

Abstract
We examined the effects of a fragment of the substrate binding domain of DnaK on protein refolding from chemically denatured states. The fragment DnaK384-638, containing a full-length substrate binding domain, tightly binds to the unfolded protein in solution. The effects of DnaK384-638 on the reactivation of β-galactosidase and luciferase were examined at low substrate concentration and low temperature, conditions in which the folding is significantly slow (several days) but the reactivation yield is higher than those in ordinary refolding conditions. In the presence of DnaK384-638, the maximum yield of active β-galactosidase was improved from 45% to 65% after a 48-h refolding reaction. Spectroscopic experiments showed that DnaK384-638 bound to partially structured monomers of β-galactosidase and consequently suppressed aggregation. DnaK384-638 accelerated the refolding of luciferase to attain equilibrium in 8 h. On the other hand, DnaK386-561, which has no affinity for the substrate, had no chaperone activity for the reactivation of these proteins. These results indicate that the substrate binding of DnaK384-638 facilitates slow protein refolding.
The molecular chaperone DnaK, a member of the HSP70 family in Escherichia coli, is involved in the folding and assembly of newly synthesized polypeptide chains and in preventing the aggregation of stress-denatured proteins. The chaperone activity of DnaK is controlled by ATP (1, 2). When ADP is bound to the N-terminal ATPase domain (residues 1–384), the adjacent C-terminal substrate binding domain (residues 385–638) tightly binds the substrate. ATP bound to the ATPase domain induces a change in the substrate binding domain to a low affinity state for the substrate. The 26-kDa substrate binding domain of DnaK, which contains the β-sheet domain and the transitional helix of the substrate binding domain, harbors the substrate binding cleft (residues 393–507) and a 14-kDa domain from residues 508 to 638, of which the first 100 residues are α-helical and appear to act as a lid covering the substrate binding cleft (3). Substrate binding occurs by a dynamic mechanism in a two-layered closing device involving independent action of an α-helical lid and an arch. Mayer et al. (4) suggested that the ADP- and ATP-bound states of DnaK differ in the frequency of conformational changes that occur in the α-helical lid and the β-domain that cause an opening of the substrate binding cavity. Previous studies showed that ATP and the cochaperone DnaJ, which stimulate the ATPase activity, are necessary to facilitate the refolding of proteins from chemically denatured states (5, 6).
The activity of GroEL, another major chaperone in E. coli, is also controlled by the hydrolysis of ATP and a cochaperone (7, 8). However, the monomeric substrate binding domain, minichaperone, alone can assist the refolding of several proteins (9–13). The chaperone activity of the minichaperone comes from the weak binding of the folding intermediate of the substrate proteins (Kd = 200 μM, ref. 14). Based on these results, we examined the chaperone activity of the substrate binding domain of DnaK. The fragment of the substrate binding domain DnaK384-638 binds peptide substrates with the same high affinity as ADP-bound full-length DnaK (Kd = 0.93–11 μM, refs. 15 and 16). The majority of DnaK384-638 molecules are in the closed conformation in which the substrate binding cavity is covered by the α-helical lid. The chaperone activity of DnaK384-638 was examined for the ability to refold β-galactosidase and luciferase from chemically denatured states. These proteins have been used as model substrates for chaperones (5, 6, 17) because their yield of spontaneous refolding from the chemically denatured state is very low because of aggregation. On the other hand, recent studies showed that the yields of spontaneous refolding of these proteins are improved under slow refolding conditions at low temperature and low protein concentration (18, 19). We found that DnaK384-638 further improved the yield of refolding by suppressing aggregation and accelerated the refolding rate under these conditions. This result indicates that substrate binding is essential to the minimal mechanism of the chaperone activity of DnaK.
Materials and Methods
Materials.
