using this protocol. One subject described transient dizziness, which was resolved within 30 min. The study volunteers were maintained on a 60- to 70-ml daily intake of 2H2O, with a goal of maintaining ≈1.5–2% body water enrichment (assuming total body water turnover of ≈3.5 liters per day in healthy, ambulatory subjects). Weekly urine and saliva samples were obtained during the 9-week 2H2O administration protocol for measurement of body 2H2O enrichment.
Isolation of blood monocytes and granulocytes.
Blood CD14+ monocytes were isolated from Ficoll-Hypaque gradients of blood by use of anti-CD14 magnetic beads (Miltenyi Biotec, Auburn, CA) in subjects who had naive-phenotype T cells isolated (n = 9). The monocyte fraction was resuspended in 200 μl of PBS, and DNA was isolated. Blood granulocytes were isolated from the buffy coat of Ficoll-Hypaque gradients by standard techniques.
Kinetics of naive-phenotype T lymphocytes in blood.
Healthy subjects (n = 9) received 2H2O for 9 weeks. Blood was collected at baseline and at weeks 5 and 9 of 2H2O administration. Naive-phenotype T cells were isolated by multiparameter fluorescence-activated cell sorting (FACS), as described (12). Briefly, sort purification was with a dual laser (argon 310 nm, argon 488 nm) FACSVantage (Becton Dickinson). CD4+ or CD8+ T cells with the CD45RA+CD62L+ phenotype were retrieved as naive-phenotype T cells (12).
Other Procedures.
Incubation of DNA with 2H2O in vitro.
As a test for nonenzymatic exchange of labeled water into DNA that is not undergoing biochemical replication, we incubated 100% 2H2O with intact DNA (salmon sperm, Sigma) for 24–48 h. DNA was then hydrolyzed to nucleosides for analysis of dR isotopic enrichment by GC/MS.
Calculations.
Fractional replacement rates of cells were calculated by use of the precursor–product relationship (13, 14, 26). The isotopic enrichment of a completely (or nearly completely) turned-over tissue was used as a measure of true precursor (dNTP, Fig. 1) enrichment for the cells of interest:
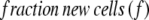 |
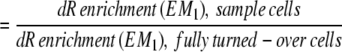 |
 |
Results
Animal Studies: Body Water Enrichments Attained.
The steady-state body 2H2O enrichments attained in rats by GC/MS measurements were in the range of 2.8% (range, 2.2–3.2%); in mice, they were slightly lower (2.0–2.4%). The dilution between 2H2O enrichment of drinking water and body water represents metabolic water produced by fuel oxidation as well as any residual moisture in the dry chow. 2H2O enrichments in body water were stable over time in animals maintained on 2H2O in their drinking water (Fig. 2A). We also measured the rate of 2H2O clearance from body water pools of mice after discontinuing 2H2O in the drinking water (Fig. 2B). Within 1 week, plasma 2H2O enrichments had fallen by >85% and by week 2, 2H2O enrichments were essentially zero. Thus, uncontaminated die-away curves of 2H-labeled cellular DNA can be monitored beginning ≈2 weeks after stopping labeled drinking water (see below).
Fig. 2.

(A) Body 2H2O enrichments over time in rats drinking 4% 2H2O. Urine was collected every 2 weeks during 2H2O administration for up to 12 weeks. Data shown are means ± SD (n = 5–7 per time point). (B) Die-away curves of body 2H2O enrichments after cessation of 4% 2H2O intake in mice. Urine was collected at time 0 and then weekly for 3 weeks. (C) Enrichments of bone marrow dR (from dA isolated from DNA) in mice maintained on 4% 2H2O in drinking water for up to 11 weeks (n = 8–10 per time point). Animals were killed after 5, 7, and 11 weeks of 2H2O administration. EM1: excess M + 1 mass isotopomer enrichment. (D) Comparison of isotopic enrichments of dR (PTA derivative) from HPLC-isolated nucleosides of bone marrow cells. Mice (n = 13) had been maintained on 4% D2O in drinking water for 21 days. Data shown are means ± SD. *, P < 0.05 vs. dA and dG.
DNA Labeling in Rapid-Turnover Tissues.
