

Abstract
The high-density lipoprotein (HDL) receptor, scavenger receptor, class B, type I (SR-BI), mediates both the selective uptake of lipids, mainly cholesterol esters, from HDL to cells and the efflux of cholesterol from cells to lipoproteins. The mechanism underlying these lipid transfers is distinct from classic receptor-mediated endocytosis, but it remains poorly understood. To investigate SR-BI's mechanism of action and in vivo function, we developed a high-throughput screen to identify small molecule inhibitors of SR-BI-mediated lipid transfer in intact cells. We identified five compounds that in the low nanomolar to micromolar range block lipid transport (BLTs), both selective uptake and efflux. The effects of these compounds were highly specific to the SR-BI pathway, because they didn't interfere with receptor-mediated endocytosis or with other forms of intracellular vesicular traffic. Surprisingly, all five BLTs enhanced, rather than inhibited, HDL binding by increasing SR-BI's binding affinity for HDL (decreased dissociation rates). Thus, the BLTs provide strong evidence for a mechanistic coupling between HDL binding and lipid transport and may serve as a starting point for the development of pharmacologically useful modifiers of SR-BI activity and, thus, HDL metabolism.
The high-density lipoprotein (HDL) receptor, scavenger receptor, class B, type I (SR-BI), plays an important role in controlling the structure and metabolism of HDL (1, 2). Studies in mice have shown that alterations in SR-BI expression can profoundly influence several physiologic systems, including those involved in biliary cholesterol secretion, female fertility, red blood cell development, atherosclerosis, and the development of coronary heart disease (3–11). SR-BI controls HDL metabolism by mediating the cellular selective uptake of cholesteryl esters and other lipids from plasma HDL (1, 2). During selective uptake (12–14), HDL binds to SR-BI, and its lipids, primarily neutral lipids such as cholesteryl esters in the core of the particles, are transferred to the cells. The lipid-depleted particles subsequently are released back into the extracellular space. Although the mechanism of SR-BI-mediated selective lipid uptake and the subsequent intracellular transport of these lipids have only just begun to be explored (2, 15, 16), they clearly differ fundamentally from the pathway of receptor-mediated endocytosis via clathrin-coated pits and vesicles used by the low-density lipoprotein (LDL) receptor to deliver cholesterol esters from LDL to cells (17). SR-BI also can mediate cholesterol efflux from cells to HDL, although the physiological significance of SR-BI-mediated lipid efflux to lipoproteins is uncertain (18).
To generate reagents that can provide new insight into the mechanism of SR-BI-mediated selective lipid transfer, we have performed a high-throughput screen of a chemical library to identify potent small molecule inhibitors of SR-BI-mediated lipid transport. We report here five chemicals that block lipid transport, BLT-1–BLT-5, and describe their effects on SR-BI activity in cultured cells. All five BLTs inhibited SR-BI-mediated selective lipid uptake from HDL and efflux of cellular cholesterol to HDL. One of these, BLT-1, was particularly potent, inhibiting lipid transport in the low nanomolar concentration range. Unexpectedly, all five BLTs enhanced HDL binding to SR-BI by increasing the binding affinity. Thus, the BLTs provide strong evidence for the mechanistic coupling between HDL binding and lipid transport and should prove helpful in the analysis of the mechanism of action and function of SR-BI.
Methods
Lipoproteins and Cells.
Human HDL was isolated and labeled with 125I (125I-HDL); 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI, Molecular Probes; DiI-HDL); or [3H]cholesteryl oleyl ether ([3H]CE, [3H]CE-HDL) (1, 19–22). LDL receptor-deficient Chinese hamster ovary cells (ldlA-7) that express low levels of endogenous SR-BI (23), ldlA-7 cells stably transfected to express high levels of murine SR-BI (ldlA[mSR-BI]) (1), Y1-BS1 murine adrenocortical cells that express high levels of SR-BI after induction with adrenocorticotropic hormone (ACTH) (24), monkey kidney BS-C1 cells (25), and HeLa cells (26) were maintained as previously described.
High-Throughput Screen.
