

Abstract
Aneuploidies are common chromosomal defects that result in growth and developmental deficits and high levels of lethality in humans. To gain insight into the biology of aneuploidies, we manipulated mouse embryonic stem cells and generated a trans-species aneuploid mouse line that stably transmits a freely segregating, almost complete human chromosome 21 (Hsa21). This “transchromosomic” mouse line, Tc1, is a model of trisomy 21, which manifests as Down syndrome (DS) in humans, and has phenotypic alterations in behavior, synaptic plasticity, cerebellar neuronal number, heart development, and mandible size that relate to human DS. Transchromosomic mouse lines such as Tc1 may represent useful genetic tools for dissecting other human aneuploidies.
Down syndrome (DS) is a complex genetic condition arising from an altered dosage of wild-type genes on human chromosome 21 (Hsa21). One approach to the molecular genetics and pathology of DS has been to model the aberrant gene dosage of trisomy 21 in the mouse by transgenesis with single Hsa21 genes or yeast artificial chromosomes. This approach has highlighted potential loci of interest (1, 2). Alternatively, mouse aneuploidies have been used to model DS. Approximately two thirds of the orthologs of the 243 known Hsa21 genes (current gene estimate, ENSEMBL database) lie on mouse chromosome (Mmu) 16, whereas the remainder are distributed between Mmu10 and Mmu17 (3, 4). Thus, trisomies of Mmu16 have been studied as potential models of DS. Mice with full trisomy Mmu16 are not viable after birth, and because Mmu16 carries genes with orthologs on Hsa21 and at least three other chromosomes, mouse trisomy 16 is equivalent to partial trisomy of four human chromosomes (Hsa3, 16, 21, and 22). Therefore, the most widely used models of DS are the partial, or segmental, trisomy strains Ts65Dn and Tc1Cje, which are trisomic for portions of Mmu16 containing only Hsa21 orthologs (5–7).
An alternative model is provided by “transchromosomic” (trans-species aneuploid) mouse strains in which mice carry an extra human chromosome and are thus trisomic only for the genes on this chromosome. Such a transchromosomic strain for Hsa21 has several advantages for modelling DS. In contrast to transgenic methods to place Hsa21 genes into mice, this approach potentially reflects more closely the 3:2 dosage difference present between trisomic and disomic individuals through the introduction of only one extra copy of each Hsa21 gene. Additionally, the complete genomic sequence can be included, including upstream and downstream regulatory elements of unusually large genes (8) or those with complex regulatory elements and multiple transcripts (9). Unlike other methods of stable gene transfer, the transchromosomic approach should not interrupt endogenous mouse sequences. Furthermore, because individual human chromosomes have orthologs on more than one mouse chromosome, aneuploidy of an individual mouse chromosome is not equivalent to the human situation and only partially represents it, whereas placing an entire human chromosome into mice would model a full human trisomy.
The first transchromosomic mice were created by Oshimura and colleagues, who placed freely segregating portions of Hsa2, 14, or 22 into mouse embryonic stem (ES) cells using microcell-mediated chromosome transfer (MMCT) (10, 11). The ES cells were used to make chimeras, and germline transmission was achieved with a ~2-Mb Hsa2 fragment and a 1.5-Mb Hsa14 fragment (10, 11). Using a similar process, irradiation MMCT (XMMCT), to construct a mouse model for human trisomy 21, we generated a panel of transchromosomic male mouse ES cell lines, each carrying a freely segregating Hsa21 or portions thereof (12). When injected into mouse blastocysts, these cell lines gave high percentage contributions in the resultant chimeras; however, they failed to achieve germline transmission of Hsa21. This is consistent with previous findings that an aneuploid chromosome often will not transmit through the male germline (12, 13). Oshimura and colleagues later reported stable germline transmission of an Hsa21 fragment of ~5 Mb that carried an internal deletion and contained genes with homology to Mmu16 only (14, 15). Here, we take this technology forward and report the germline transmission of an almost complete Hsa21 and analysis of the resulting mouse strain, Tc1, which models aspects of human DS.
Generating the Tc1 transchromosomic mouse strains
We took the approach of reproducing the human-mouse transchromosomic cell lines on a female background, through further rounds of XMMCT into the female MPI VI ES cell line (16). We analyzed the resultant transchromosomic ES cell lines for human DNA content, using fluorescence in situ hybridization (FISH) to detect Hsa21 and reverse FISH to detect other human chromosomes (12). Five MPI VI–derived cell lines were identified, with a single freely segregating Hsa21 as the only human contribution (17) (fig. S1A). These cell lines were further assessed for the presence or absence of a panel of Hsa21 markers (Fig. 1A and table S1). Cell line 91-1 contained the most of Hsa21 with two gaps: the first bounded by the markers CXADR (at position 17,807,195) and D21S1922 (at 21,220,691) with a maximum size of 3.4 Mb, and the second was bounded by IFNAR1 (at 33,649,973) and RUNX1 (at 35,115,486) with a maximum size of 1.5 Mb (Fig. 1A). Therefore, 91-1 appears to contain at least 42 Mb (90%) of the complete 46.9 Mb of Hsa21, and we estimate that this includes ~92% of all known Hsa21 genes (17).
