

Abstract
Purpose.
Docosahexaenoic acid (DHA22:6n3), the principal n3-polyunsaturated fatty acid (PUFA) in the retina, has been shown to have a pronounced anti-inflammatory effect in numerous in vivo and in vitro studies. Despite the importance of vascular inflammation in diabetic retinopathy, the anti-inflammatory role of DHA22:6n3 in cytokine-stimulated human retinal vascular endothelial cells (hRVECs) has not been addressed.
Methods.
Cytokine-induced expression of cell adhesion molecules (CAMs) was assessed by Western blot. The effect of DHA22:6n3 on cytokine-induced nuclear factor (NF)-κB signaling was analyzed by Western blot analysis and electrophoretic mobility shift assay (EMSA).
Results.
Stimulation of hRVECs with VEGF165, TNFα, or IL-1β for 6 to 24 hours caused significant induction of intracellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 expression. Pretreatment of the cells with 100 μM of BSA-bound DHA22:6n3 for 24 hours remarkably inhibited cytokine-induced CAM expression. IL-1β, TNFα, and VEGF165 induced nuclear translocation and binding of p65 and p50 NF-κB isoforms to the VCAM-1 promoter. DHA22:6n3 pretreatment inhibited cytokine-induced NF-κB binding by 25% to 40%. Moreover, DHA22:6n3 diminished IL-1β induced phosphorylation of the inhibitor of nuclear factor (NF)-κB (I-κBα), thus preventing its degradation.
Conclusions.
IL-1β, TNFα, and VEGF165 induced CAM expression in hRVECs through activation of the NF-κB pathway. DHA22:6n3 inhibited cytokine induced CAM expression through suppression of NF-κB nuclear translocation and upstream I-κBα phosphorylation and degradation. DHA22:6n3 could be an important anti-inflammatory agent in the face of increased cytokine production and CAM expression in the diabetic retina.
The early stage of diabetic retinopathy has been recognized to result from a chronic inflammatory condition involving attachment to and transmigration of leukocytes through the retinal microvasculature.1–3 Several inflammatory pathways are active in the early stages of diabetic retinopathy. Proinflammatory cytokines including TNFα4–7 and IL-1β8 are elevated in the extracellular matrix, endothelium, vessel walls, and vitreous of eyes of patients with proliferative diabetic retinopathy; and in the retinas of rats after 2 months of diabetes. Moreover, inhibition of TNFα and IL-1β signaling with a TNFα receptor/Fc construct2 or with ILRa9 significantly reduced leukocyte adhesion and endothelial cell (EC) injuries. Vascular endothelial cell growth factor (VEGF) has also been strongly implicated in the pathogenesis of both background and proliferative diabetic retinopathy.10–13 Increased intraocular VEGF levels, as well as VEGF receptor 1 and 2 were detected in the rat and human diabetic retina.10–18 In addition to its well-known mitogenic and angiogenic activity, VEGF was recently recognized as a proinflammatory cytokine.19,20 As such, VEGF induces inter-cellular adhesion molecule (ICAM)-1 expression on endothelial cells19 and specific inhibition of the VEGF pathway inhibits ICAM-1 expression, leukocyte adhesion, blood–retinal barrier breakdown, and neovascularization in streptozotocin (STZ)-induced diabetic rats.19 These data suggest an important role for TNFα, IL-1β, and VEGF (and their receptors) in the activation of signaling pathways leading to endothelium injury preceding the development of diabetic retinopathy. Despite these findings, the effect of inflammatory cytokines on human retinal endothelial cells has not been well studied.
Inflammatory cytokines function through their receptors, to initiate a series of signal transduction events that generally lead to the phosphorylation and degradation of inhibitor of nuclear factor (NF)-κB (I-κB) followed by the translocation and activation of NF-κB in the nucleus.21 NF-κB is an important transcription factor controlling the expression of an array of inflammatory response genes including adhesion molecules.21 Activation of NF-κB (p65 and p50) has been well documented in diabetes, especially in the retinal vasculature of diabetic patients and in animal models.19,22 In vitro high glucose has been shown to cause the activation of NF-κB in bovine retinal endothelial cells and pericytes.22,23 The role of NF-κB in response to inflammatory cytokines in hRVECs was the subject of the present study.
n3-PUFAs, such as DHA22:6n3 and EPA20:5n3, have long been recognized to modulate the inflammatory response and are widely applied clinically as an adjuvant immunosuppressant in the treatment of inflammatory disorders (reviewed in Refs. 24,25). Several studies in human umbilical vein endothelial cells (HUVECs),26,27 human saphenous vein endothelial cells,28,29 and glomerular endothelial cells30 have demonstrated that n3 PUFAs and their products can effectively inhibit TNFα- and IL-1β-induced CAM expression. Retinal vascular endothelial cells have unusually high levels of PUFAs.31 The response to fatty acids could be modified in retinal endothelial cells compared with endothelial cells from other organs. Indeed, we have previously demonstrated that hRVECs respond with much higher potency to n6 PUFA than do HUVECs.32 Whether DHA22:6n3 plays an anti-inflammatory role in the regulation of TNFα- and IL-1β-mediated induction of CAM expression in hRVECs similar to other endothelial cells has not been studied and represents the main focus of this study. Moreover, the effect of n3 PUFA on VEGF-induced CAM expression is not known and will be addressed in this study.