On day 0, ldlA[mSR-BI] cells were plated at 15,000 cells per well in clear bottom, black wall, 384-well black assay plates (Costar) in 50 μl of medium A (Ham's F12 supplemented with 2 mM L-glutamine, 50 units/ml penicillin/50 μg/ml streptomycin, and 0.25 mg/ml G418) supplemented with 10% FBS (medium B). On day 1, cells were washed once with medium C [medium A with 1% (wt/vol) BSA and 25 mM Hepes, pH 7.4, but no G418] and refed with 40 μl of medium C. Compounds (16,230 from the DiverSet E, ChemBridge Corp., San Diego) dissolved in 100% DMSO were individually, robotically “pin” transferred (40 nl) by a pin-based compound transfer robot (http://iccb.med.harvard.edu) to the wells to give a nominal concentration of 10 μM (0.01% DMSO). After 1 h of incubation at 37°C, DiI-HDL (final concentration of 10 μg of protein per ml) in 20 μl of medium C was added. Two hours later, fluorescence was measured at room temperature (≈2 min per plate) with an Analyst plate reader (rhodamine B dichroic filter; excitation, 525 nm; emission, 580 nm; Molecular Devices), both before removing the incubation medium (to test for autofluorescence and quenching) and after the medium removal and four washes with 80 μl of PBS/1 mM MgCl2/0.1 mM CaCl2 to determine cellular uptake of DiI. All compounds were sampled in duplicate on different plates, and each screen included ldlA-7 and ldlA[mSR-BI] cells in the presence or absence of a 40-fold excess of unlabeled HDL, but with no added compounds, as controls.
Assays.
For the assays, all media and buffers contained 0.5% DMSO and 0.5% BSA to maintain compound solubility. Cells were preincubated with BLTs for 1 h (or 2.5 h for transferrin, epidermal growth factor, and cholera toxin uptake experiments), and all of the experiments were performed at 37°C unless otherwise noted. Detailed characterization of the BLTs and their effects was performed with compounds whose identities and purities were confirmed by LC-MS.
Lipid uptake from HDL, cholesterol efflux to HDL, and HDL binding assays.
Assays for the uptake of lipids from DiI-HDL and [3H]CE-HDL, efflux of [3H]cholesterol from labeled cells, and 125I-HDL binding were performed as described (1, 18, 20). In some experiments, values were normalized so that 100% of control represents activity in the absence of compounds and 0% represents activity determined in the presence of a 40-fold excess of unlabeled HDL or, for Y1-BS1 cells, in the presence of a 1:500 dilution of the KKB-1 blocking antibody (generous gift from Karen Kozarsky, Glaxo/SmithKline Beecham, King of Prussia, PA) (20). The amounts of cell-associated [3H]cholesteryl oleyl ether are expressed as the equivalent amount of [3H]CE-HDL protein (ng) to permit direct comparison of the relative amounts of 125I-HDL binding and [3H]CE uptake.
The rates of HDL dissociation from cells were determined by incubation of the cells with 125I-HDL (10 μg of protein per ml, 2 h, 37°C) with and without BLTs. Then either the medium was replaced with the same medium in which a 40-fold excess of unlabeled HDL was substituted for the 125I-HDL, or a 40-fold excess of unlabeled HDL was added to the labeled incubation medium. The amounts of cell-associated 125I-HDL then were determined as a function of time. The two methods gave similar results.
Fluorescence microscopic analysis of intracellular trafficking and cytoskeletal organization.
Receptor-mediated endocytosis of Alexa-594-labeled transferrin or FITC-labeled epidermal growth factor (Molecular Probes) by HeLa cells (27) and uptake of Alexa-594-labeled holo-cholera toxin (kind gift of Wayne Lencer, Children's Hospital Boston and Harvard Medical School) by BSC-1 cells were detected by fluorescence microscopy. The intracellular transport of the temperature-sensitive glycoprotein of vesicular stomatitis virus (VSV-Gts045), fused at its carboxyl terminus to enhanced GFP (VSV-Gts045-EGFP) from the endoplasmic reticulum to the plasma membrane, after a shift from 40 to 32°C for 2 h, was determined by fluorescence microscopy (Y. Feng, S. Yu, T. Lasell, A. Jadhav, E. Macia, P. Chardin, P. Melancon, M. Roth, T. Mitchison, and T.K., unpublished data). The effects of the compounds on the distribution of actin using rhodamine-labeled phalloidin and tubulin using the FITC-labeled DM1α monoclonal antibody (Sigma) in ldlA[mSR-BI] cells were determined as described (25) by fluorescence microscopy using an air ×63 objective (Nikon).
Flow cytometric analysis of SR-BI cell surface expression.
Cells were incubated for 3 h (medium C) with or without BLTs at their IC95CE concentrations and harvested with PBS containing 2 mM EDTA and compounds, and the levels of SR-BI surface expression in unfixed cells were determined at 4°C by flow cytometry with the KKB-1 antibody (19).
Results
High-Throughput Screening for Inhibitors of SR-BI-Mediated Selective Lipid Uptake.