The isotopic enrichment of dR in dA isolated from bone marrow DNA was used as a marker of maximal labeling to calculate fractional replacement rates of other cells (Eq. 1). Bone marrow cells reached peak dR enrichments within 7–10 days and remained at relatively stable levels for the subsequent 8–10 weeks (Fig. 2C). The two derivatives, which differ in the number of potentially labeled C–H bonds in dR (see above, Fig. 1B) were compared. The isotopic enrichments from the PTA derivative were consistently higher (e.g., 9.5–10%) than with the dRATA derivative (7.0–8.0%, not shown) at mean body 2H2O enrichments of 2.6%. The amplification factor from body water labeling in dR was, therefore, ≈3.5- to 4.0-fold (i.e., 3.5–4.0% for each percent enrichment in 2H2O) for the PTA derivative and 3.0-fold for the dRATA derivative. It should be noted that the maximal EM1 enrichment in dR, calculated by using combinatorial principles described (16), is 11.7% at a body water enrichment of 2.6% because of the presence of EM2 and EM3 species (2.5% and 0.4%, respectively). Thus, plateau values observed here were ≈75% of the theoretical maximum, which is consistent with a predicted dilution caused by the purine nucleoside salvage pathway, after correction for glycolytic/gluconeogenic flux (16).
Fetal tissues in mice also were collected after maternal intake of 4% 2H2O from preconception to the time of delivery. Isotopic enrichment (using the PTA derivative) of dA from DNA in cardiac muscle was 8.5 ± 0.1%; from skin cells, it was 8.5 ± 0.2%; from hepatocytes, 9.1 ± 0.1%; and from brain cells, it was 8.1 ± 0.2%. Maternal 2H2O enrichment was 2.4%. The ratio of dR:2H2O enrichments in fetal tissues was, therefore, ≈3.5–3.8 (PTA derivative).
Comparison of dA to dG, dC, and dT Enrichments.
The isotopic enrichments of dR derivatized from dA, dG, dC, and dT were compared after HPLC isolation of the nucleosides from hydrolysates of mouse bone marrow cells (13). The dA and dG enrichments (10.33 ± 0.88% and 9.99 ± 1.04%, respectively) were not significantly different, and both were higher than dT (7.99 ± 0.68%) or dC (6.40 ± 0.41%) (Fig. 2D).
Absence of Label Incorporation into DNA Incubated in Vitro with 2H2O.
To exclude the possibility of nonbiosynthetic incorporation of 2H2O into dR of DNA (i.e., exchange), we incubated salmon sperm DNA in vitro with 100% 2H2O. No label was measurable in dR after 24–48 h of incubation (data not shown).
Proliferation of MECs in Response to Hormonal Stimuli.
To test the sensitivity of the 2H2O-labeling technique to changes in cell proliferation rates, we studied two settings known to stimulate MEC division: pregnancy and administration of estrogen after ovariectomy (19). After 25 days of labeling in nonpregnant female mice, newly divided cells represented 52.4 ± 14.5% (EM1 = 0.0342 ± 0.0101; bone marrow EM1 = 0.0653 ± 0.0064) of total MEC present (k = 0.029 d−1, t1/2 = 28.5 days, Fig. 3A). Pregnancy increased the MEC proliferative fraction to 88.5% (EM1 = 0.0557 ± 0.008; bone marrow = 0.0634 ± 0.0077; k = 0.085d−1; Fig. 3A, P < 0.01 vs. nonpregnant animals) and the total MEC DNA content of the mammary gland (17.6 ± 11.6 vs. 90.0 ± 43.0 μg DNA; P < 0.01). The total number of MEC that had divided exhibited an ≈9-fold increase (9.3 ± 6.5 to 80.8 ± 41.2 μg newly synthesized DNA; P < 0.001) compared with placebo-treated animals (Fig. 3A Lower).
Fig. 3.

(A) MEC proliferation in pregnant mice compared with nonpregnant controls after 25 days of 2H2O administration. Mating and analyses are as described in the text. (Upper) Fraction of newly divided cells (f) present. (Lower) Absolute DNA synthesis (μg). (B) Dose–response curve of MEC proliferation vs. estradiol dose administered to ovariectomized rats. Estradiol was administered by s.c. miniosmotic pump, as described in the text. f, fraction of newly divided cells present after 14 days of 2H2O administration (n = 5 per group).
A dose–response relationship was observed between the dose of implanted estradiol and MEC proliferation in ovariectomized rats labeled for 1–2 weeks in the range between 0 and 100 μg pellets (MEC EM1 increasing from 0.0049 ± 0.0014 to 0.0251 ± 0.0043 at 100 μg pellets, Fig. 3B). No further increase in proliferation was observed at >100 μg doses. Body 2H2O enrichments and bone marrow EM1 enrichments were not significantly different among estradiol treatment groups (not shown).