Materials and Methods
Reagents
DMEM and F12 culture medium, antibiotics, fetal bovine serum, and trypsin were obtained from Invitrogen (Carlsbad, CA). Commonly used chemicals and reagents were from Sigma-Aldrich Chemical Co. (St. Louis, MO). TNFα and IL-1β were from R&D Systems (Minneapolis, MN). VEGF165 was purchased from Calbiochem (San Diego, CA).
Cell Culture and Fatty Acid Treatment
Primary cultures of hRVECs obtained from at least three donors from the tissue provided by National Disease Research Interchange (Philadelphia, PA) were prepared and cultured, as previously described.32 Passages 3 to 6 were used in the experiments. For experimental treatments, cells were transferred to serum-free medium for 18 to 24 hours before addition of the stimulatory agents. A dose–response curve was first established for each cytokine using the cells from each donor in the range of 0 to 10 ng/mL for TNFα, 0 to 2 ng/mL for IL-1β, and 0 to 50 ng/mL for VEGF165. The dose at which the maximum stimulation was achieved was used in all the consequent experiments.
Treatment of hRVECs with fatty acids was performed as follows. Fatty acid stocks were prepared by dissolving fatty acids (NuCheck Prep, Inc., Elysian, MN) in 100% ethanol, to a final concentration of 100 mM fatty acid. The fatty acid stock solutions were diluted in serum-free medium to reach concentrations of 100 μM in the presence of 20 μM of bovine serum albumin (BSA; charcoal-treated, solvent-extracted, fatty acid-free; Serologics Inc., Norcross, GA). The fatty acid-to-albumin molar ratio was maintained at 5:1. Cells were incubated for the times indicated in the Results section. The concentration of fatty acids used was within the physiological range and was confirmed by propidium iodide staining not to cause apoptosis (data not shown). Equivalent amounts of BSA alone were added to control plates. Palmitic16:0 acid was chosen as a lipid control in this study based on the following considerations. The most abundant fatty acids in retinal endothelial cells are palmitic16:0, stearic18:0, linoleic18:2n6, and arachidonic20:4n6 acids and DHA22:6n3 (Ref. 31 and Chen et al., unpublished observations, 2004). We have previously demonstrated that linoleic18:2n6 and arachidonic20:4n6 acids induce CAM expression in hRVECs.32 Palmitic16:0 acid is comparable to DHA22:6n3 in the abundance level in retinal endothelial cells,31 and it does not have the proinflammatory properties of n6 PUFA.32
SDS-PAGE and Western Blot Analysis
Cells were lysed in the lysis buffer (50 mM HEPES [pH 7.5], 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 1% Triton X-100, and 10% glycerol) with freshly added protease inhibitor cocktail (Sigma-Aldrich) and phosphatase inhibitors (1 mM Na3VO4, 100 μM glycerophosphate, 10 mM NaF, and 1 mM Na4PPi). Proteins were resolved by SDS-PAGE and transferred to nitrocellulose, immunoblotted using appropriate antibodies followed by secondary horseradish-peroxidase–conjugated antibody (Bio-Rad). Immunoreactive bands were visualized by enhanced chemiluminescence (ECL kit; GE Healthcare, Piscataway, NJ). Blots were quantitated by scanning densitometry using ImageJ software, ver. 1.29 (available by ftp at zippy.nimh.nih.gov/or at http://rsb.info.nih. gov/nih-image; developed by Wayne Rasband, National Institutes of Health, Bethesda, MD).
Electrophoretic Mobility Gel Shift Assay
The double-stranded oligonucleotides containing the NF-κB binding sequence derived from human vascular cell adhesion molecule (VCAM)-1 promoter were designed and synthesized as follows: 5′TGCCCTGGGTTTCCCCTTGAAGGGATTTCCCTC-3′ and 3′-GACCCAAAGGGGAACTTCCCTAAAGGGAGGCGG-5′ (NF-κB binding domains are shown in bold). The oligonucleotides were annealed and labeled in the presence of [32P]dCTP with a random primer kit from Invitrogen, according to manufacturer’s protocol. For binding reactions, nuclear extracts (6 μg) were incubated in 25 μL of total reaction volume with 32P-labeled NF-κB oligonucleotides for 20 minutes at room temperature. DNA-protein complexes were resolved on 6% nondenaturing polyacrylamide gels, and the bands were examined by autoradiography and quantitated using phosphorescent imaging (PhosphorImager; Molecular Dynamics, Sunnyvale, CA). Incubation of the nuclear extracts with excess cold NF-κB oligonucleotides was used to confirm the specificity of binding activity.
Statistical Analysis
There are two variability levels involved in this study—experimental variability within the cells from the same donor and the interdonor variability. We statistically analyzed the experimental variability within the cells from the same donor and confirmed, but did not include in the analysis, the results from three different donors. Data are expressed as the mean ± SD from one donor. ANOVA was used for comparing data obtained from independent samples. The Bonferroni procedure was used to control type I errors. Significance was established at P < 0.05.
Results
Effect of TNFα, IL-1β, and VEGF165 on Adhesion Molecule Expression in hRVECs
The inflammatory responsiveness of primary human retinal endothelial cells to the three proinflammatory cytokines (TNFα, IL-1β, and VEGF165) known to be increased in diabetic eyes was first assessed with CAM expression as a measure. The doses of cytokines used in this study were selected based on the dose–response curves in the cells from each donor, as described in the Methods section (data not shown). TNFα (5 ng/mL) and IL-1β (1 ng/mL) acutely stimulated the expression of ICAM-1 and VCAM-1 (Fig. 1A). Recombinant VEGF165 (20 ng/mL), an important angiogenesis factor in diabetic retinopathy, also markedly induced ICAM-1 and VCAM-1 in hRVECs (Fig. 1B). The VEGF165 induction of ICAM-1 and VCAM-1 was time dependent, with VCAM-1 expression peaking at 24 hours and ICAM-1 expression persisting for up to 48 hours. There was no significant effect of cytokine stimulation on E-selectin expression at the time points checked (Fig. 1).