Cellular uptake and accumulation of the fluorescent lipophilic dye DiI from DiI-labeled HDL (DiI-HDL) is a reliable surrogate for SR-BI-dependent selective uptake of the cholesteryl esters in HDL (1). To identify small molecule inhibitors of SR-BI-mediated selective lipid uptake, we screened 16,320 compounds representing the DiverSet E of the ChemBridge library collection for their abilities to block the cellular uptake of DiI from DiI-HDL. The compounds were tested at a nominal concentration of 10 μM in a 384-well plate assay using ldlA[mSR-BI] cells that express a high level of mSR-BI (1). Fig. 1 shows results from a representative assay plate along with controls (no compounds, addition of excess unlabeled HDL or use of untransfected ldlA-7 cells). Compounds that quenched (Q) or enhanced the intrinsic fluorescence of DiI-HDL were not examined further. Approximately 200 compounds that reproducibly blocked DiI uptake in a first round of screening were retested. Five of the most effective with IC50DiIs in the micromolar range or lower (Fig. 2A) were designated BLT-1–BLT-5 and further characterized. Strikingly, the most potent of these, BLT-1 and BLT-2, inhibited in the nanomolar range and are structurally related (Table 1). Inhibition of DiI uptake did not require de novo protein synthesis, because pretreatment of cells for 30 min with 100 μg/ml cycloheximide did not diminish their inhibitory effects (data not shown). Finally, there were essentially no effects of the BLTs on the low background level of uptake of DiI or [3H]CE by untransfected ldlA-7 cells expressing minimal amounts of SR-BI (data not shown).
Fig 1.

High-throughput screen for inhibitors of SR-BI-mediated DiI uptake from DiI-HDL. Example of a fluorescent readout obtained from a single 384-well plate during the first round of the high-throughput screen. SR-BI-expressing ldlA[mSR-BI] cells were plated into 384-well plates, and the effect of compounds (≈10 μM) on the uptake of DiI from DiI-HDL (10 μg of protein per ml) was determined by using a high-speed fluorescence plate reader. Columns 1–20 show results (fluorescence in arbitrary units) from 16 independent wells per column (different colored symbols) from a single plate, representing a total of 320 compounds. Controls without compounds are wells containing ldlA[mSR-BI] cells in the absence or presence of a 40-fold excess of unlabeled HDL, or containing untransfected ldlA-7 cells (very low SR-BI expression). Wells containing an inhibitory compound named BLT-1 and wells with compounds that quenched DiI-HDL fluorescence (Q) are indicated.
Fig 2.

Concentration dependence of the inhibition by BLTs of SR-BI-mediated lipid transfer between HDL and cells. ldlA[mSR-BI] cells were incubated with the indicated concentrations of BLTs, and their effects on DiI uptake from DiI-HDL (A), [3H]CE uptake from [3H]CE-HDL (B), and the efflux of [3H]cholesterol from cells to HDL (C) were determined. The 100% of control values were 50.6 ng of HDL protein equivalents per well (384-well plates) (A) and 3,908 ng of HDL protein equivalents per mg of cellular protein (B). In C, the data were normalized such that the maximum amount of [3H]cholesterol transferred from cells to HDL in the absence of compounds (55.7% of total) was set to 100%. The 0% value corresponds to the efflux of [3H]cholesterol transferred from ldlA[mSR-BI] cells to HDL without BLTs and in the presence of saturating inhibitory amounts of the specific anti-SR-BI blocking antibody KKB-1 (15% of total) (20). The efflux of [3H]cholesterol from ldlA-7 cells measured in the absence or presence of KKB-1 was 15% and 10% of total cellular [3H]cholesterol, respectively.
Table 1.