The expression vector pDKC carrying DnaK384-638 and an attached 6×His tag was kindly provided by W. F. Burkholder (Massachusetts Institute of Technology, Cambridge) and M. E. Gottesman (Columbia University, New York). The expression vector for DnaK386-561 was constructed by PCR amplification and cloned into a pRSETA vector (Invitrogen). A QuikChange site-directed mutagenesis kit (Strategene) was used to introduce the mutation M469C into the target gene (using a pair of completely complementary primers) and to amplify the full-length plasmid. DnaK384-638, DnaK384-638M469C, and DnaK386-561 were expressed and affinity-purified by using a Ni-NTA resin column (Qiagen, Chatsworth, CA). CD spectra of the mutant were identical to those of DnaK384-638, indicating that the native structure is maintained in the mutant. β-Galactosidase from E. coli was obtained from Toyobo (Osaka). Recombinant Photinus pyralis luciferase was purchased from Promega. 5-[2(Iodoacetyl)aminoethyl]aminonaphthalene-1-sulfonate (IAEDANS) and the reduced and carboxylmethylated α-lactalbumin (RCMLA) were purchased from Sigma. Other reagents were of the highest grade commercially available and were used without further purification.
Fluorescent Labeling of Proteins.
The N-terminal amino group of RCMLA was labeled with FITC in a 4-h reaction at pH 8.0, and excess FITC was removed by using a PD10 column (Amersham Pharmacia). The final protein concentration was determined with the BCA protein assay, and the concentration of FITC was determined by absorbance at 490 nm (68,000 M−1⋅cm−1). The labeling ratio for the fluorescent derivatives of RCMLA was confirmed to be 1.0. The molecular mass of FITC-RCMLA was measured by Bruker Reflex III matrix-assisted laser desorption ionization–time-of-flight MS. The obtained molecular mass was almost identical to the theoretical molecular mass of FITC-RCMLA, indicating that 1 mol of FITC was attached per mol of RCMLA. The thiol-reactive fluorescence probe IAEDANS was attached to DnaK384-638M469C. To prevent aggregation, the reaction was performed with the protein immobilized on the nickel-NTA resin. IAEDANS (final concentration 5 mM) was added to the suspension in 50 mM Tris (pH 8.2), 150 mM NaCl, containing 2 mM DTT with stirring in the dark at 25°C for 1 h. The resin was washed to remove the excess IAEDANS, and 5-[2(acetyl)aminoethyl]aminonaphthalene-1-sulfonate (AEDANS)-modified DnaK384-638 was then eluted with imidazole buffer. The final protein concentration was determined with the BCA protein assay, and the concentration of attached AEDANS was determined by absorbance at 337 nm (6,100 cm−1). The labeling ratios for the fluorescent derivatives of the mutants were confirmed to be 1.0.
Refolding Experiments.
Experiments measuring the refolding of β-galactosidase (18) and luciferase (19) were performed as described with a minor alteration as follows. Luciferase activity was measured by using the assay system from Promega, and the light reaction was followed at 550 nm with a spectral bandwidth of 30 nm by using a Shimadzu RF2000 spectrofluorometer with its light source turned off.
Spectroscopic Measurements.
Fluorescence anisotropy measurements for FITC-RCMLA were made with a Shimadzu RF2000 spectrofluorimeter with an excitation wavelength of 490 nm and an emission wavelength of 520 nm. DnaK forms an oligomer via the substrate binding domain in the presence of the salt (20), and we used 10 mM Tris (pH 7.0) as the solvent for the DnaK fragment. The oligomer was not detected in native PAGE under these conditions. Anisotropy values of FITC-RCMLA were measured repeatedly at 3-min intervals in the presence of DnaK384-638 at 15°C. The anisotropy was gradually increased, and stable values were obtained about 30 min after mixing. The slow change in the fluorescence polarization might reflect a rearrangement of the substrate conformation after the initial binding to DnaK384-638. The static light scattering measurement was performed by using an Otsuka DLS7000 (Osaka). The measurements were done at a fixed scattering angle of 90° and a wavelength of 488 nm from an argon laser operating at 75 mW output power. For light scattering measurements, urea-denatured β-galactosidase was diluted in refolding buffer in a glass tube at 10°C. The solutions used for the measurement were filtered through a 0.2-μm membrane filter. Fluorescence measurements of AEDANS-DnaK384-638 were made with a Shimadzu RF2000 spectrofluorimeter with an excitation wavelength of 320 nm. The average emission wavelength (〈λ〉) was calculated by using the equation
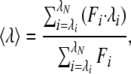 |
where F is the fluorescence intensity, and λ is the wavelength. The CD spectroscopic measurement was performed with a Jasco J-720 spectropolarimeter (Tokyo).