Figure 1.
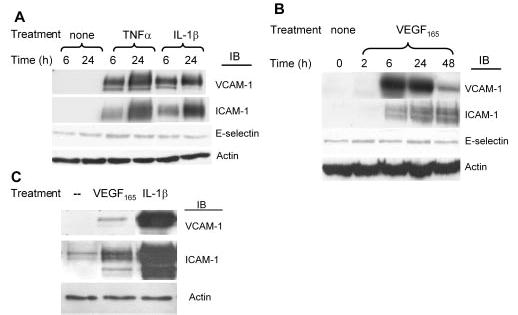
TNFα, IL-1β, and VEGF165 induced expression of cell adhesion molecules in hRVECs. hRVECs were serum starved overnight and stimulated with 5 ng/mL TNFα, 1 ng/mL IL-1β (A), or 20 ng/mL VEGF165 (B) for different times, as indicated. Western blot exposure time in (B) was at least 30 times longer than that in (A), to illustrate the details of the VEGF response. For comparison of the actual potency of doses at which the maximum response is achieved for VEGF165 (20 ng/mL for 6 hours) and IL-1β (1 ng/mL for 6 hours), the samples were loaded on the same gel and exposed for the same time as shown in (C). The induction of the adhesion molecules VCAM-1, ICAM-1, and E-selectin was assessed by immunoblot analyses. Equal amounts of protein were added to each lane, as confirmed by actin levels. Representative results from at least three independent experiments are shown.
The Western blot analyses in Figure 1A and 1B are shown at different sensitivity levels to illustrate the details of the response to each cytokine; however, they do not provide information on the relative potency of IL-1β, TNFα, and VEGF. To compare the potency of angiogenesis factor VEGF165 with classic inflammatory cytokines, such as IL-1β, we treated hRVECs with the doses at which the maximum activation was achieved for each cytokine in the parallel plates and analyzed the samples on the same gel. As shown in Figure 1C, VEGF165 has much weaker cytokine activity when compared with the inflammatory cytokine IL-1β.
Effect of DHA22:6n3 on TNFα-, IL-1β-, and VEGF165-Induced CAM Expression
Because DHA22:6n3 is the most abundant retinal n3-PUFA, we evaluated the potential modulating effect of DHA22:6n3 on the inflammatory response in retinal endothelial cells. Pretreatment of hRVECs with DHA22:6n3 (100 μM of BSA-bound DHA22:6n3 for 24 hours) significantly inhibited IL-1β- and TNFα-induced VCAM-1 expression by approximately 40% and 50%, respectively (Figs. 2A, 2B). In contrast, pretreatment with a lipid control (palmitate16:0) did not exhibit a significant effect on cytokine-induced VCAM-1 expression (Figs. 2A, 2B). Similarly, DHA22:6n3 pretreatment inhibited VEGF165-induced VCAM-1 and ICAM-1 expression, whereas palmitic16:0 acid pretreatment had no such effect (Fig. 2C).
Figure 2.
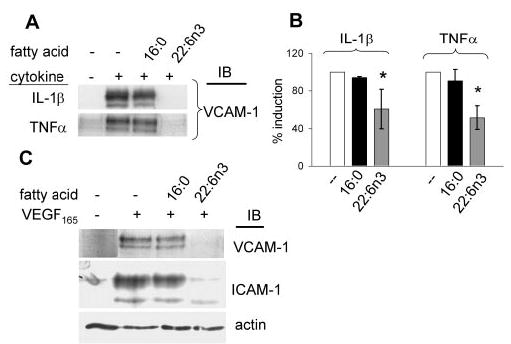
Inhibition of cytokine-induced CAM expression by DHA22:6n3 pretreatment. hRVECs were serum starved overnight and then treated with 100 μM palmitate16:0 or DHA22:6n3 for 24 hours. Cells were then stimulated with 1 ng/mL IL-1β, 5 ng/mL TNFα (A), or 20 ng/mL VEGF165 (C) for 6 hours. The induction of VCAM-1 and ICAM-1 was assessed by immunoblot analyses. (B) Quantitative compilation of the data on VCAM-1 induction in hRVECs stimulated with 1 ng/mL IL-1β or 5 ng/mL TNFα, with or without pretreatment with 100 μM palmitate16:0 or DHA22:6n3 from three independent experiments. *P < 0.05 compared with the control.
Role of NF-κB in Regulating Cytokine-Induced Adhesion Molecule Expression in hRVECs
To investigate the role of NF-κB in cytokine-induced adhesion molecule expression in hRVECs, a double-stranded DNA probe containing the specific NF-κB binding site from human VCAM-1 promoter was used in electrophoretic mobility shift assay (EMSA) to study the activation and binding of NF-κB to the promoters of adhesion molecules. As shown in Figures 3A, all three cytokines induced NF-κB binding to the VCAM-1 promoter. VEGF165 induced a delayed NF-κB activation in the nucleus, with the NF-κB induced shifts starting from 1 hour and peaking at 2 hours (Fig. 3A). Moreover, Western blot analysis showed that two specific isoforms of the NF-κB family, p65 and p50, accumulated in the nucleus after stimulation with IL-1β and TNFα (Fig. 3B). Phosphorylation of p65 at Ser(536), necessary for optimal transactivation of NF-κB,33 was also observed in the nucleus of IL-1β- and TNFα-stimulated cells (Fig. 3B). Likewise, VEGF165 induced translocation of p50 and p65 into the nucleus (Fig. 3B), although with no obvious p65 phosphorylation observed (data not shown).