Effects of BLTs on SR-BI activity
| n |
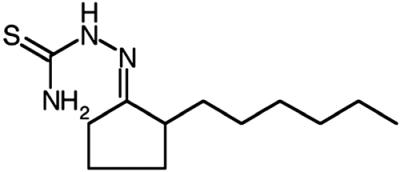 BLT-1, mean ± SD |
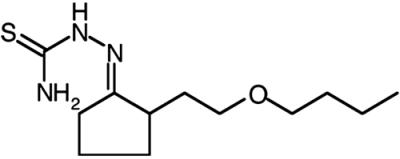 BLT-2, mean ± SD |
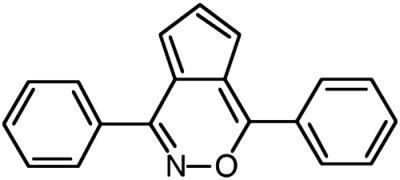 BLT-3, mean ± SD |
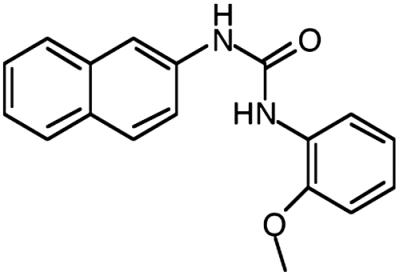 BLT-4, mean ± SD |
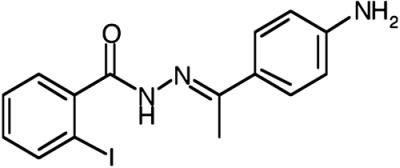 BLT-5, mean ± SD |
No BLT, mean ± SD | |
|---|---|---|---|---|---|---|---|
| IC50, μM | |||||||
| DiI-HDL uptake [3H]CE-HDL uptake | 3 | 0.06 ± 0.04 | 0.35 ± 0.18 | 0.51 ± 0.15 | 2.0 ± 1.0 | 7.1 ± 3.7 | — — |
| ldlA[mSR-BI] cells | 6 | 0.11 ± 0.08 | 0.24 ± 0.1 | 2.3 ± 1.5 | 3.9 ± 0.76 | 13.8 ± 8.5 | — — |
| Y1-BS1 cells | 2 | 0.38 | 0.41 | 1.7 | 4.4 | 8.0 | — — |
| [3H]cholesterol efflux | 3 | 0.15 ± 0.09 | 0.47 ± 0.23 | 17.2 ± 4.0 | 54.9 ± 35.2 | 75.3 ± 40.1 | — — |
| 125I-HDL binding | 3 | 0.088 ± 0.05 | 0.25 ± 0.13 | 46.5 ± 49.3 | 24.9 ± 14.8 | 18.0 ± 3.7 | — — |
| Binding parameters | |||||||
| Apparent Kd, μg⋅ml−1 | 3 | 4.7 ± 0.05 | 6.0 ± 6.0 | 8.0 ± 4.0 | 8.9 ± 2.3 | 12.0 ± 1.6 | 16.6 ± 1.5 |
| Koff, min−1 | 2 | 0.06 | 0.062 | 0.08 | 0.082 | 0.079 | 0.11 |
| Bmax, % | 95.8 ± 10.1 | 93.0 ± 20.5 | 85.8 ± 15.8 | 79.9 ± 15.9 | 92.1 ± 36.8 | 100.0 ± 18.4 |
All experiments were with ldlA[mSR-BI] cells except where noted.
n = 5.
SD is not applicable.
The IC50CEs for inhibition of uptake of the more physiologic lipid [3H]cholesteryl oleyl ether ([3H]CE) from [3H]CE-HDL by ldlA[mSR-BI] cells were similar to those for DiI uptake (Fig. 2B and Table 1). The inhibition of [3H]CE uptake was reversible (1 h of incubation with compounds followed by a 3- to 6-h washout period and a subsequent 2-h incubation with [3H]CE-HDL, data not shown). Moreover, the compounds also blocked the uptake of [3H]CE by Y1-BS1 adrenocortical cells that express high levels of SR-BI (24) (Table 1), indicating that the inhibitory effects by the compounds are not cell-type specific. Experiments in which the cells or the labeled HDL were preincubated with the compounds indicated that the cells, rather than the HDL, were the target of the compounds (data not shown).
Inhibition of Selective Lipid Uptake by BLTs Is Specific.
We examined the specificity of BLT inhibition by testing the effects of the BLTs on several other cellular properties at concentrations that inhibit [3H]CE uptake by 95% (IC95CE) (Fig. 3). None of the BLTs disrupted the integrity of the actin and tubulin networks. They also did not inhibit the uptake or alter the intracellular distribution of the fluorescently labeled endocytic receptor ligands transferrin and epidermal growth factor. The BLTs also failed to inhibit the uptake of fluorescently labeled cholera toxin from the cell surface to perinuclear regions through a pathway believed to depend in part on cholesterol- and sphingolipid-rich lipid rafts (28). Moreover, BLTs did not interfere with the secretory pathway, as assessed by analysis of the transport of VSV-Gts045-EGFP. Thus, BLTs do not induce general defects in clathrin-dependent and -independent intracellular membrane trafficking or in the organization of the cytoskeleton and are, by these criteria, specific inhibitors of SR-BI-dependent lipid uptake.
Fig 3.

Effects of BLT-1 on intracellular membrane trafficking and cytoskeletal organization. Cells were incubated for 3 h in the absence (Upper) or presence (Lower) of 50 μM BLT-1, and epifluorescence light microscopy was used to monitor the following cellular activities: clathrin-dependent endocytosis of fluorescently labeled transferrin (A and B; HeLa cells) and epidermal growth factor (C and D; HeLa cells); clathrin-independent endocytosis of fluorescently labeled cholera toxin (E and F; BSC-1 cells); and transport of the temperature-sensitive fluorescent membrane protein VSV-Gts045-EGFP from the endoplasmic reticulum to the cell surface (G and H; BSC-1 cells). In addition, the intracellular distributions of the actin cytoskeleton (I and J; ldlA-[mSRBI] cells visualized with rhodamine-labeled phalloidin) and the tubulin network (K and L; BSC-1 cells visualized with fluorescently labeled antibodies specific to α-tubulin) were determined. BLT-1 and the other BLTs (data not shown) had no effects on any of these cellular properties or activities.