Fig. 1.
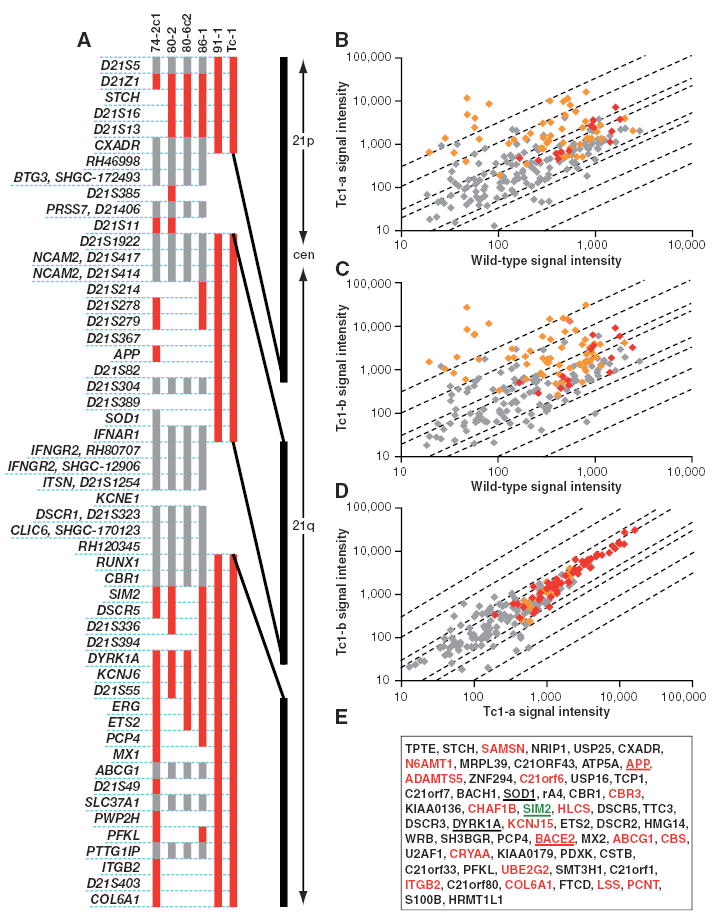
DNA and expression analysis of transchromosomic cell lines and mice carrying Hsa21. (A) Hsa21 content in five transchromosomic ES cell lines (74-2c1, 80-2, 80-6c2, 86-1, and 91-1) and Tc1 mouse DNAs. Cell line and mouse names are shown above the vertical bars that indicate the presence of the Hsa21 markers listed on the left. Markers are shown in order but are not spaced relative to their distance apart on Hsa21 (positions are given in table S1). Red denotes the presence of the marker, gray denotes not scored, and a blank space denotes a negative score, i.e., the marker was not present. CXADR was shown to be present by microarray results from Tc1 embryos. The vertical black line on the right is a scaled representation of the physical map of Hsa21, showing the relative spacing and maximum sizes of the two gaps. cen, centromere. (B to E) RNA samples from two Tc1 whole embryos and one wild-type littermate (E14.5) were hybridized to Affymetrix HG-U133A GeneChips. Array data were scaled to a target intensity of 500 before analysis. (B and C) Scatter plots showing Hsa21 gene signal intensities of embryos (B) Tc1-a and (C) Tc1-b against the wild-type littermate. (D) Scatter plot showing Hsa21 gene signal intensities of embryo Tc1-a against Tc1-b as a hybridization control. In each scatter plot, red points represent genes called present in both embryos, orange points represent genes called present in one of the two embryos, and gray points represent genes below the threshold for detection in both embryos. Diagonal lines indicate the thresholds for twofold, threefold, tenfold, and thirtyfold differences between the two embryos. (E) Genes with increased expression in Tc1 embryos compared to wild-type embryos. Red lettering indicates those found with the reduced stringency search method. Underlined genes were also found by RT-PCR or immunoblot. Expression of one gene, SIM2 (green), was detected by RT-PCR but not by microarray analysis.