Figure 3.
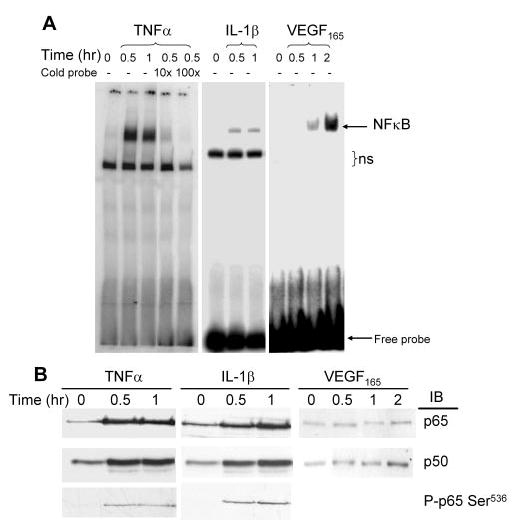
Inflammatory cytokines activated NF-κB signaling in hRVECs. (A) hRVECs were serum starved overnight and then treated with TNF-α (10 ng/mL), IL-1β (1 ng/mL), and VEGF165 (20 ng/mL). Nuclear extracts were prepared and EMSAs were performed with probes containing a specific NF-κB-binding motif to the human VCAM-1 promoter. Arrows: the NF-κB-induced shift; ns, nonspecific binding. The specificity of NF-κB binding was confirmed by addition of cold DNA at 10× and 100× concentration. (B) Equal amounts of nuclear extracts were loaded for Western blot analysis with anti-p65, p50, and P-p65Ser(536). Representative results from at least three independent experiments are shown.
Effect of DHA22:6n3 Pretreatment on Cytokine-Induced NF-κB Binding to the VCAM-1 Promoter
To address the molecular mechanism underlying the inhibitory effect of DHA22:6n3, we analyzed whether DHA22:6n3 acts through inhibition of NF-κB signaling to attenuate adhesion molecules expression. VEGF165-induced binding to the VCAM-1 promoter at 2 hours was decreased approximately 40% by pretreatment with DHA22:6n3, compared with carrier (BSA) and lipid (palmitic16:0 or linoleic18:2n6 acid) controls (Fig. 4A). Similarly, DHA22:6n3 pretreatment inhibited IL-1β-induced NF-κB binding to the VCAM-1 promoter by 25% compared with palmitate16:0-treated controls (Figs. 4B, 4C). The decrease in NF-κB binding was concomitant with a decrease in the nuclear level of p65 and p50 in DHA22:6n3-treated cells (Fig. 4D), implying that DHA22:6n3 decreases IL-1β induced nuclear translocation of p65 and p50, thus attenuating their binding to the VCAM-1 promoter.
Figure 4.
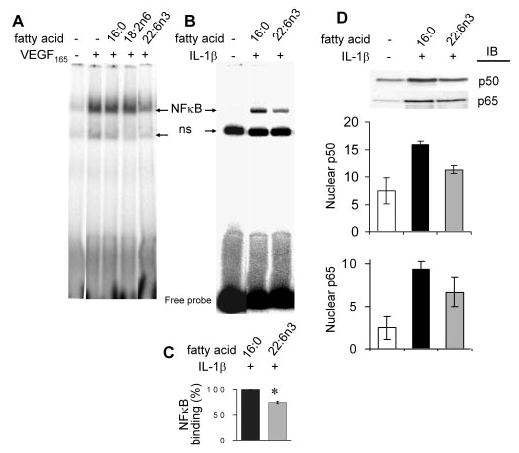
DHA22:6n3 inhibited VEGF165 and IL-1β induced NF-κB signaling. (A) hRVECs were serum starved overnight and treated with 100 μM palmitate16:0, linoleic acid18:2n6, or DHA22:6n3 for 24 hours. Cells were then stimulated with 20 ng/mL VEGF165 for 2 hours, and nuclear extracts were prepared. EMSA assay was performed with probes containing the specific NF-κB-binding motif to the human VCAM-1 promoter. (B) hRVECs were treated with 100 μM palmitate16:0 or DHA22:6n3 for 24 hours and then stimulated with 1 ng/mL IL-1β for 30 minutes, and EMSA was performed as before. Arrows: the NF-κB induced shift; ns, a nonspecific band. (C) Quantitative compilation of the results obtained in three independent experiments. *P < 0.05 compared with palmitate16:0 control. (D) Equivalent amounts of nuclear extracts as shown in (B) were analyzed with anti-p65 and p50 antibodies by Western blot and quantitated. Representative results from two independent experiments are shown. The result was repeated in the cells from three different donors (data not shown).
Effect of DHA22:6n3 Pretreatment on I-κBα Phosphorylation and Degradation
We next addressed the specific upstream step that DHA22:6n3 acts on to inhibit cytokine-induced NF-κB signaling, by evaluating the phosphorylation of one of the NF-κB inhibitors (I-κBα) and its ubiquitin-mediated proteosome degradation. DHA22:6n3 pretreatment attenuated IL-1β-induced I-κBα phosphorylation compared with the palmitate16:0-treated control (Figs. 5A, 5B). Attenuation of I-κBα phosphorylation prevents I-κBα degradation, thereby retaining NF-κB in the inactive NF-κB–I-κBα complex in the cytosol (Figs. 5A, 5B). VEGF165, even at the highest dose used, was not as potent as IL-1β (Fig. 1C). VEGF165-induced I-κBα phosphorylation and degradation was below the sensitivity level of I-κBα and phospho-I-κBα antibodies used in this study (data not shown).