BLTs Inhibit SR-BI-Mediated Cholesterol Efflux from Cells to HDL.
In addition to mediating selective lipid uptake from HDL, SR-BI can facilitate the efflux of unesterified, or free, cholesterol (FC) from cells to HDL particles (18). To determine whether the BLTs could inhibit this SR-BI-mediated lipid transport activity, we labeled cells with [3H]cholesterol and measured its efflux to unlabeled HDL in the presence or absence of the BLTs (Fig. 2C and Table 1). All BLTs inhibited SR-BI-mediated cholesterol efflux with relative potencies (IC50FCs) similar to those for [3H]CE uptake; however, in the cases of BLT-3, BLT-4, and BLT-5, the IC50FCs for efflux were higher than those for uptake, suggesting that the BLTs may have uncovered possible differences in the mechanisms of uptake and efflux. In ldlA[mSR-BI] cells, the BLTs had little effect on the SR-BI-independent efflux (not inhibited by the specific anti-SR-BI blocking antibody KKB-1; ref. 20) (data not shown). Similarly, in untransfected ldlA-7 cells expressing relatively low levels of endogenous SR-BI, total and SR-BI-dependent (e.g., KKB-1-inhibitable) cholesterol efflux was substantially lower than in ldlA[mSR-BI] cells. The BLTs were able to inhibit the low SR-BI-dependent cholesterol efflux in ldlA-7 cells, but they had no inhibitory effect on the similarly low SR-BI-independent efflux (data not shown).
BLTs Do Not Change the Surface Expression of SR-BI.
To determine whether BLTs inhibited SR-BI function by reducing its cell surface expression, we measured surface expression by using the KKB-1 anti-mSR-BI antibody (20) and flow cytometry. Fig. 4 shows that, after 3 h of incubation at their IC95CEs (corresponding to 1 μM for BLT-1 and BLT-2 and 50 μM for BLT-3–BLT-5), the BLTs did not alter the expression of mSR-BI on the surfaces of ldlA[mSR-BI] cells.
Fig 4.

Cell surface expression of SR-BI. ldlA[mSR-BI] and ldlA-7 cells were treated for 3 h with or without BLTs at their corresponding IC95CE concentrations (1 μM for BLT-1 and BLT-2 and 50 μM for BLT-3–BLT-5) followed by determination of surface expression levels of SR-BI by flow cytometry. A–C show histograms of the surface expression for ldlA[mSR-BI] cells without BLTs, ldlA[mSR-BI] cells with 1 μM BLT-1, and ldlA-7 cells without BLTs, respectively. D summarizes the results in ldlA[mSR-BI] cells for all five BLTs, with the value determined without compounds set to 100%. n, number of independent determinations.
BLTs Enhance Binding of HDL to SR-BI.
We initially expected that the BLTs would function by inhibiting HDL binding to SR-BI. However, when cells were incubated with a subsaturating concentration of either [3H]CE-HDL or 125I-labeled HDL (125I-HDL) (10 μg of protein per ml) and increasing amounts of compound (Fig. 5), the decreases in [3H]CE uptake (solid lines, no symbols, data from Fig. 2B) and [3H]cholesterol efflux (dashed lines, data from Fig. 2C) were accompanied by corresponding increases in 125I-HDL binding (Fig. 5, solid lines). The concentration dependence of 125I-HDL binding was determined in the presence or absence of BLTs at their IC95CE concentrations (Fig. 6 and Table 1). The BLTs did not substantially alter the number of binding sites (Bmax), but rather induced small, yet significant, increases in the affinity of SR-BI for HDL (lower apparent Kd). Furthermore, the BLTs reduced the rates of dissociation of 125I-HDL from SR-BI (Table 1), indicating that the tighter binding induced by the BLTs was due, at least in part, to a decrease in the dissociation rate.
Fig 5.

Effects of BLTs on SR-BI-mediated cholesterol ether uptake from HDL, cellular cholesterol efflux to HDL, and HDL binding. The effects of the indicated concentrations of BLTs on SR-BI-mediated uptake of [3H]CE from [3H]CE-HDL (solid lines, no symbols), efflux of [3H]cholesterol from cells to HDL (dashed lines), or binding of 125I-HDL to cells (solid lines, symbols) were determined by using ldlA[mSR-BI] cells. To simplify comparisons, we define the lowest observed [3H]CE uptake and [3H]cholesterol efflux values (from Fig. 2) as 0% and the values in the absence of BLTs as 100%. The 100% control value for the 125I-HDL binding in the absence of BLTs was 403 ng of HDL protein per mg of cell protein.