Colon Epithelial Cell Proliferation.
CHA has well established stimulatory effects on colon epithelial cell proliferation, perhaps by activation of protein kinase C (27). Accordingly, we evaluated the sensitivity of the 2H2O method for measuring stimulation of colonocyte proliferation by CHA. A dose–response relationship between CHA dose (0%, 0.2%, and 0.5% CHA in diet) and proliferation of the basal (proliferative) zone of the colonic crypt was observed (Fig. 4), with fractional replacement of 22.3%, 38.8%, and 62.5% per day, respectively, on the three doses of CHA (EM1 increasing from 0.0263 ± 0.0041 to 0.0480 ± 0.0083 and 0.0738 ± 0.0116, respectively). BrdUrd incorporation confirmed the dose-related stimulation observed by 2H2O incorporation (percent BrdUrd-positive cells 1.4-fold higher on 0.2% CHA than on 0% CHA and 2.4-fold higher on 0.5% CHA than on 0% CHA). There was much greater variability for the BrdUrd method than the 2H2O measurements, however (not shown).
Fig. 4.

Colon epithelial cell proliferation (proliferative zone of crypt) in response to dietary CHA. f, fraction of newly divided cells present. Cells were isolated from the proliferative (bottom) zone of the colon crypt. 2H2O was administered for 3 days. Data shown are means ± SD (n = 4 per group).
VSMC Proliferation in Mouse Aorta.
Female mice were labeled with 4% 2H2O for 3 weeks. Mean somatic growth (weight change) was 44% between weeks 3 and 6 (6.4 g), 1% from weeks 15 to 18 (0.25 g), and 3% from weeks 18 to 21 (1.0 g). VSMC proliferation rates were 5.3 ± 2.2% (weeks 3–6), 3.4 ± 1.7% (weeks 15–18), and 4.9 ± 3.5% (weeks 18–21). Calculated fractional replacement rates were 0.0026 ± 0.0011 d−1, 0.0017 ± 0.0008 d−1, and 0.0024 ± 0.0018 d−1, respectively. VSMC half-lives were calculated, therefore, to range between 267 and 408 days. Die-away curves of VSMC DNA also were measured after discontinuing 2H2O administration. Between weeks 2 and 8 off of 2H2O (when body 2H2O enrichments were unmeasurable, Fig. 2B), VSMC dR enrichments fell by 10% (from 0.0040 to 0.0036), reflecting a fractional removal rate of 0.0025 d−1, which is consistent with the estimates from 2H2O incorporation.
Proliferation of Mature Adipocyte-Enriched Cells from Adipose Tissue of Control and ob/ob Mice.
Fractional proliferation rates of the mature adipocyte-enriched (stromal-vascular cell-depleted) fraction isolated from adipose tissue depots of mice are shown (Fig. 5A). The proliferation rate was low (≈1–1.5% new cells produced per day) in lean control mice, with different anatomic depots exhibiting different rates. Proliferation rates were higher in the stromal-vascular-enriched compartment (≈5% new cells produced per day, not shown) than in the mature adipocyte-enriched fraction, which is consistent with the model that preadipocytes divide and differentiate from the stromal-vascular compartment (28). Fractional proliferation rates were higher in ad libitum-fed, obese ob/ob mice (Fig. 5A, 2.5–3.3% new cells per day). After correction for the markedly increased adipocyte pool size in the ob/ob mice (calculated from DNA recovered from the adipocyte-enriched fraction), absolute rates of cell proliferation were even more elevated in ob/ob mice compared with lean controls (Fig. 5B).
Fig. 5.

Cell proliferation in the mature adipocyte-enriched (stromal-vascular cell-depleted) fraction isolated from adipose depots of control (n = 7) and ob/ob (n = 4) mice. Animals received 4% 2H2O in drinking water for 21 days. Controls weighed 18.5 ± 0.2 g (means ± SE) at the start of 2H2O administration and 20.8 ± 0.3 g at the end; ob/ob weighed 26.3 ± 0.6 g and 35.7 ± 0.9 g, respectively. (A) Fraction of newly divided cells (f). *, P < 0.05 vs. controls. (B) Absolute cell proliferation rate (calculated from total DNA recovery multiplied by f. *, P < 0.05 vs. controls.
Human Studies: Body Water Enrichments Attained.