To generate transchromosomic chimeras, ES cells were injected into host blastocysts and the resulting chimeras were mated to mice from the C57BL/6J strain. Germline transmission was achieved from one female chimera, carrying the 91-1 transchromosomic ES cell line. This chimera had only one litter of two pups, a male (Tc1-01) and a female (Tc1-02). These progeny both retained a freely segregating Hsa21 with the same Hsa21 profile as the parental ES cell line, 91-1 (Fig. 1A and fig. S1B). This transchromosomic mouse strain was designated Tc(Hsa21)1TybEmcf, hereafter referred to as Tc1 (18).
In an attempt to establish the Tc1 strain on a number of genetic backgrounds, mouse Tc1-01 was mated either to mice that were an F1 hybrid between C57BL/6J and 129S8 mice [F1(C57BL/6Jx129S8)] or to inbred BALB/c, C3H/He, and C57BL/6J mice. As the inbred line backcrosses progressed and the genetic background became more homogenous, transmission of the human chromosome diminished, to the point where these colonies could no longer be maintained (table S2). Subsequently, only the F1(C57BL/6Jx129S8) colony was retained, with a stable transmission frequency of >40% of progeny inheriting Hsa21 from their mothers, with occasional transmission through the male germ-line (table S2). We have aged male and female Tc1 mice up to 20 months of age, and at this time they are healthy and mobile with no signs of debilitation. They are thus likely to live well beyond 20 months of age.
Chromosome retention and gene expression
In previous studies in which chimeric mice have been engineered to carry human chromosome fragments, the human fragment was observed to be differentially retained in different organs and on different genetic backgrounds (14, 19). Thus, we used quantitative polymerase chain reaction (PCR) to determine the level of Hsa21 retention in various tissues from adult Tc1 male mice and observed that retention of Hsa21 ranged from 55 ± 6% in the spinal cord to 24 ± 3% in the spleen (table S3). For two tissues, brain and spleen, we also undertook interphase FISH using an Hsa21 paint and a mouse X chromosome probe as a control to directly count the percentage of nuclei carrying Hsa21 (table S4). Our results showed 66 ± 7% of brain nuclei and 49 ± 5% of spleen nuclei are positive for Hsa21, a higher percentage of positive cells than estimated by quantitative PCR. Mosaicism is also seen in human DS (20).
We next undertook a large-scale analysis of gene expression from Hsa21 in Tc1 mice. A comparison of whole embryo RNAs [at embryonic day (E) 14.5] from two Tc1 and one wild-type littermate (Fig. 1, B to D), on Affymetrix human HG-U133A arrays, demonstrated the expression of Hsa21 genes along the entire length of the chromosome, from TPTE in the p arm to HRMT1L1 at the distal end of the q arm (Fig. 1E). A total of 205 Hsa21 probe sets (representing 131 genes) were present on the arrays, of which 51 (representing 39 genes) were classified as increased (P < 0.01) in both Tc1 versus lit-termate comparisons. This method showed an increased signal from only 9 non-Hsa21 probe sets out of a possible total of 22,078. A reduced stringency search (17) brought the total number of Hsa21 genes with detectable expression over and above the littermate background to 58 (Fig. 1E). It remains possible that other Hsa21 genes are also expressed in Tc1 mice but were not detected, either because they are not expressed at sufficiently high levels in E14.5 embryos or because cross-reaction between the mouse homolog and the human gene on the microarray may have precluded detection. We also examined the expression of selected human genes previously implicated in various DS neuronal phenotypes (APP, SOD1, SIM2, DYRK1A, and BACE2) in Tc1 tissues by reverse transcription (RT)–PCR and/or immunoblotting (17). In all cases, gene expression was detected in Tc1 tissues (fig. S2).
Deficits in synaptic plasticity and learning
Because a primary aspect of DS is mental retardation (3), we assessed potential neural deficits in Tc1 mice using behavioral tests of learning and memory. We also examined hippocampal long-term potentiation (LTP), a form of synaptic plasticity that is altered in the Ts65Dn mouse (21, 22) and that provides a putative physiological substrate for hippocampus-dependent learning and memory (23). Field recordings of evoked potentials in the dentate gyrus of the hippocampus revealed no differences between wild-type and Tc1 mice in input-output curves (Fig. 2A and fig. S3) or paired-pulse interactions (Fig. 2, B and C), suggesting normal basal synaptic transmission and inhibitory tone. However, Tc1 mice exhibited significantly reduced LTP when compared with wild-type littermates (Fig. 2, D and E). To assess whether this deficit correlated with an effect on hippocampus-dependent memory, we tested Tc1 mutants in a novel-object recognition task (24, 25). Wild-type mice spent significantly more time exploring a novel object than two familiar objects that had been presented in a training session 10 min previously (Fig. 3A). Tc1 mice, however, failed to show significantly greater exploration of the novel object as compared with the familiar objects.