Fig 6.

Effects of BLT-1 on the concentration dependence of 125I-HDL binding to ldlA[mSR-BI] cells. The binding of 125I-HDL to ldlA[mSR-BI] cells was determined in duplicate at the indicated concentrations of HDL in the presence (blue) or absence (black) of 1 μM BLT-1 (IC95CE). Each value was corrected for binding of 125I-HDL in the presence of 40-fold excess of unlabeled HDL to ldlA [mSR-BI] cells in the presence of BLT-1.
Discussion
In this study, we report the discovery and characterization of BLT-1–BLT-5, small molecules that inhibit the transfer of lipids between HDL and cells mediated by the HDL receptor SR-BI. BLTs inhibited both cellular selective lipid uptake of HDL cholesteryl ether and efflux of cellular cholesterol to HDL. The inhibitory effects of the BLTs were specific, because they required SR-BI and they did not interfere with several clathrin-dependent and -independent endocytic pathways, the secretory pathway, or the actin or tubulin cytoskeletal networks. Strikingly, inhibition of lipid transfer by BLTs was accompanied by enhanced HDL binding affinity (reduced dissociation rates).
BLTs and other inhibitors or activators of SR-BI should be useful for the analysis of the molecular and cellular mechanisms of SR-BI activity and the study of the physiologic functions of SR-BI by its pharmacologic manipulation in vivo. Our studies with the BLTs support a two-step mechanism of SR-BI activity, productive binding followed by lipid transfer (19, 29), and suggest that these two steps may be mechanistically linked because they result in coordinated decreases in lipid transfer and increases in binding affinity. They also suggest that there may be differences in the mechanisms of SR-BI-mediated lipid uptake and cholesterol efflux (20, 30), because the IC50 values for uptake and efflux for each of three BLTs (BLT-3–BLT-5) differed.
The mechanistic coupling of HDL binding and lipid transport observed here is consistent with the productive binding model for SR-BI interaction with HDL (29). A number of studies have explored the influence of the protein composition of HDL particles on binding and lipid transfer (26, 29, 31–33). They showed that variations in the structure/composition of HDL particles could affect the efficiency of selective uptake or efflux relative to lipoprotein binding and thus support the concept of productive binding. In addition, CD36, a close homolog of SR-BI, mediates high-affinity HDL binding but not efficient lipid transfer (19, 34). Thus, HDL binding per se to a member of this superfamily of receptors is not sufficient for efficient lipid transport; rather, the binding must be productive. That is, SR-BI and HDL must bind in a precisely aligned fashion, have the capacity to undergo appropriate conformational changes on binding, or both.
BLTs might block productive binding by inducing (or preventing) interactions between HDL and its receptor, or interfering with critical conformational changes such that lipid transfer could not occur. A direct consequence of these altered interactions could be the tighter HDL binding observed when the cells were treated with BLTs. Alternatively, the BLTs might not interfere with productive binding and lipid transfer, but rather might simply inhibit dissociation of HDL from SR-BI, thereby preventing the multiple rounds of HDL binding, lipid transfer, dissociation of the smaller lipid-depleted particle, and binding of a new HDL particle that are required for efficient selective uptake. If this were the case, in the presence of BLTs the initially bound HDL particles would competitively inhibit the binding of other particles. Another possibility is that direct BLT inhibition of the lipid transfer step might prevent the size reduction of HDL that accompanies selective uptake. Previous studies have shown that larger, lipid-rich HDLs bind more tightly to SR-BI than smaller, relatively lipid-poor HDLs (33, 35). Thus, BLT inhibition of the lipid transfer step would lead to larger HDL particles bound more tightly to SR-BI at steady state, resulting in a slower dissociation rate and apparently higher binding affinity. Finally, it seems unlikely that the BLTs would inhibit lipid transfer step(s) occurring distally to the receptor itself [e.g., between the plasma membrane and internal cellular compartments significantly downstream from the association between HDL and SR-BI (16)], given the strong inverse correlation between the effects of BLTs on binding and lipid transport. Future identification of the molecular target(s) of the BLTs, whether SR-BI or other proteins or lipids, should provide insights into the mechanisms underlying the activities of both the BLTs and SR-BI.