Plateau body water enrichments were variable but were generally between 1.5–2.0% in human subjects during 50–70 ml of 2H2O intake per day for up to 10 weeks after the initial 24-h priming protocol (Fig. 6A). These plateau enrichments were consistent with a daily body water turnover of ≈3.5 liters per day (or ≈10% per day of the estimated body water pool of ≈35 liters). Delabeling kinetics of body 2H2O also were consistent with this calculated body water turnover rate. Near-zero 2H2O enrichments in body water were observed within 3–4 weeks after discontinuing 2H2O intake (not shown).
Fig. 6.

(A) Body 2H2O enrichments in human subjects (n = 21) maintained on 50–70 ml of 2H2O intake per day for up to 10 weeks. (B) Correlation between blood monocyte and granulocyte dR enrichments in humans (n = 9) after 5–10 weeks of 2H2O intake. Blood cells were isolated as described in the text; DNA was isolated and hydrolyzed, and the enrichment of dR in dA was measured. (C) Ratio of dR:body 2H2O enrichments from monocytes and granulocytes of human subjects (n = 9) after 5–10 weeks of 2H2O intake. The PTA derivative of dA was analyzed. Data are means ± SD.
Blood monocyte and granulocyte enrichments.
The steady-state enrichments of dR in dA isolated from blood monocyte and granulocyte DNA were highly correlated (Fig. 6B). Values for both cells were ≈3.5 times the concurrent body 2H2O enrichments (Fig. 6C), similar to the values observed in fully turned-over rodent tissues.
Naive-Phenotype T Lymphocyte Kinetics.
Enrichments of naive-phenotype (CD45RA+CD62+) T cells were determined after 5 and 9 weeks of 2H2O administration in healthy human subjects (n = 8) and compared with enrichments in blood granulocytes and monocytes from the same subjects to calculate fractional replacement rates (Fig. 7). The fraction of newly divided naive-phenotype CD4+ T cells was 3.0 ± 1.7% (T cell EM1 = 0.0018 ± 0.0012; monocyte EM1 = 0.0600 ± 0.0012) after 5 weeks and 5.6 ± 3.6% after 9 weeks (T cell EM1 = 0.0032 ± 0.0019; monocyte EM1 = 0.0598 ± 0.0016). For CD8+ T cells, the values were 2.7 ± 1.3% (EM1 = 0.0017 ± 0.0009) and 4.3 ± 2.3% (0.0025 ± 0.0011), respectively. Fractional replacement rate constants were calculated to be ≈0.1% per day, or a half-life of ≈700 days.
Fig. 7.

Time course of labeling in naive-phenotype T cells isolated by FACS from healthy human subjects (n = 8) receiving 2H2O for 10 weeks. Naive-phenotype T cells were isolated from blood as described (12). Fraction of newly divided cells (f) is shown. Data are shown as means ± SEM.
Discussion
We describe here an in vivo method for measuring the rates of proliferation and replacement of slow-turnover cells that can be used safely in human subjects. Because controversy still exists concerning the rate of proliferation of many important slow-turnover cell types, including neurons, VSMC, adipocytes, and naive-phenotype T cells, the ability to measure cell division rates in these tissues may prove useful for resolving basic questions in human biology.
The technique is supported by internal as well as external validation. A number of conditions or agents known to be associated with increased proliferation of specific cells, e.g., the effects of pregnancy or estradiol on MEC, CHA on colonocytes, and ob/ob mutation on adipocytes, all confirmed increased proliferation when measured by the 2H2O incorporation method. Dose–response relationships were apparent for estradiol and for CHA (Figs. 3 and 4). Also, BrdUrd incorporation paralleled 2H2O results, although the BrdUrd method exhibited greater variability. Moreover, labeled VSMC die-away curves after wash-out of 2H2O from body water provided another confirmation of the 2H2O incorporation technique, independent of the model described here (Fig. 1). The absence of 2H2O exchange into dR of nonreplicating DNA in vitro excludes the occurrence of nonspecific labeling, independent of DNA replication. Removal of the base moiety from the derivative used for mass spectrometric analysis of deoxyribonucleoside enrichment reduces artifacts potentially induced by physiologic variability in purine or pyrimidine base metabolism (7, 8). Restriction of analyses to dR from purine deoxyribonucleosides further reduces physiologic variability because of the relatively constant contribution from the de novo nucleotide synthesis pathway to the dR moiety of replicating DNA described for purines (7, 8, 13) and is supported by data presented here (Fig. 2D).