Fig. 2.
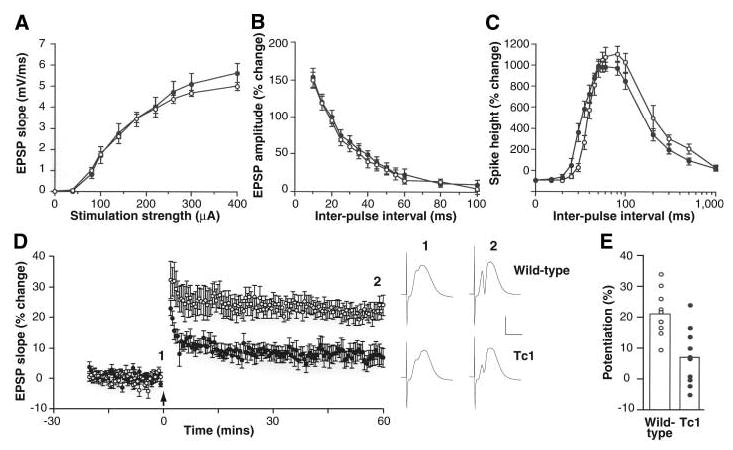
Short-term plasticity is normal but LTP is impaired in the dentate gyrus of Tc1 mice. (A) Input-output curves, (B) paired-pulse facilitation of the field excitatory postsynaptic potential (EPSP), and (C) paired-pulse modulation of the population spike are similar in wild-type (open circles) and Tc1 (solid circles) littermates. (D) LTP averaged for a group of 9 wild-type mice (open circles) and 10 Tc1 littermates (solid circles). The arrow marks the tetanus (six series of six trains of six stimuli at 400 Hz, with 200 ms between trains and 20 s between series). One hour after the tetanus, the magnitude of LTP was 21.9 ± 2.6% in wild-type mice, compared with 7.4 ± 2.9% in Tc1 mutants (P < 0.001, 2-tailed t test). Sample potentials from a single wild-type and a single Tc1 mouse, recorded just before (1) and 60 min after (2) the tetanus, are displayed on the right. Calibration, 5 mV, 5 ms. (E) The magnitude of LTP, measured 55 to 60 min after induction, is displayed for each wild-type mouse (open circles) and each Tc1 mutant (solid circles). The open histobars indicate the means for each group. All error bars represent SEM.
Fig. 3.
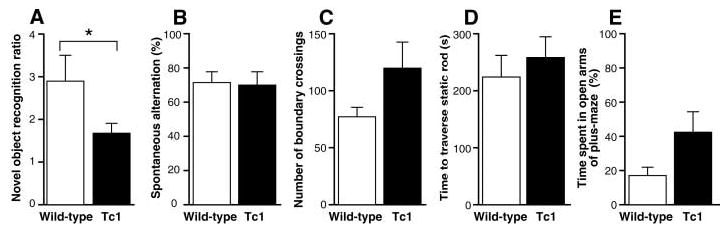
Tc1 mice have a hippocampus-dependent learning and memory deficit. (A) An index of novel-object exploration (the ratio of time spent exploring a novel object to mean time spent exploring a familiar object) in 12 wild-type (open) and 10 Tc1 (solid) littermates reveals a deficit in novel-object recognition in Tc1 mice (*, P < 0.05). Wild-type mice took significantly more time exploring a novel object than objects A and B, which had been presented in a training session 10 min previously (object A, 27.7 ± 16.9 s; object B, 31.8 ± 25.0 s; novel object, 69.0 ± 35.2 s; P < 0.05 for the novel object compared to either object A or B but not significant for object A compared to object B). (B) Five Tc1 mice and seven wild-type littermates performed above chance in the spontaneous alternation T-maze, but levels of alternation are not significantly different across genotypes. (C) Ten Tc1 mice displayed a nonsignificant trend toward hyperactivity in an open field compared with 12 wild-type littermates. (D) Testing on a static rod task revealed no significant motor deficit in five Tc1 mice as compared with seven wild-type littermates. (E) Five Tc1 mice did not spend significantly more time in the open arms of an elevated plus maze than seven wild-type littermates, although a nonsignificant trend may reflect overall increased levels of activity. All error bars represent SEM.