Results
Structure of the Substrate Binding Domain of DnaK.
We used DnaK384-638, containing a full-length substrate binding domain, and the shorter fragment DnaK386-561 in this study. The 3D structure of DnaK389-607 in complex with a model peptide substrate was solved by x-ray crystallography (Fig. 1a, ref. 3), and the structure of the remaining C-terminal domain from residues 606 to 638 was solved as a random coil by NMR (21). The substrate binding domain of DnaK consists of a β-subdomain (amino acids 393–501) and a C-terminal α-helical subdomain (amino acids 509–607). The peptide substrate is bound in a cavity formed by two pairs of inner and outer loops, and the peptide backbone is enclosed in an arch formed by residues Met-404 and Ala-429. The 3D structure of the shorter fragment, DnaK386-561, was solved by NMR (Fig. 1b, ref. 22). Whereas the conformation of the β domain is almost identical, the C-terminal helix in the NMR structure is considerably displaced from the position of the same helix in the crystal structure. This NMR study also showed that the substrate model peptide was not bound to DnaK386-561 because the substrate binding site is blocked by self-binding. These results indicate that the location of the α-helix in the NMR structure represents one of the loose conformations with low affinity for substrate, and the helical domain pivots on the β domain when the conformation changes.
Fig 1.

3D structure of the substrate binding domain of DnaK. The secondary structure representation was drawn with molscript (33) and RASTER 3D (34). (a) DnaK389-607 complexed with a model peptide substrate solved by x-ray crystallography (3) with a space-filling model of Met-404 and Ala-429 and the position of the mutation M469C that was labeled with IAEDANS. (b) DnaK386-561 solved by NMR (22).
We examined the interaction of DnaK384-638 and DnaK386-561 with RCMLA, the model polypeptide of denatured protein. A previous study using gel filtration analysis (23) showed that the fragment of the substrate binding domain of human HSP70 bound RCMLA. Consistently, the native PAGE of the solution of DnaK384-638 and RCMLA showed a new band from the complex of DnaK384-638 and RCMLA. In contrast, no evidence for the interaction of DnaK386-561 with RCMLA was obtained by native PAGE, which is consistent with the previous NMR result indicating a lack of affinity for the peptide. In the experiments in the following section, DnaK386-561 was used as a control for DnaK384-638 with no affinity to substrate. The affinity of DnaK384-638 for substrate was estimated by measuring the fluorescence anisotropy of the FITC attached to RCMLA. An increase in the measured anisotropy as a function of macromolecules is a measure of macromolecule binding. An equilibrium binding isotherm between DnaK384-638 and RCMLA was constructed by fluorescence anisotropy assays titrating a fixed concentration of FITC-RCMLA with increasing concentrations of DnaK384-638 (Fig. 2). The anisotropy of FITC was increased with increasing concentrations of DnaK384-638, reflecting the binding of RCMLA. The apparent Kd was estimated to be 5 μM from fitting the titration curve with the simple binding model.
Fig 2.