An attractive feature of the 2H2O/mass spectrometric technique is its operational simplicity. Deuterated water simply has to be added to drinking water, preferably after an initial priming dose. Therefore, it is almost no extra trouble or effort to extend a study for as long as is optimal, e.g., several months or longer. The 2H2O is also relatively inexpensive to administer because of the slow turnover of body water. The ease of sampling bodily water enrichment, by serial sampling of urine, blood, or saliva, is an important advantage for out-patient or field studies. The ≈3.5- to 4.0-fold amplification between body 2H2O enrichment and dR enrichment (because of the multiple C–H bonds in dR that can exchange with body water during deoxyribonucleotide synthesis; Fig. 1B) and the fact that this amplification is predictable (Figs. 2 and 6) represent important practical advantages. Finally, the availability of fully or nearly fully turned-over tissues (e.g., bone marrow cells in rodents and circulating monocytes or granulocytes in humans) during long-term labeling protocols allows the use of fully turned-over cells as internal standards for calculating fractional replacement rates. These features of simplicity, flexibility, reproducibility, and predictability make this approach well suited for routine applications.
Some basic questions about the kinetics of slow-turnover cell types were addressed here. Whether aortic VSMC are replaced within the lifetime of a rodent or whether all VSMC proliferation could be explained by somatic growth has been uncertain. The observation that VSMC proliferate even in weight- and length-stable, nongrowing adult female rodents implies that replacement of VSMC occurs (although with a very slow turnover: t1/2 = 300–400 days). This observation was possible only because the long-term labeling required for such a slow turnover cell population is easily performed with 2H2O administration. Similar considerations apply for the estimates of naive-phenotype T cell turnover in humans. The true replacement rate of this cell population seems to be in the range of 0.1% per day, or a half-life of ≈2 years. Because the turnover rate of naive-phenotype T cells is so low, previous measurements using 24–48 h of [2H]glucose labeling (12) were less certain than measurements of more rapidly turning-over T cell populations and may have included transitional proliferation (i.e., cells activated and in transition to memory/effector surface phenotype).
Of note, the technique was easily and safely performed in human subjects. An important finding technically was that the plateau isotopic enrichments in fully replaced cells (e.g., granulocytes or monocytes after 5–10 weeks of labeling) exhibited a similar relationship and amplification factor to body 2H2O enrichments as were observed in rodents.
In conclusion, a technique is now available for measuring the in vivo proliferation rates of both rapidly and slowly dividing cells in humans and experimental animals. Here, we describe some applications for cell types such as breast epithelial cells, colonocytes, VSMC, adipose-tissue cells, and T lymphocytes. Many other applications of this technique can be envisioned.
Abbreviations
dNTP, deoxyribonucleotide-triphosphate
dR, deoxyribose
dA, deoxyadenosine
dG, deoxoguanosine
GC/MS, gas chromatographic/mass spectrometric
PTA, pentane-tetraacetate
MEC, mammary epithelial cells
CHA, cholic acid
VSMC, vascular smooth muscle cells
Mohri, H., Tung, K., Ramrotnam, B., Furlan, S., Monard, S., Markowitz, M., Harley, A., Kost, R., Cesar, D., Abe, K., et al. (2000) Seventh Conference on Retroviruses and Opportunistic Infections, Feb. 2000, San Francisco, CA, abstr. 652.
Kim, J., Neese, R. & Hellerstein, M. (2000) FASEB J. 14, A718 (abstr.).
Vance, B., Levine, B., Cesar, D., Abe, K., Telford, B., Hellerstein, M. & Gress, R. (2000) Am. Soc. Hematol. Forty-second Annual Meeting, Dec. 3, 2000, San Francisco, CA.
Antelo, F., Neese, R. & Hellerstein, M. (2000) FASEB J. 14, A214 (abstr.).
Chu, A., Ordonez, E. & Hellerstein, M. (2002) Arterioscler. Thromb. Vasc. Biol. 22,878A (abstr.).
Misell, L., Thompson, J., Antelo, F., Chou, Y.-C., Nandi, S., Neese, R. & Hellerstein, M. (2000) FASEB J. 14, A786 (abstr.).