In contrast, a test of hippocampus-dependent short-term memory, the spontaneous alternation T-maze (26), did not reveal a significant difference between wild-type and Tc1 littermates (Fig. 3B). This finding suggests that Tc1 mice are able to retain hippocampus-dependent memory for up to a minute but show deficits in retaining memories over longer periods in the novel-object recognition task. To investigate potential behavioral confounds in these learning and memory tests, we subjected wild-type and Tc1 mice to tests of generalized activity, motor coordination, and anxiety. Compared with wild-type littermates, Tc1 mice showed a trend toward hyperactivity, which is a behavioral characteristic of DS, in an open-field test in which the number of boundaries crossed by an animal is scored. However, this trend did not reach significance (Fig. 3C) (P = 0.07) and did not manifest itself in abnormal levels of exploration during the novel-object recognition task (fig. S4). There was no indication of a deficit in moving along a static rod, a standard test of motor coordination (Fig. 3D). Hyperactivity may have contributed to a nonsignificant trend toward increased exploration of open arms in the elevated plus maze, a test of anxiety (27) (Fig. 3E). These findings suggest that Tc1 mice have deficits in both hippocampal synaptic plasticity and hippocampus-dependent learning and memory.
Cerebellar neuron counts and brain histopathology
As there is a decrease in cell density in the internal granule layer of the cerebellum in the Ts65Dn mouse (28–30) and total brain volume is reduced in DS, in particular in the cerebellum (31–33), we counted cerebellar granule neurons in four different cerebellar areas of four wild-type and four Tc1 littermates (table S5). The density of neurons was significantly lower in Tc1 mice (13,189 ± 2198 neurons per mm2) compared with wild-type mice (15,611 ± 2034 neurons per mm2) (P < 0.003). Comparison of one anatomically corresponding cerebellar lobe (VIII) across four wild-type and four Tc1 littermates showed similarly significant differences [16,515 ± 1516 neurons per mm2 (wild-type) versus 13,894 ± 1071 neurons per mm2 (Tc1); P = 0.03] (table S5). Preliminary analyses of brain histopathology with standard histological and immunohistochemical techniques, including immunostaining for neural markers (MAP2, neurofilament 200, synaptophysin, and glial fibrillary acidic protein), the microglial marker Iba-1, Tau (AT270 and AT8), and βA4 amyloid showed no anatomical, or cyto-architectural defects in brain sections of seven wild-type and seven Tc1 littermates aged between 9 and 21 months.
Heart development
Congenital heart defects occur in ~40% of DS individuals (3). We therefore compared heart development in E14.5 Tc1 embryos and their wild-type litter-mates. A perimembranous ventricular septal defect, representing a failure of fusion between the ventricular septum and the proximal outflow tract cushions, was seen in the majority of Tc1 mice studied (7 out of 11); this was associated with an overriding aorta (where the aorta straddles the ventricular septum) in a single case (Fig. 4, A, B, and G). In another fetus, the atrioventricular cushions were unfused, resulting in an atrioventricular septal defect (Fig. 4, C, D and G). A similar condition represents the most common cardiac defect seen in human babies with Down syndrome (34). In addition, some of the fetuses with ventricular septal defects also presented with the heart tilted onto its right side, when compared with wild-type littermates (Fig. 4, E and F). A minority of Tc1 fetuses (3 out of 11) showed no obvious cardiac defects (Fig. 4G).
Fig. 4.
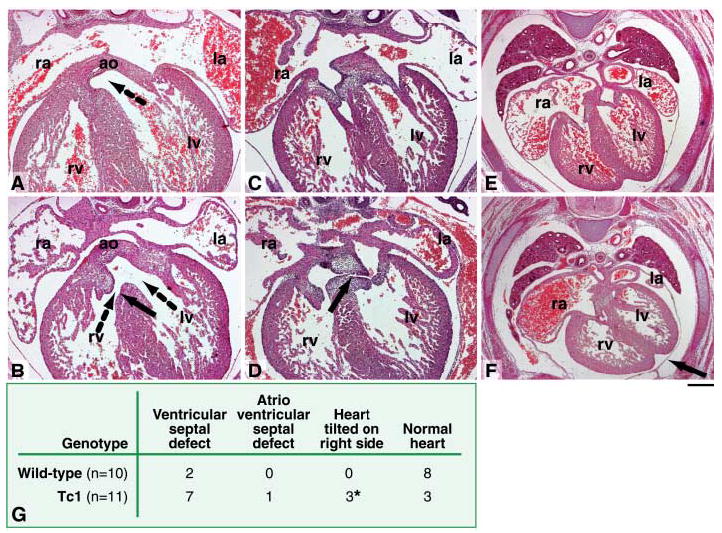
Cardiac defects in Tc1 fetuses. Transverse sections of E14.5 Tc1 and wild-type littermates, stained with hematoxylin and eosin. (A) Normal ventricular septation in a wild-type fetus, showing fusion of the interventricular septum with the proximal regions of the outflow tract cushions. The dotted arrow shows communication between the aorta and the left ventricle. (B) A perimembranous ventricular septal defect (solid arrow) in a Tc1 fetus, allowing the flow of blood between the right and left ventricles. In this case, the aorta is overriding the ventricular septum, allowing blood to enter the aorta from both the right and left ventricles (dotted arrows). (C) Normal atrioventricular septation in a wild-type fetus, showing fusion of the inferior and superior atrioventricular cushions and the primary atrial septum with the superior cushion. (D) Atrioventricular septal defect in a Tc1 fetus, showing that the inferior and superior atrioventricular cushions remain unfused (black arrow). (E) Normal positioning of the heart in a wild-type fetus. (F) In a Tc1 fetus the heart is tilted on to its right side, causing the interventricular sulcus to point to the left (black arrow). Scale bar, (A to D) 100 μm; (E and F) 200 μm. ao, aorta; la, left atrium; lv, left ventricle; ra, right atrium; rv, right ventricle. (G) Incidence of cardiac defects in Tc1 fetuses and their wild-type littermates. The asterisk indicates mice that also had ventricular septal defects.