Titration of FITC-RCMLA with DnaK384-638. The measured increase (Δanisotropy) from the initial anisotropy value is plotted on the y axis. The titration was performed with 0.25 μM FITC-RCMLA in 10 mM Tris, pH 7.0, at 15°C. Lines through the data points are the results of the fit with a simple binding model. The fitting was done by using the nonlinear least-squares method with kaleida graph.
Effect of DnaK384-638 on the Reactivation of β-Galactosidase.
The effect of DnaK384-638 on the refolding of the tetrameric enzyme β-galactosidase from a urea-induced unfolded state was examined. Nichtl et al. (18) investigated the mechanism of spontaneous refolding of β-galactosidase and showed that the regain of activity strictly depends on the urea concentration present during refolding. In their study, refolding was achieved by first incubating 3.4 μM enzyme (monomer concentration) for 30 min at 10°C to minimize aggregation, followed by a shift to 20°C to reconstitute the active oligomer. Reactivation was achieved only in the presence of 0.8–1.6 M urea in which the aggregation of the refolding is minimized. The folding process was revealed to proceed as follows: folding of a partially structured monomer (M) from a denatured monomer (Mu) precedes association, and occurs very quickly (on the order of a second). The first rate-limiting step is the bimolecular association of partially structured monomers to form a partially folded dimer (D′) with a rate constant of 4.3 × 103 M−1⋅s−1. The second rate-limiting unimolecular folding step leads to formation of dimers (D), with a rate constant of 0.5 × 10−3 s−1. The rate of the final reaction, the formation of tetramers (T) from D, is fast. These refolding reactions can be summarized by the following model:
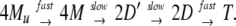 |
The overall folding rate of β-galactosidase was extremely slow and required 48 h to reach equilibrium. We examined the effect of DnaK384-638 on this slow refolding of β-galactosidase. Fig. 3a shows the reactivation yields of β-galactosidase in the presence or absence of 10 μM DnaK384-638 at various concentrations of urea. In the absence of DnaK384-638, a maximum yield of 45% was attained after a 48-h incubation at 20°C in 1.4 M urea. As shown by the closed circles in Fig. 3a, DnaK384-638 improved the yield of active β-galactosidase in the late stages of reactivation and a maximum yield of 65% was obtained in 1 M urea, whereas little effect on activation was achieved in the early stage. The shift in the urea concentration for maximum yield is caused by the decreased activity of DnaK384-638 at higher urea concentrations because the affinity of DnaK384-638 for RCMLA decreased with increasing urea concentrations (data not shown).
Fig 3.

The reactivation yields of β-galactosidase. β-Galactosidase was denatured with 8 M urea and diluted into refolding buffer (0.1 M sodium phosphate/1 mM MgCl2/5 mM EDTA). Refolding was achieved by incubation for 30 min at 10°C followed by a shift to 20°C to reconstitute the active tetramer. The final concentration of β-galactosidase was 3.4 μM (monomer concentration). (a) The reactivation yields at various concentrations of urea in the absence (○) and presence (•) of 10 μM DnaK384-638. At these urea concentrations, DnaK384-638 was confirmed to be in the native conformation by CD. (b) Concentration dependence of the yield of the reactivity of β-galactosidase at 0.73 M urea in the presence of DnaK384-638 (○) and DnaK386-561 (•). The extent of the reactivation was measured 24 h after refolding.
To determine the effect of the DnaK384-638 concentration on reactivation, refolding experiments were performed in refolding buffer containing different amounts of DnaK384-638 at 0.73 M urea (Fig. 3b). The yield of active β-galactosidase after a 24-h incubation increased from 27% to 43% when the concentration of DnaK384-638 reached 10 μM and did not increase significantly when the concentration of DnaK384-638 was raised further. This saturation concentration is in the same range as that for binding to RCMLA shown in Fig. 2. On the other hand, DnaK386-561 had no activity for reactivation and did not improve the yield of active β-galactosidase. These results indicate that the effect of DnaK384-683 on the reactivation of β-galactosidase was induced by the interaction of the polypeptide binding site of this domain and not by interactions in other regions.