Figure 5.
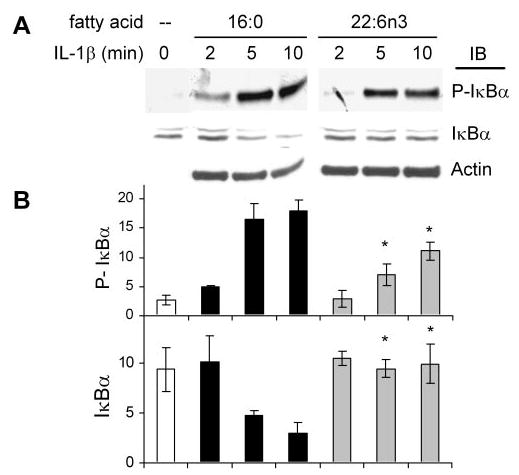
Inhibition of IL-1β-induced I-κBα phosphorylation and degradation by DHA22:6n3 pretreatment. hRVECs were serum starved over-night and treated with 100 μM palmitate16:0 or DHA22:6n3 for 24 hours. Cells were then stimulated with 1 ng/mL IL-1β for the indicated times. Western blot analyses were performed to detect I-κBα phosphorylation and degradation. Representative blots (A) and quantitative compilation of the data from three independent experiments (B) are shown. *P < 0.05 compared with the same time point in palmitate16:0 control.
Discussion
In chronic inflammatory conditions, endothelial cells actively recruit blood-borne leukocytes, such as monocytes and T lymphocytes to the underlying tissue, in response to the activation by cytokines and growth factors. This process is mediated by the increased expression of adhesion molecules on both leukocytes and endothelial cells. The early stages of diabetic retinopathy have been recognized as a chronic inflammatory disease.1–3 Upregulation of inflammatory cytokines, especially TNFα,5–7 IL-1β,8,9 and VEGF,10–12 along with their corresponding receptors have been well documented in the eyes of diabetic patients and in diabetic animal models. However, the effect of these principal cytokines on human retinal endothelial cell adhesion molecule expression, especially VCAM-1, a specific vascular inflammatory marker, has not been tested. This study demonstrated, for the first time that the inflammatory cytokines upregulated in the diabetic eye induce expression of adhesion molecules in cultures of human retinal endothelial cells. Our data further demonstrated the anti-inflammatory effect of the principal n3-PUFA in the retina, DHA22:6n3, on cytokine-triggered inflammatory signaling.
NF-κB, an essential nuclear factor in the regulation of inflammatory signaling,21,34 has been shown to be involved in the development of diabetic microvascular complications.19,22 Our data indicate that retinal endothelial cells contain the cognate receptors for TNFα, IL-1β, and VEGF165 and that activation of these receptors leads to increased DNA binding activity of NF-κB. The major NF-κB isoforms activated by the inflammatory cytokines in hRVECs were p65 and p50, which normally form a p65/p50 heterodimer to mediate DNA binding. Previous studies have demonstrated increased accumulation of the p50, but not the p65, isoform of NF-κB in nuclei of retinal endothelial cells from diabetic animals,35 and only p65 has been shown to be increased in retinal pericyte nuclei but not in endothelial cells from patients with diabetic retinopathy patients and/or in cells from STZ-induced diabetic rats.23 The apparent differences could come from the different systems used. The results in our study of cultured human retinal endothelial cells emphasize that both p65 and p50 are important DNA-binding transcription factors activated by proinflammatory cytokines to mediate VCAM-1 and ICAM-1 expression.
Our data agree with those in other reports showing that ICAM-1 is a critical adhesion molecule elevated in diabetic retinas and that VEGF165 is a proinflammatory cytokine capable of inducing its expression.19,20 A significant finding in this study is that VEGF165, IL-1β, and TNFα also induced VCAM-1 expression in hRVECs underscoring the likely importance of VCAM-1 in mediating leukostasis in human diabetic retinal vasculature.
There was a very minor, if any, increase in E-selectin expression after 6 and 24 hours of cytokine stimulation in hRVECs. Studies in endothelial cells from other organs have demonstrated E-selectin activation after cytokine stimulation. The apparent discrepancy could be explained by organ specificity. A recent study of human microvascular endothelial cells from different organs demonstrated organ-specific CAM expression and activation patterns.36 Although retinal endothelial cells were not analyzed in that study, brain microvascular endothelial cells, which are very close to retinal endothelial cells in structure and fatty acid composition, had the lowest E-selectin response to cytokine activation.
n3-PUFAs have long been recognized to modulate inflammatory responses and are widely applied clinically as an adjuvant immunosuppressant in the treatment of various inflammatory disorders.24,25 Numerous studies have shown that treatment with n3-PUFAs inhibits adhesion molecule and cytokine expression induced by inflammatory agents.24,26–30 Our study of human retinal endothelial cells strongly supports an important role for DHA22:6n3 as an anti-inflammatory agent in ameliorating endothelial cell response to cytokines in these primary target cells. The dose of DHA22:6n3 used in this study was within a normal physiological range. Higher doses of DHA22:6n3 may have a more prominent effect on NF-κB signaling; however, we chose to use the low dose because high concentrations of fatty acids in general could have adverse effects on the cells and may be proapoptotic. Moreover, concentrations higher than the levels achievable in vivo would not provide information relevant to diabetic retinopathy. The fact that DHA22:6n3, even at this low dose, attenuates the effect of such potent cytokines as TNFα and IL-1β by as much as 25% to 50% is of great clinical importance.