Craniofacial morphology
Craniofacial abnormalities are seen in DS and in the Ts65Dn and Ts1Cje mouse models (35, 36). We were unable to find differences between Tc1 mice and wild-type littermates in analyses of facial bone morphology using light microscopy; all visible cranial and facial bone shapes of the Tc1 heads were found to be normal, with no obvious morphological variation from wild-type litter-mates. The overall dimensions of the Tc1 skulls were indistinguishable from these wild-type controls. Scanning electron micrographs were prepared of the teeth to accurately compare Tc1 with wild-type dentitions. Tooth shapes, positions, sizes, and cusp patterns were all found to be identical. However, these analyses would not have been able to detect the skull dysmorphology described in Ts65Dn and Ts1Cje mice (29, 35, 36). We therefore carried out micro–computerized tomography (CT) scans of 10 wild-type and 10 Tc1 littermate heads, comparing distances between anatomical landmarks similar to those used by Richtsmeier, Reeves, and colleagues (fig. S5). All measurements taken of the cranium indicated there were no significant differences between wild-type and Tc1 littermates, although it is possible that there may be differences that could be revealed by complex mathematical modeling approaches, such as Euclidean distance matrix analysis as used by Richtsmeier, Reeves, and colleagues. While the majority of measurements of the mandibles were also similar, Tc1 mandibles were significantly smaller between the coronoid process and the mandibular angle and between the coronoid process and the most superior point on the incisor alveolar rim (fig. S5 and table S6). The mandible is known to be smaller in DS individuals than in the euploid population (29, 35, 36).
T lymphocyte activation
The T cell receptor (TCR)–induced activation of T lymphocytes has been reported to be defective in DS (37), and thus we examined this in Tc1 mice. We consistently found that both CD4+ and CD8+ T cells from both spleen and lymph nodes of Tc1 mice up-regulated CD25 and CD69 to a lesser extent than wild-type cells, in response to stimulation either through the TCR alone with antibodies to CD3ɛ or in combination with antibodies to the costimulatory receptor CD28 (fig. S6).
The Tc1 mouse as a model of DS
DS manifests with phenotypes common to all affected individuals, such as mental retardation, and phenotypes that are variable between individuals, such as atrioventricular septal defects of the heart (3). We have generated a strain of trans-species aneuploid mouse that carries an almost complete human chromosome and recapitulates features seen in humans with DS and in other DS mouse models, including changes in behavior, synaptic plasticity, cerebellar neuronal number, heart development, and mandible size. Altered LTP, for example, has been seen in Ts65Dn mice, whereas DS-like heart defects have not been reported in these or Ts1Cje animals but are seen in Tc1 mice and in chimeric mice carrying a 5-Mb noncontiguous Hsa21 fragment (3, 19, 21, 22). Thus, for some aspects of DS, transchromosomic mice may be a better model of the human condition than the mouse chromosome segmental trisomies. Nonetheless, because the wide-scale effects of introducing human proteins into mouse protein complexes in transchromosomic animals are unknown, further work will be required to establish how well transchromosomic mice will model human aneuploidy conditions.