Effects of DnaK384-638 on the Aggregation of β-Galactosidase.
To confirm the activity of DnaK384-683, light scattering experiments were performed to detect suppression of the aggregation of β-galactosidase. When urea-denatured β-galactosidase was diluted in refolding buffer to a final protein concentration of 8.6 μM (monomer concentration) and a urea concentration of 0.73 M at 10°C, the scattering intensity was increased, indicating the presence of a significant population of the aggregate (Fig. 4). The scattering intensity rapidly increased just after the start of refolding, indicating that aggregation is mainly caused by the association between partially structured monomers, because partially structured dimers are formed several minutes after refolding begins (18). On the other hand, the closed circle plots in Fig. 4 show that the extent of aggregation was significantly reduced when 10 μM DnaK384-638 was present in the refolding buffer. To control the affinity for unfolded protein, DnaK386-561 was substituted for DnaK384-638. As shown by x plots in Fig. 4, the extent of the suppression of the aggregation was reduced, indicating that the role of the polypeptide binding site of DnaK384-638 is significant for this activity.
Fig 4.

Aggregation of β-galactosidase in the absence (○) and presence of 10 μM DnaK384-638 (•) or 10 μM DnaK386-561 (x). β-Galactosidase denatured with 8 M urea was diluted into refolding buffer at 10°C. The final concentrations of β-galactosidase and urea were 8.6 μM (monomer concentration) and 0.73 M, respectively. Relative light scattering at 488 nm was measured at a fixed angle of 90°.
We examined the interaction between the folding intermediate of β-galactosidase and DnaK384-638 by fluorescence. The thiol-reactive fluorescence probe IAEDANS, an environment-sensitive fluorophore, was attached to DnaK384-638M469C. As shown in Fig. 1a, M469C is located in the vicinity of the substrate binding cavity, and substrate binding is detected from the change in fluorescence. The fluorescence spectra of AEDANS-M469C in refolding buffer were measured before and after the urea-denatured β-galactosidase was diluted into the buffer. As shown in Fig. 5, the maximum wavelength of fluorescence of AEDANS-M469C in refolding buffer was blue-shifted just after the start of refolding (within 30 s) with the shift of the average emission wavelength (〈λ〉) from 490 to 487 nm. This finding indicates that the fluorescence probe was in a hydrophobic environment caused by substrate binding. In the initial refolding stage, most of the β-galactosidase molecules are either partially structured monomers or are in an aggregate, and the change in the AEDANS-M469C fluorescence is induced during binding to either of these forms. To examine the affinity of DnaK384-638 for the β-galactosidase aggregates, we examined the fluorescence change of AEDANS-M468C when it was mixed into the refolding buffer 30 min after the start of refolding, in which a considerable portion of the β-galactosidase molecules are aggregated. In this case, a blue-shift in the fluorescence of AEDANS-M469C was not observed, indicating that DnaK384-638 was not bound to the aggregate form of β-galactosidase. Consequently, DnaK384-638 bound to the partially structured monomers of β-galactosidase in the refolding process and thus reduced aggregation.
Fig 5.

The effect of the binding of β-galactosidase on the emission spectra of AEDANS-M469C in refolding buffer. β-Galactosidase denatured with 8 M urea was diluted 50-fold into refolding buffer, in which 0.2 μM AEDANS-M469C and 0.8 M urea were present at 10°C. The final concentration of β-galactosidase was 17.2 μM.
Effect of DnaK384-638 on the Folding Rate of Luciferase.