Experiments with SR-BI-deficient or SR-BI-overexpressing mice have provided important insights into SR-BI function and potential roles of SR-BI and HDL metabolism in pathophysiology, including female infertility, aberrant red blood cell development, and cardiovascular disease (6, 7, 10, 15). BLTs and other small molecules isolated by using the approach described here may prove helpful in further studying the physiology of these systems. In this regard it is noteworthy that BLT-1 has been shown to interfere with the early stages of brain development in zebrafish (36). It may be worthwhile to explore the potential role of disrupted lipid metabolism in this system. Given the widespread consequences of genetically manipulating SR-BI expression in mice, pharmacologic manipulation may have potential therapeutic value.
Acknowledgments
We thank Dr. Xiang-Ju Gu for his advice and participation in the early stages of the development of the high-throughput screen, Dr. Karen Kozarsky for generously providing the KKB-1 antibody, Shangzhe Xu for technical assistance, Dr. Yan Feng (Harvard Institute of Chemistry and Cell Biology, Harvard Medical School) for advice and the VSV-Gts045-EGFP construct, and Dr. Wayne Lencer (Children's Hospital Boston, Harvard Medical School) for the fluorescently labeled cholera toxin. We thank Dr. Tim Mitchison (Harvard Institute of Chemistry and Cell Biology) for helpful discussions. We also thank Ian Levesque for administrative help. We are especially grateful to members of the Harvard Institute of Chemistry and Cell Biology for generously permitting us to use their facilities for the development and performance of the screen and for their advice and cooperation. In particular we thank Jim Follen and David Hayes for performing the robotic pin transfer. This work was supported by National Institutes of Health Grants HL52212, HL66105, and HL64737 (to M.K.) and GM62566 (to T.K.).
Abbreviations
HDL, high-density lipoprotein
SR-BI, scavenger receptor, class B, type I
LDL, low-density lipoprotein
BLT, chemical that blocks lipid transport
DiI, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate
CE, cholesteryl oleyl ether
FC, free cholesterol
mSR-BI, murine SR-BI
VSV-G, vesicular stomatitis virus glycoprotein
EGFP, enhanced GFP
IC, inhibitory concentration
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Acton S., Rigotti, A., Landschulz, K. T., Xu, S., Hobbs, H. H. & Krieger, M. (1996) Science 271 518-520. [DOI] [PubMed] [Google Scholar]
- 2.Krieger M. (1999) Annu. Rev. Biochem. 68 523-558. [DOI] [PubMed] [Google Scholar]
- 3.Trigatti B., Rayburn, H., Viñals, M., Braun, A., Miettinen, H., Penman, M., Hertz, M., Schrenzel, M., Amigo, L., Rigotti, A. & Krieger, M. (1999) Proc. Natl. Acad. Sci. USA 96 9322-9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kozarsky K. F., Donahee, M. H., Glick, J. M., Krieger, M. & Rader, D. J. (2000) Arterioscler. Thromb. Vasc. Biol. 20 721-727. [DOI] [PubMed] [Google Scholar]
- 5.Arai T., Wang, N., Bezouevski, M., Welch, C. & Tall, A. R. (1999) J. Biol. Chem. 274 2366-2371. [DOI] [PubMed] [Google Scholar]
- 6.Holm T. M., Braun, A., Trigatti, B. L., Brugnara, C., Sakamoto, M., Krieger, M. & Andrews, N. C. (2002) Blood 99 1817-1824. [DOI] [PubMed] [Google Scholar]
- 7.Miettinen H. E., Rayburn, H. & Krieger, M. (2001) J. Clin. Invest. 108 1717-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ueda Y., Gong, E., Royer, L., Cooper, P. N., Francone, O. L. & Rubin, E. M. (2000) J. Biol. Chem. 275 20368-20373. [DOI] [PubMed] [Google Scholar]
- 9.Kozarsky K. F., Donahee, M. H., Rigotti, A., Iqbal, S. N., Edelman, E. R. & Krieger, M. (1997) Nature 387 414-417. [DOI] [PubMed] [Google Scholar]
- 10.Braun A., Trigatti, B. L., Post, M. J., Sato, K., Simons, M., Edelberg, J. M., Rosenberg, R. D., Schrenzel, M. & Krieger, M. (2002) Circ. Res. 90 270-276. [DOI] [PubMed] [Google Scholar]