The specific mechanisms underlying the anti-inflammatory effect of n3 PUFA have been intensively sought for decades. Several possible mechanisms have been suggested, including the displacement of the major substrate for the synthesis of proinflammatory eicosanoids, arachidonic acid20:4n6, from sn-2 position in membrane phospholipids37; direct activation of the nuclear receptors, such as peroxisome proliferators-activated receptors (PPARs)38–44; or modification of specific plasma membrane domains called lipid rafts or caveolae.45,46 All these pathways are likely to play a role in DHA22:6n3 effects in hRVECs.
In our study, DHA22:6n3 not only attenuated the nuclear translocation and DNA binding of p50/p65 NF-κB isoforms, but also inhibited the upstream I-κBα phosphorylation and degradation (Fig. 6). This implies that DHA22:6n3 acts at least up-stream of I-κBα to inhibit inflammatory signaling in hRVECs. The more specific upstream steps where DHA22:6n3 acts are under investigation. However, modification of lipid composition of caveolae–lipid rafts and caveolae–lipid raft signaling components, similar to that observed in T cells,45,46 represents a very plausible possibility (Fig. 6).
Figure 6.
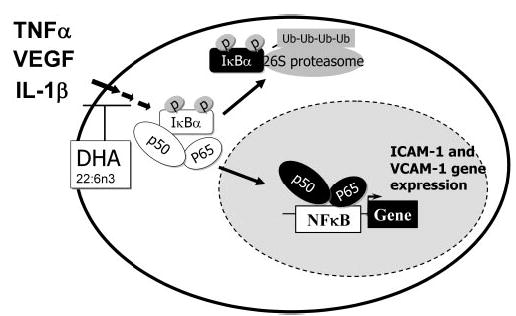
Cytokine-induced NF-κB activation. Cytokine binding to the cognate receptors recruits signaling molecules to initiate a cascade of signal transduction that leads to the phosphorylation of I-κB, causing its ubiquitin (Ub)-mediated degradation and thus releasing NF-κB (p65 and p50) from the cytosol, which transports into the nucleus and binds to the NF-κB-dependent genes to mediate expression. DHA22:6n3 treatment inhibits the signaling pathways upstream of I-κBα phosphorylation and possibly acts at the plasma membrane receptor level.
In summary, our data demonstrate that three major inflammatory cytokines, known to be upregulated in diabetic eyes—TNFα, IL-1β, and VEGF165—induce VCAM-1 and ICAM-1 expression through activating the NF-κB pathway in primary human retinal endothelial cells. DHA22:6n3 plays a central role in antagonizing cytokine-induced adhesion molecule expression by attenuating NF-κB signaling in the early steps in inflammation in hRVECs. Our data suggest that DHA22:6n3 is a principal anti-inflammatory agent in the face of activated cytokine production in the diabetic retina.
Footnotes
Supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK43220 (DBJ) and DK065014 (JVB), the Juvenile Diabetes Research Foundation 2-2005-97 (JVB), U.S. Department of Agriculture Grant 2003-5200-3400 (DBJ), and the Michigan Agricultural Experimental Station (DBJ).
Disclosure: W. Chen, None; W.J. Esselman, None; D.B. Jump, None; J.V. Busik, None
References
- 1.Schroder S, Palinski W, Schmid-Schonbein GW. Activated monocytes and granulocytes, capillary nonperfusion, and neovascularization in diabetic retinopathy. Am J Pathol. 1991;139:81–100. [PMC free article] [PubMed] [Google Scholar]
- 2.Joussen AM, Poulaki V, Mitsiades N, et al. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. FASEB J. 2002;16:438–440. doi: 10.1096/fj.01-0707fje. [DOI] [PubMed] [Google Scholar]
- 3.Adamis AP. Is diabetic retinopathy an inflammatory disease? Br J Ophthalmol. 2002;86:363–365. doi: 10.1136/bjo.86.4.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Limb GA, Chignell AH, Green W, LeRoy F, Dumonde DC. Distribution of TNF alpha and its reactive vascular adhesion molecules in fibrovascular membranes of proliferative diabetic retinopathy. Br J Ophthalmol. 1996;80:168–173. doi: 10.1136/bjo.80.2.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Limb GA, Soomro H, Janikoun S, Hollifield RD, Shilling J. Evidence for control of tumour necrosis factor-alpha (TNF-alpha) activity by TNF receptors in patients with proliferative diabetic retinopathy. Clin Exp Immunol. 1999;115:409–414. doi: 10.1046/j.1365-2249.1999.00839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Limb GA, Webster L, Soomro H, Janikoun S, Shilling J. Platelet expression of tumour necrosis factor-alpha (TNF-alpha), TNF receptors and intercellular adhesion molecule-1 (ICAM-1) in patients with proliferative diabetic retinopathy. Clin Exp Immunol. 1999;118:213–218. doi: 10.1046/j.1365-2249.1999.01067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spranger J, Meyer-Schwickerath R, Klein M, Schatz H, Pfeiffer A. TNF-alpha level in the vitreous body. Increase in neovascular eye diseases and proliferative diabetic retinopathy [in German] Med Klin (Munich) 1995;90:134–137. [PubMed] [Google Scholar]