Next, we examined the effect of DnaK384-638 on the reversible folding of luciferase to investigate its effect on the folding rate. Herbst et al. (19) investigated the folding mechanism of luciferase in detail. The kinetic intermediates I1 and I2 rapidly formed on dilution from a completely unfolded state at 5 M GuHCl. The rate-limiting step is that from I1 to Ifast, which appears only as kinetic intermediates between I2 and the native conformation. Refolding reactions can be summarized by the followed model:
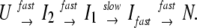 |
I1 and I2 appear also as the equilibrium intermediates at 1 M and 3 M GuHCl, respectively, and these were shown to be prone to aggregation. Therefore, a first-order folding reaction and a higher-order aggregate reaction compete in the refolding of luciferase. These authors found that the refolding of luciferase at low temperature (5°C) and low protein concentration (61 nM) is reversible because the higher-order reaction is decelerated with decreasing temperature and protein concentration. Under these conditions, the folding rate was significantly slow and equilibrium was attained after incubation for several days.
Fig. 6 shows the effect of the kinetics of the reactivation of luciferase from a completely unfolded state at 5°C. In the plot, the closed circles indicate that the luciferase folding rate was increased in the presence of 10 μM DnaK384-638, and refolding was complete after 8 h. On the other hand, the open squares in the plot show that DnaK386-561 has no effect on the kinetics of refolding. These results indicate that the folding rate of luciferase was accelerated by interaction with DnaK384-638 by avoiding the kinetic trap from I1 to Ifast. The putative model of this mechanism is as follows: When I1 of luciferase binds to the closed conformation of DnaK384-638, I1 is converted to Ifast. In the absence of the ATPase domain, the closed conformation of DnaK384-638 might occasionally convert to the open conformation by thermal fluctuation and the substrate may be slowly released. Mayer et al. (4) estimated the koff values for the substrate model peptide from DnaK in the ADP-bound state were in the range of 5.5 × 10−2 to 66 × 10−2 min−1, suggesting that substrates bound to DnaK384-638 would dissociate faster than the refolding rate of luciferase. Therefore, the folding rate of luciferase might be accelerated by DnaK384-638 because the frequency of the conformational conversion of DnaK384-638 is faster than the rate-limiting step from I1 to Ifast. Recurrent transitions in the conformation of DnaK384-638 may limit the rate of protein folding, and the slow refolding can be mediated by this action.
Fig 6.

Kinetics of reactivation of luciferase from the GuHCl-denatured state in the absence (○) and the presence of 10 μM DnaK384-638 (•) or 10 μM DnaK386-561 (□). Luciferase denatured in 5 M GuHCl was diluted 100-fold into refolding buffer (100 mM potassium phosphate, pH 7.8/1 mM EDTA/1 mM DTT) at 5°C. The final protein concentration was 61 nM. The activity of native luciferase incubated in refolding buffer containing 50 mM GuHCl is also shown (x).
Discussion
Molecular chaperones assist the refolding of proteins by interacting with folding intermediates to avoid kinetic traps, and consequently, by suppressing aggregation of the substrate. By the same mechanism, DnaK384-638 facilitates the slow refolding of β-galactosidase and luciferase without the function of ATPase. This finding indicates that substrate binding is essential to the minimal mechanism of the chaperone activity of DnaK. An ATP-induced conformational change in full-length DnaK enhances the kon value to facilitate faster protein folding. Similar results have been reported for the mechanism of mitochondrial HSP70 (mtHSP70), which assists protein translocation into organelles. Two operating principles have been suggested for the role of mtHSP70. The power stroke model (24) proposes that mtHSP70 pulls the precursor protein through the translocation pore by an ATP-induced conformational change. In the Brownian ratchet model (25), the role of mtHSP70 is simply to block backsliding through the pore. Because experimental evidence for both models have been obtained (26, 27), a dual role for mtHSP70 has been suggested (28, 29). Polypeptide binding is common to the minimal mechanism of HSP70 proteins, and the ATP-induced conformational change plays a role in improving its function.