- 11.Mardones P., Quinones, V., Amigo, L., Moreno, M., Miquel, J. F., Schwarz, M., Miettinen, H. E., Trigatti, B., Krieger, M., VanPatten, S., et al. (2001) J. Lipid Res. 42 170-180. [PubMed] [Google Scholar]
- 12.Glass C., Pittman, R. C., Weinstein, D. B. & Steinberg, D. (1983) Proc. Natl. Acad. Sci. USA 80 5435-5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glass C., Pittman, R. C., Civen, M. & Steinberg, D. (1985) J. Biol. Chem. 260 744-750. [PubMed] [Google Scholar]
- 14.Stein Y., Dabach, Y., Hollander, G., Halperin, G. & Stein, O. (1983) Biochim. Biophys. Acta 752 98-105. [DOI] [PubMed] [Google Scholar]
- 15.Krieger M. (2001) J. Clin. Invest. 108 793-797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uittenbogaard A., Everson, W. V., Matveev, S. V. & Smart, E. J. (2002) J. Biol. Chem. 277 4925-4931. [DOI] [PubMed] [Google Scholar]
- 17.Brown M. S. & Goldstein, J. L. (1986) Science 232 34-47. [DOI] [PubMed] [Google Scholar]
- 18.Ji Y., Jian, B., Wang, N., Sun, Y., Moya, M. L., Phillips, M. C., Rothblat, G. H., Swaney, J. B. & Tall, A. R. (1997) J. Biol. Chem. 272 20982-20985. [DOI] [PubMed] [Google Scholar]
- 19.Gu X., Trigatti, B., Xu, S., Acton, S., Babitt, J. & Krieger, M. (1998) J. Biol. Chem. 273 26338-26348. [DOI] [PubMed] [Google Scholar]
- 20.Gu X., Kozarsky, K. & Krieger, M. (2000) J. Biol. Chem. 275 29993-30001. [DOI] [PubMed] [Google Scholar]
- 21.Acton S. L., Scherer, P. E., Lodish, H. F. & Krieger, M. (1994) J. Biol. Chem. 269 21003-21009. [PubMed] [Google Scholar]
- 22.Pitas R. E., Innerarity, T. L., Weinstein, J. N. & Mahley, R. W. (1981) Arteriosclerosis (Dallas) 1 177-185. [DOI] [PubMed] [Google Scholar]
- 23.Kingsley D. M. & Krieger, M. (1984) Proc. Natl. Acad. Sci. USA 81 5454-5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rigotti A., Edelman, E. R., Seifert, P., Iqbal, S. N., DeMattos, R. B., Temel, R. E., Krieger, M. & Williams, D. L. (1996) J. Biol. Chem. 271 33545-33549. [DOI] [PubMed] [Google Scholar]
- 25.Kapoor T. M., Mayer, T. U., Coughlin, M. L. & Mitchison, T. J. (2000) J. Cell Biol. 150 975-988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Temel R. E., Walzem, R. L., Banka, C. L. & Williams, D. L. (2002) J. Biol. Chem. 277 26565-26572. [DOI] [PubMed] [Google Scholar]
- 27.Spiro D. J., Boll, W., Kirchhausen, T. & Wessling-Resnick, M. (1996) Mol. Biol. Cell 7 355-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lencer W. I., Hirst, T. R. & Holmes, R. K. (1999) Biochim. Biophys. Acta 1450 177-190. [DOI] [PubMed] [Google Scholar]
- 29.Liu T., Krieger, M., Kan, H. Y. & Zannis, V. I. (2002) J. Biol. Chem. 277 21576-21584. [DOI] [PubMed] [Google Scholar]
- 30.de la Llera-Moya M., Rothblat, G. H., Connelly, M. A., Kellner-Weibel, G., Sakr, S. W., Phillips, M. C. & Williams, D. L. (1999) J. Lipid Res. 40 575-580. [PubMed] [Google Scholar]
- 31.Thuahnai S. T., Lund-Katz, S., Williams, D. L. & Phillips, M. C. (2001) J. Biol. Chem. 276 43801-43808. [DOI] [PubMed] [Google Scholar]
- 32.Pilon A., Briand, O., Lestavel, S., Copin, C., Majd, Z., Fruchart, J. C., Castro, G. & Clavey, V. (2000) Arterioscler. Thromb. Vasc. Biol. 20 1074-1081. [DOI] [PubMed] [Google Scholar]
- 33.de Beer M. C., Durbin, D. M., Cai, L., Jonas, A., de Beer, F. C. & van der Westhuyzen, D. R. (2001) J. Lipid Res. 42 309-313. [PubMed] [Google Scholar]
- 34.Connelly M. A., Klein, S. M., Azhar, S., Abumrad, N. A. & Williams, D. L. (1999) J. Biol. Chem. 274 41-47. [DOI] [PubMed] [Google Scholar]
- 35.Liadaki K. N., Liu, T., Xu, S., Ishida, B. Y., Duchateaux, P. N., Krieger, J. P., Kane, J., Krieger, M. & Zannis, V. I. (2000) J. Biol. Chem. 275 21262-21271. [DOI] [PubMed] [Google Scholar]
- 36.Peterson R. T., Link, B. A., Dowling, J. E. & Schreiber, S. L. (2000) Proc. Natl. Acad. Sci. USA 97 12965-12969. [DOI] [PMC free article] [PubMed] [Google Scholar]