- 8.Kowluru RA, Odenbach S. Role of interleukin-1beta in the development of retinopathy in rats: effect of antioxidants. Invest Ophthalmol Vis Sci. 2004;45:4161–4166. doi: 10.1167/iovs.04-0633. [DOI] [PubMed] [Google Scholar]
- 9.Kowluru RA, Odenbach S. Role of interleukin-1beta in the pathogenesis of diabetic retinopathy. Br J Ophthalmol. 2004;88:1343–1347. doi: 10.1136/bjo.2003.038133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adamis AP, Miller JW, Bernal MT, et al. Increased vascular endothelial growth factor levels in the vitreous of eyes with proliferative diabetic retinopathy. Am J Ophthalmol. 1994;118:445–450. doi: 10.1016/s0002-9394(14)75794-0. [DOI] [PubMed] [Google Scholar]
- 11.Murata T, Nakagawa K, Khalil A, et al. The relation between expression of vascular endothelial growth factor and breakdown of the blood-retinal barrier in diabetic rat retinas. Lab Invest. 1996;74:819–825. [PubMed] [Google Scholar]
- 12.Aiello LP, Pierce EA, Foley ED, et al. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci USA. 1995;92:10457–10461. doi: 10.1073/pnas.92.23.10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qaum T, Xu Q, Joussen AM, et al. VEGF-initiated blood-retinal barrier breakdown in early diabetes. Invest Ophthalmol Vis Sci. 2001;42:2408–2413. [PubMed] [Google Scholar]
- 14.Ellis EA, Guberski DL, Somogyi-Mann M, Grant MB. Increased H2O2, vascular endothelial growth factor and receptors in the retina of the BBZ/Wor diabetic rat. Free Radic Biol Med. 2000;28:91–101. doi: 10.1016/s0891-5849(99)00216-6. [DOI] [PubMed] [Google Scholar]
- 15.Aiello LP, Avery RL, Arrigg PG, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med. 1994;331:1480–1487. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- 16.Amin RH, Frank RN, Kennedy A, Eliott D, Puklin JE, Abrams GW. Vascular endothelial growth factor is present in glial cells of the retina and optic nerve of human subjects with nonproliferative diabetic retinopathy. Invest Ophthalmol Vis Sci. 1997;38:36–47. [PubMed] [Google Scholar]
- 17.Frank RN, Amin RH, Eliott D, Puklin JE, Abrams GW. Basic fibroblast growth factor and vascular endothelial growth factor are present in epiretinal and choroidal neovascular membranes. Am J Ophthalmol. 1996;122:393–403. doi: 10.1016/s0002-9394(14)72066-5. [DOI] [PubMed] [Google Scholar]
- 18.Obrosova IG, Minchenko AG, Marinescu V, et al. Antioxidants attenuate early up regulation of retinal vascular endothelial growth factor in streptozotocin-diabetic rats. Diabetologia. 2001;44:1102–1110. doi: 10.1007/s001250100631. [DOI] [PubMed] [Google Scholar]
- 19.Joussen AM, Poulaki V, Qin W, et al. Retinal vascular endothelial growth factor induces intercellular adhesion molecule-1 and endothelial nitric oxide synthase expression and initiates early diabetic retinal leukocyte adhesion in vivo. Am J Pathol. 2002;160:501–509. doi: 10.1016/S0002-9440(10)64869-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Usui T, Ishida S, Yamashiro K, et al. VEGF164–165 as the pathological isoform: differential leukocyte and endothelial responses through VEGFR1 and VEGFR2. Invest Ophthalmol Vis Sci. 2004;45:368–374. doi: 10.1167/iovs.03-0106. [DOI] [PubMed] [Google Scholar]
- 21.Monaco C, Paleolog E. Nuclear factor kappaB: a potential therapeutic target in atherosclerosis and thrombosis. Cardiovasc Res. 2004;61:671–682. doi: 10.1016/j.cardiores.2003.11.038. [DOI] [PubMed] [Google Scholar]
- 22.Kowluru RA, Koppolu P, Chakrabarti S, Chen S. Diabetes-induced activation of nuclear transcriptional factor in the retina, and its inhibition by antioxidants. Free Radic Res. 2003;37:1169–1180. doi: 10.1080/10715760310001604189. [DOI] [PubMed] [Google Scholar]
- 23.Romeo G, Liu WH, Asnaghi V, Kern TS, Lorenzi M. Activation of nuclear factor-kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes. 2002;51:2241–2248. doi: 10.2337/diabetes.51.7.2241. [DOI] [PubMed] [Google Scholar]
- 24.Calder PC. N-3 polyunsaturated fatty acids and inflammation: from molecular biology to the clinic. Lipids. 2003;38:343–352. doi: 10.1007/s11745-003-1068-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calder PC. Polyunsaturated fatty acids, inflammation, and immunity. Lipids. 2001;36:1007–1024. doi: 10.1007/s11745-001-0812-7. [DOI] [PubMed] [Google Scholar]
- 26.Kim I, Moon SO, Kim SH, et al. Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J Biol Chem. 2001;276:7614–7620. doi: 10.1074/jbc.M009705200. [DOI] [PubMed] [Google Scholar]
- 27.Mishra A, Chaudhary A, Sethi S. Oxidized omega-3 fatty acids inhibit NF-kappaB activation via a PPARalpha-dependent pathway. Arterioscler Thromb Vasc Biol. 2004;24:1621–1627. doi: 10.1161/01.ATV.0000137191.02577.86. [DOI] [PubMed] [Google Scholar]