Both the substrate binding domains of DnaK and GroEL facilitate protein folding by simply binding to the substrate. The remarkable difference is that the Kd value of RCMLA for DnaK384-638 (5 μM) is lower than that reported for GroEL minichaperone (200 μM, ref. 14). This difference reflects the distinct roles of each chaperone in folding processes in vivo. The DnaK system acts at an early folding step during refolding in the in vivo refolding process, and the GroEL system acts at a late step (30). Therefore, the substrate of DnaK is an unstructured nascent polypeptide undergoing rapid motions, but the substrate of GroEL is a sticky, partially structured polypeptide. Consistent results were reported in a NMR study demonstrating that DnaK binds the extended conformation of a peptide, whereas GroEL binds the same peptide in helical conformation (31). These results suggest that a high affinity for the polypeptide is intrinsically necessary for DnaK to bind the rapidly moving nonsticky substrate. On the other hand, a weak complex to the folding intermediate plays an important role in the activity of minichaperone. The intermediate is released and follows the proper folding pathway after the formation of the complex due to the driving force provided by the partial folding of the solvent-exposed polypeptide domains, which lower the affinity. In a fusion protein MC7 containing a heptameric ring composed of seven minichaperones, the chaperone activity was enhanced by providing a large continuous surface to accommodate a diverse range of non-native protein conformations differing in size and in structural and chemical properties (32). The avidity mediated by this multisite ring structure is another important key feature of GroEL function. To prevent irreversible binding with this sticky substrate, GroEL arrests the substrates with an accumulation of weak interactions at the seven substrate binding sites.
Acknowledgments
We thank Prof. A. R. Fersht for providing laboratory facilities, Dr. W. F. Burkholder and Prof. M. E. Gottesman for providing pDKC, and Mr. T. Kuroita (Toyobo.co., Ltd.) for technical assistance. Financial support for this study was provided by the Nissan Science Foundation.
Abbreviations
AEDANS, 5-[2(acetyl)aminoethyl]aminonaphthalene-1-sulfonate
IAEDANS, 5-[2(iodoacetyl)aminoethyl]aminonaphthalene-1-sulfonate
RCMLA, reduced and carboxylmethylated α-lactalbumin
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Schröder H., Langer, T., Hartl, F. U. & Bukau, B. (1993) EMBO J. 12 4137-4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szabo A., Langer, T., Schröder, H., Flanagan, J., Bukau, B. & Hartl, F. U. (1994) Proc. Natl. Acad. Sci. USA 91 10345-10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu X., Zhao, X., Burkholder, W. F., Gragerov, A., Ogata, C. M., Gottesman, M. E. & Hendrickson, W. A. (1996) Science 272 1606-1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayer M. P., Schröder, H., Rüdiger, S., Paal, K., Laufen, T. & Bukau, B. (2000) Nat. Struct. Biol. 7 586-592. [DOI] [PubMed] [Google Scholar]
- 5.Buchberger A., Schröder, H., Buttner, M., Valencia, A. & Bukau, B. (1994) Nat. Struct. Biol. 1 95-101. [DOI] [PubMed] [Google Scholar]
- 6.Schumacher R. J., Hansen, W. J., Freeman, B. C., Alnemri, E., Litwack, G. & Toft, D. O. (1996) Biochemistry 35 14889-14898. [DOI] [PubMed] [Google Scholar]
- 7.Ellis R. J. & Hartl, F. U. (1996) FASEB J. 10 20-26. [DOI] [PubMed] [Google Scholar]