References
- 1.Sawada S., Asakura, S., Daimon, H. & Furihata, C. (1995) Mutat. Res. 344 109-116. [DOI] [PubMed] [Google Scholar]
- 2.Waldman F. M., Chew, K., Ljung, B. M., Goodson, W., Hom, J., Duarte, L. A., Smith, H. S. & Mayall, B. (1991) Mod. Pathol. 4 718-722. [PubMed] [Google Scholar]
- 3.Gratzner H. G. (1982) Science 218 474-475. [DOI] [PubMed] [Google Scholar]
- 4.Asher E., Payne, C. M. & Bernstein, C. (1995) Leuk. Lymph. 19 107-119. [DOI] [PubMed] [Google Scholar]
- 5.Rocha B., Penit, C., Baron, C., Vasseur, F., Dautigny, N. & Freitas, A. A. (1990) Eur. J. Immunol. 20 1697-1708. [DOI] [PubMed] [Google Scholar]
- 6.Hellerstein M. (1999) Immunol. Today 20 438-441. [DOI] [PubMed] [Google Scholar]
- 7.Reichard P. (1988) Annu. Rev. Biochem. 57 349-374. [DOI] [PubMed] [Google Scholar]
- 8.Cohen A., Barankiewicz, J., Lederman, H. M. & Gelfand, E. W. (1983) J. Biol. Chem. 258 12334-12340. [PubMed] [Google Scholar]
- 9.Hirsch J. & Knittle, J. (1970) Fed. Proc. 29 1516-1521. [PubMed] [Google Scholar]
- 10.Miller W., Faust, I. & Hirsch, J. (1984) J. Lipid Res. 25 226-247. [PubMed] [Google Scholar]
- 11.Cairns J. (1975) Nature 255 197-200. [DOI] [PubMed] [Google Scholar]
- 12.McCune J., Hanley, M., Cesar, D., Halvorsen, R., Hoh, R., Schmidt, D., Deeks, S., Siler, S., Neese, R. & Hellerstein, M. (2000) J. Clin. Invest. 105 R1-R8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Macallan D. C., Fullerton, C. A., Neese, R. A., Haddock, K., Park, S. S. & Hellerstein, M. K. (1998) Proc. Natl. Acad. Sci. USA 95 708-713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hellerstein M., Hanley, M., Cesar, D., Siler, S., Papageorgopoulos, C., Wieder, E., Schmidt, D., Hoh, R., Neese, R., Macallan, D., et al. (1999) Nat. Med. 5 83-89. [DOI] [PubMed] [Google Scholar]
- 15.Katz J. & Rognstad, R. (1976) Curr. Top. Cell. Regul. 10 237-289. [DOI] [PubMed] [Google Scholar]
- 16.Hellerstein M. K. & Neese, R. A. (1999) Am. J. Physiol. 276 E1146-E1162. [DOI] [PubMed] [Google Scholar]
- 17.Neese R., Siler, S., Cesar, D., Antelo, F., Turner, S., Chu, A., Misell, L., Tehrani, S., Shah, P., Hashemi, Z. & Hellerstein, M. (2001) Anal. Biochem. 298 189-195. [DOI] [PubMed] [Google Scholar]
- 18.Patterson B., Zhao, G. & Klein, S. (1998) Metab. Clin. Exp. 47 706-712. [DOI] [PubMed] [Google Scholar]
- 19.Nandi S., Guzman, R. & Yang, J. (1995) Proc. Natl. Acad. Sci. USA 92 3650-3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Todd D., Singh, A. J., Greiner, D. L., Mardes, J. P., Rossini, A. A. & Bortell, R. (1999) J. Immunol. Methods 224 111-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brasitus T. & Keresztes, R. (1983) Differentiation (Berlin) 24 239-244. [DOI] [PubMed] [Google Scholar]
- 22.Chang W.-C., Chapkin, R. & Lupton, J. (1997) Carcinogenesis 18 721-730. [DOI] [PubMed] [Google Scholar]
- 23.Travo P., Barret, G. & Burnstock, G. (1980) Blood Vessels 17 110-116. [DOI] [PubMed] [Google Scholar]
- 24.Rodbell M. (1964) J. Biol. Chem. 239 375-380. [PubMed] [Google Scholar]
- 25.Jones P. & Leatherdale, S. (1991) Clin. Sci. 80 277-280. [DOI] [PubMed] [Google Scholar]
- 26.Waterlow J. C., Garlick, P. J. & Millward, D. J., (1978) Protein Turnover in Mammalian Tissues and in the Whole Body (North Holland, Amsterdam).
- 27.Craven P., Pfanstiel, J. & DeRubertis, F. (1987) J. Clin. Invest. 79 532-541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sul H.-S., Smas, C., Wang, D. & Chen, L. (1998) Prog. Nucleic Acid Res. Mol. Biol. 60 317-345. [DOI] [PubMed] [Google Scholar]