To dissect the molecular genetics of DS, human phenotype-karyotype correlations of the smallest region of overlap have been made for different traits in rare individuals with partial trisomies of Hsa21. Thus, DS critical regions (DSCRs) have been delineated that are described as carrying the gene(s) underlying specific traits, and the major effect gene(s) for several traits have been thought to lie within a few Mb of the marker D21S55 (4, 29, 38, 39). However, in recent studies, Reeves and colleagues studied mice carrying engineered portions of the Ts65Dn chromosome and showed that genes lying within the DSCR alone were not sufficient and were largely unnecessary to generate the craniofacial dysmorphologies found in Ts65Dn mice (29). We found an intermediate situation in which Tc1 mice have some features of a smaller mandible but not an overall diminution in cranium size; these data support the suggestion of Reeves and colleagues that individual triplicated genes contribute to a particular trisomic phenotype in combination with other genes, and this effect will depend on the function of these genes and the nature of their interactions (29).
As the generation of Tc1 mice was based on manipulation of mouse ES cells, we will be able to use chromosome engineering and DNA targeting of Hsa21 (or of endogenous mouse sequences) to examine the dosage effects of individual genes or chromosome regions on the phenotypic abnormalities. Because Tc1 mice are trisomic for genes with orthologs on Mmu16, as well as on Mmu10 and Mmu17, they should contribute both to the analysis of how DS results from trisomy 21 and to the testing of gene dosage effects for individual genes.
Conclusions
We have extended previous approaches to generating transchromosomic mice (10, 12, 14, 15) by achieving stable germline transmission of an almost complete human chromosome. This methodology is also relevant for studies of human artificial chromosomes as vectors. Technical issues arise from our studies. Both our analysis and previous studies have found that germline transmission of the human chromosome is dependent on the genetic background and sex of the transmitting parent and that the majority of transchromosomic lines will not give germline transmission, for reasons as yet unknown (12, 14). Finally, trisomy 21, which is found in ~1 out of 43 spontaneous abortions (40) and in ~1 in 750 live births (3), is just one of many aneuploidy syndromes. Aneuploidies are a common cause of human morbidity and mortality, occurring in at least 5% of all pregnancies (41). The Tc1 mouse shows that modelling whole human chromosome aneuploidy syndromes is feasible in the mouse.
Supplementary Material
Footnotes
Supporting Online Material www.sciencemag.org/cgi/content/full/309/5743/2033/DC1
Materials and Methods
Figs. S1 to S6
Tables S1 to S6
References and Notes
References
- 1.Chrast R, et al. Hum Mol Genet. 2000;9:1853. doi: 10.1093/hmg/9.12.1853. [DOI] [PubMed] [Google Scholar]
- 2.Smith DJ, et al. Nat Genet. 1997;16:28. doi: 10.1038/ng0597-28. [DOI] [PubMed] [Google Scholar]
- 3.Antonarakis SE, Lyle R, Dermitzakis ET, Reymond A, Deutsch S. Nat Rev Genet. 2004;5:725. doi: 10.1038/nrg1448. [DOI] [PubMed] [Google Scholar]
- 4.Nelson DL, Gibbs RA. Science. 2004;306:619. doi: 10.1126/science.1105226. [DOI] [PubMed] [Google Scholar]
- 5.Gardiner K, Fortna A, Bechtel L, Davisson MT. Gene. 2003;318:137. doi: 10.1016/s0378-1119(03)00769-8. [DOI] [PubMed] [Google Scholar]
- 6.Reeves RH, et al. Nat Genet. 1995;11:177. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- 7.Sago H, et al. Proc Natl Acad Sci USA. 1998;95:6256. doi: 10.1073/pnas.95.11.6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamakawa K, et al. Hum Mol Genet. 1998;7:227. doi: 10.1093/hmg/7.2.227. [DOI] [PubMed] [Google Scholar]
- 9.Kang J, et al. Nature. 1987;325:733. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 10.Tomizuka K, et al. Nat Genet. 1997;16:133. doi: 10.1038/ng0697-133. [DOI] [PubMed] [Google Scholar]
- 11.Tomizuka K, et al. Proc Natl Acad Sci USA. 2000;97:722. doi: 10.1073/pnas.97.2.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hernandez D, Mee PJ, Martin JE, Tybulewicz VL, Fisher EMC. Hum Mol Genet. 1999;8:923. doi: 10.1093/hmg/8.5.923. [DOI] [PubMed] [Google Scholar]
- 13.Hernandez D, Fisher EMC. Trends Genet. 1999;15:241. doi: 10.1016/s0168-9525(99)01743-6. [DOI] [PubMed] [Google Scholar]
- 14.Kazuki Y, et al. J Hum Genet. 2001;46:600. doi: 10.1007/s100380170028. [DOI] [PubMed] [Google Scholar]
- 15.Shinohara T, et al. Hum Mol Genet. 2001;10:1163. doi: 10.1093/hmg/10.11.1163. [DOI] [PubMed] [Google Scholar]
- 16.Voss AK, Thomas T, Gruss P. Exp Cell Res. 1997;230:45. doi: 10.1006/excr.1996.3418. [DOI] [PubMed] [Google Scholar]
- 17.Materials and methods are available as supporting material on Science Online.