- 8.Fenton W. A. & Horwich, A. L. (1997) Protein Sci. 6 743-760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zahn R., Buckle, A. M., Perrett, S., Johnson, C. M., Corrales, F. J., Golbik, R. & Fersht, A. R. (1996) Proc. Natl. Acad. Sci. USA 93 15024-15029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Altamirano M. M., Golbik, R., Zahn, R., Buckle, A. M. & Fersht, A. R. (1997) Proc. Natl. Acad. Sci. USA 94 3576-3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chatellier J., Hill, F., Lund, P. A. & Fersht, A. R. (1998) Proc. Natl. Acad. Sci. USA 95 9861-9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altamirano M. M., Garcia, C., Possani, L. D. & Fersht, A. R. (1999) Nat. Biotechnol. 17 187-191. [DOI] [PubMed] [Google Scholar]
- 13.Ben-Zvi A. P., Chatellier, J., Fersht, A. R. & Goloubinoff, P. (1998) Proc. Natl. Acad. Sci. USA 95 15275-15280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka N. & Fersht, A. R. (1999) J. Mol. Biol. 292 173-180. [DOI] [PubMed] [Google Scholar]
- 15.Burkholder W. F., Zhao, X., Zhu, X., Hendrickson, W. A., Gragerov, A. & Gottesman, M. E. (1996) Proc. Natl. Acad. Sci. USA 93 10632-10637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J. & Walker, G. C. (1998) Arch. Biochem. Biophys. 356 177-186. [DOI] [PubMed] [Google Scholar]
- 17.Freeman B. C. & Morimoto, R. I. (1996) EMBO J. 15 2969-2979. [PMC free article] [PubMed] [Google Scholar]
- 18.Nichtl A., Buchner, J., Jaenicke, R., Rudolph, R. & Scheibel, T. (1998) J. Mol. Biol. 282 1083-1091. [DOI] [PubMed] [Google Scholar]
- 19.Herbst R., Gast, K. & Seckler, R. (1998) Biochemistry 37 6586-6597. [DOI] [PubMed] [Google Scholar]
- 20.Shi L., Kataoka, M. & Fink, A. L. (1996) Biochemistry 35 3297-3308. [DOI] [PubMed] [Google Scholar]
- 21.Bertelsen E. B., Zhou, H., Lowry, D. F., Flynn, G. C. & Dahlquist, F. W. (1999) Protein Sci. 8 343-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H., Kurochkin, A. V., Pang, Y., Hu, W., Flynn, G. C. & Zuiderweg, E. R. P. (1998) Biochemistry 37 7929-7940. [DOI] [PubMed] [Google Scholar]
- 23.Freeman B. C., Myers, M. P., Schumacher, R. & Morimoto, R. I. (1995) EMBO J. 14 2281-2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glick B. S., Wachter, C., Reid, G. A. & Schatz, G. (1993) Protein Sci. 2 1901-1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simon S. M., Perskin, C. S. & Oster, G. F. (1992) Proc. Natl. Acad. Sci. USA 89 3770-3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geissler A., Rassow, J., Pfanner, N. & Voos, W. (2001) Mol. Cell. Biol. 21 7097-7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaume B., Klaus, C., Ungermann, C., Guiard, B., Neupert, W. & Brunner, M. (1998) EMBO J. 17 6497-6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bauer M. F., Hofmann, S., Neupert, W. & Brunner, M. (2000) Trends Cell Biol. 10 25-31. [DOI] [PubMed] [Google Scholar]
- 29.Pfanner N. & Truscott, K. N. (2002) Nat. Struct. Biol. 9 234-236. [DOI] [PubMed] [Google Scholar]
- 30.Langer T., Lu, C., Echols, H., Flanagan, J., Hayer, M. K. & Hartl, F. U. (1992) Nature 356 683-689. [DOI] [PubMed] [Google Scholar]
- 31.Landry S. J., Jordan, R., McMacken, R. & Gierasch, L. M. (1992) Nature 355 455-457. [DOI] [PubMed] [Google Scholar]
- 32.Chatellier J., Hill, F. & Fersht, A. R. (2000) J. Mol. Biol. 304 883-896. [DOI] [PubMed] [Google Scholar]
- 33.Kraulis P. J. (1991) J. Appl. Crystallogr. 24 946-950. [Google Scholar]
- 34.Merritt E. A. & Bacon, D. J. (1997) Methods Enzymol. 277 505-524. [DOI] [PubMed] [Google Scholar]