- 28.De Caterina R, Bernini W, Carluccio MA, Liao JK, Libby P. Structural requirements for inhibition of cytokine-induced endothelial activation by unsaturated fatty acids. J Lipid Res. 1998;39:1062–1070. [PubMed] [Google Scholar]
- 29.De Caterina R, Liao JK, Libby P. Fatty acid modulation of endothelial activation. Am J Clin Nutr. 2000;71(1 suppl):213S–23S. doi: 10.1093/ajcn/71.1.213S. [DOI] [PubMed] [Google Scholar]
- 30.Chaudhary A, Mishra A, Sethi S. Oxidized omega-3 fatty acids inhibit proinflammatory responses in glomerular endothelial cells. Nephron Exp Nephrol. 2004;97:E136–E145. doi: 10.1159/000079178. [DOI] [PubMed] [Google Scholar]
- 31.Lecomte M, Paget C, Ruggiero D, Wiernsperger N, Lagarde M. Docosahexaenoic acid is a major n-3 polyunsaturated fatty acid in bovine retinal microvessels. J Neurochem. 1996;66:2160–2167. doi: 10.1046/j.1471-4159.1996.66052160.x. [DOI] [PubMed] [Google Scholar]
- 32.Chen W, Jump DB, Grant MB, Esselman WJ, Busik JV. Dyslipidemia, but not hyperglycemia, induces inflammatory adhesion molecules in human retinal vascular endothelial cells. Invest Ophthalmol Vis Sci. 2003;44:5016–5022. doi: 10.1167/iovs.03-0418. [DOI] [PubMed] [Google Scholar]
- 33.Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30:43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 34.Kuldo JM, Ogawara KI, Werner N, et al. Molecular pathways of endothelial cell activation for (targeted) pharmacological intervention of chronic inflammatory diseases. Curr Vasc Pharmacol. 2005;3:11–39. doi: 10.2174/1570161052773898. [DOI] [PubMed] [Google Scholar]
- 35.Zheng L, Szabo C, Kern TS. Poly(ADP-ribose) polymerase is involved in the development of diabetic retinopathy via regulation of nuclear factor-kappaB. Diabetes. 2004;53:2960–2967. doi: 10.2337/diabetes.53.11.2960. [DOI] [PubMed] [Google Scholar]
- 36.Invernici G, Ponti D, Corsini E, et al. Human microvascular endothelial cells from different fetal organs demonstrate organ-specific CAM expression. Exp Cell Res. 2005;308:273–282. doi: 10.1016/j.yexcr.2005.04.033. [DOI] [PubMed] [Google Scholar]
- 37.Lee TH, Hoover RL, Williams JD, et al. Effect of dietary enrichment with eicosapentaenoic and docosahexaenoic acids on in vitro neutrophil and monocyte leukotriene generation and neutrophil function. N Engl J Med. 1985;312:1217–1224. doi: 10.1056/NEJM198505093121903. [DOI] [PubMed] [Google Scholar]
- 38.Diep QN, Touyz RM, Schiffrin EL. Docosahexaenoic acid, a peroxisome proliferator-activated receptor-alpha ligand, induces apoptosis in vascular smooth muscle cells by stimulation of p38 mitogen-activated protein kinase. Hypertension. 2000;36:851–855. doi: 10.1161/01.hyp.36.5.851. [DOI] [PubMed] [Google Scholar]
- 39.Jump DB. Dietary polyunsaturated fatty acids and regulation of gene transcription. Curr Opin Lipidol. 2002;13:155–164. doi: 10.1097/00041433-200204000-00007. [DOI] [PubMed] [Google Scholar]
- 40.Staels B, Koenig W, Habib A, et al. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPAR-gamma activators. Nature. 1998;393:790–793. doi: 10.1038/31701. [DOI] [PubMed] [Google Scholar]
- 41.Marx N, Sukhova GK, Collins T, Libby P, Plutzky J. PPARalpha activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation. 1999;99:3125–3131. doi: 10.1161/01.cir.99.24.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Delerive P, De Bosscher K, Besnard S, et al. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J Biol Chem. 1999;274:32048–32054. doi: 10.1074/jbc.274.45.32048. [DOI] [PubMed] [Google Scholar]
- 43.Ruan H, Pownall HJ, Lodish HF. Troglitazone antagonizes tumor necrosis factor-alpha-induced reprogramming of adipocyte gene expression by inhibiting the transcriptional regulatory functions of NF-kappaB. J Biol Chem. 2003;278:28181–28192. doi: 10.1074/jbc.M303141200. [DOI] [PubMed] [Google Scholar]
- 44.Delerive P, Gervois P, Fruchart J-C, Staels B. Induction of Ikappa Balpha expression as a mechanism contributing to the anti-inflammatory activities of peroxisome proliferator-activated receptor-alpha activators. J Biol Chem. 2000;275:36703–36707. doi: 10.1074/jbc.M004045200. [DOI] [PubMed] [Google Scholar]
- 45.Stulnig TM, Huber J, Leitinger N, et al. Polyunsaturated eicosapentaenoic acid displaces proteins from membrane rafts by altering raft lipid composition. J Biol Chem. 2001;276:37335–37340. doi: 10.1074/jbc.M106193200. [DOI] [PubMed] [Google Scholar]
- 46.Zeyda M, Staffler G, Horejsi V, Waldhausl W, Stulnig TM. LAT displacement from lipid rafts as a molecular mechanism for the inhibition of T cell signaling by polyunsaturated fatty acids. J Biol Chem. 2002;277:28418–28423. doi: 10.1074/jbc.M203343200. [DOI] [PubMed] [Google Scholar]