- 18.The name Tc(Hsa21)1TybEmcf, which we have shortened to Tc1, is the official name given by the Mouse Nomenclature Committee.
- 19.Shinohara T, et al. Chromosome Res. 2000;8:713. doi: 10.1023/a:1026741321193. [DOI] [PubMed] [Google Scholar]
- 20.Devlin L, Morrison PJ. Arch Dis Child. 2004;89:1177. doi: 10.1136/adc.2003.031765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kleschevnikov AM, et al. J Neurosci. 2004;24:8153. doi: 10.1523/JNEUROSCI.1766-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siarey RJ, Stoll J, Rapoport SI, Galdzicki Z. Neuropharmacology. 1997;36:1549. doi: 10.1016/s0028-3908(97)00157-3. [DOI] [PubMed] [Google Scholar]
- 23.Bliss TV, Collingridge GL. Nature. 1993;361:31. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 24.Broadbent NJ, Squire LR, Clark RE. Proc Natl Acad Sci USA. 2004;101:14515. doi: 10.1073/pnas.0406344101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hammond RS, Tull LE, Stackman RW. Neurobiol Learn Mem. 2004;82:26. doi: 10.1016/j.nlm.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 26.Lalonde R. Neurosci Biobehav Rev. 2002;26:91. doi: 10.1016/s0149-7634(01)00041-0. [DOI] [PubMed] [Google Scholar]
- 27.Lister RG. Psychopharmacology. 1987;92:180. doi: 10.1007/BF00177912. [DOI] [PubMed] [Google Scholar]
- 28.Baxter LL, Moran TH, Richtsmeier JT, Troncoso J, Reeves RH. Hum Mol Genet. 2000;9:195. doi: 10.1093/hmg/9.2.195. [DOI] [PubMed] [Google Scholar]
- 29.Olson LE, Richtsmeier JT, Leszl J, Reeves RH. Science. 2004;306:687. doi: 10.1126/science.1098992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olson LE, et al. Dev Dyn. 2004;230:581. doi: 10.1002/dvdy.20079. [DOI] [PubMed] [Google Scholar]
- 31.Aylward EH, et al. Arch Neurol. 1997;54:209. doi: 10.1001/archneur.1997.00550140077016. [DOI] [PubMed] [Google Scholar]
- 32.Jernigan TL, Bellugi U. Arch Neurol. 1990;47:529. doi: 10.1001/archneur.1990.00530050049011. [DOI] [PubMed] [Google Scholar]
- 33.Raz N, et al. Neurology. 1995;45:356. doi: 10.1212/wnl.45.2.356. [DOI] [PubMed] [Google Scholar]
- 34.Maslen CL. Curr Opin Cardiol. 2004;19:205. doi: 10.1097/00001573-200405000-00003. [DOI] [PubMed] [Google Scholar]
- 35.Richtsmeier JT, Baxter LL, Reeves RH. Dev Dyn. 2000;217:137. doi: 10.1002/(SICI)1097-0177(200002)217:2<137::AID-DVDY1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 36.Richtsmeier JT, Zumwalt A, Carlson EJ, Epstein CJ, Reeves RH. Am J Med Genet. 2002;107:317. doi: 10.1002/ajmg.10175. [DOI] [PubMed] [Google Scholar]
- 37.Scotese I, et al. Pediatr Res. 1998;44:252. doi: 10.1203/00006450-199808000-00019. [DOI] [PubMed] [Google Scholar]
- 38.Delabar JM, et al. Eur J Hum Genet. 1993;1:114. doi: 10.1159/000472398. [DOI] [PubMed] [Google Scholar]
- 39.Korenberg JR, et al. Proc Natl Acad Sci USA. 1994;91:4997. doi: 10.1073/pnas.91.11.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.L. Y. Hsu, in Genetic Disorders and the Fetus, A. Milunsky, Ed. (Johns Hopkins Univ. Press, Baltimore, MD, 1998), pp. 179–248.
- 41.Hassold T, Hunt P. Nat Rev Genet. 2001;2:280. doi: 10.1038/35066065. [DOI] [PubMed] [Google Scholar]
- 42.We thank the Wellcome Trust, the Medical Research Council, the Leukaemia Research Fund, the Barts and the London Charitable Foundation, and the British Heart Foundation for support, A. Voss for the MPI VI ES cell line, P. Choquet for help with CT scans, R. Young for graphics, and R. Reeves for helpful comments. Gene array data are deposited at European Molecular Biology Laboratory–European Bioinformatics Institute with accession number E-MEXP-409.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.