

Abstract
Despite extensive deposition of putatively neurotoxic amyloid-β (Aβ) protein in the brain, it has not been possible to demonstrate an association of Aβ deposits with neuronal loss in Alzheimer's disease (AD), and neuronal loss is minimal in transgenic mouse models of AD. Using triple immunostaining confocal microscopy and analyzing the images with the cross-correlation density map method from statistical physics, we directly compared Aβ deposition, Aβ morphology, and neuronal architecture. We found dramatic, focal neuronal toxicity associated primarily with thioflavin S-positive fibrillar Aβ deposits in both AD and PSAPP mice. These results, along with computer simulations, suggest that Aβ develops neurotoxic properties in vivo when it adopts a fibrillar β-pleated sheet conformation.
The primary pathologic features of Alzheimer's disease (AD) are amyloid deposition, neurofibrillary tangle formation, and neuronal loss. There is substantial indirect evidence implicating amyloid-β (Aβ) protein in the pathological cascade leading to neuronal loss in AD (1). Presenilin-1 (PS1), presenilin-2, and amyloid precursor protein (APP) mutations causing familial AD and the apolipoprotein E ɛ4 allele risk factor for AD all increase plasma, fibroblast, or brain levels of Aβ or Aβx-42/43 in AD and transgenic mice (2–10). However, paradoxically, in human AD, the total amount of extracellular Aβ has little or no correlation with the amount of neuronal loss, which exceeds 50% in vulnerable regions of the cortex (11–13). Although quantitative stereological studies of neuron number in CA1 of APP23 (APPSw overexpressing) mice show a small decrement in neuronal number, no neuronal loss was detected in APP23 mice in the cortex (despite high levels of amyloid deposits) or in the hippocampus or cortex of PDAPP (APPV717F), Tg2576 (APPKM670−1NL), and PSAPP (APPSw × PS1M146L) mice (14–17), arguing that, in contrast to in vitro data, Aβ deposits are not neurotoxic in vivo.
Aβ deposits have a variety of morphologies, reflecting different amounts of fibrillar, β-pleated sheet conformation, ranging from “diffuse” deposits with little β-pleated sheet to dense core compact deposits that can be stained with dyes such as thioflavin S (ThioS) or Congo red; the extent of associated gliosis and synaptic loss is associated with the morphology of the Aβ deposits. We now test the hypothesis that only a subset of Aβ deposits is biologically toxic. In our approach we adopt a statistical physics method, i.e., the density map method, which is typically used in condensed matter physics to study local molecular organization, and which we have recently applied successfully to study the microcolumnar structure in brains of AD patients and patients with Lewy body dementia (18). We generalize the density map method to study the relationship between two different populations and then apply this method, which we call the cross-correlation density map (CCDM) method, to study the local neuronal organization surrounding Aβ-immunoreactive deposits. We find evidence that a specific, relatively small subset of Aβ deposits—compact, fibrillar ThioS deposits—are significantly more neurotoxic than non-ThioS plaques in Alzheimer's association cortex. We further test the hypothesis that ThioS-positive plaques are neurotoxic by examining PSAPP transgenic mice. The results of the analysis clearly indicate that ThioS-positive plaques are selectively neurotoxic. Furthermore, in deposits in which the ThioS-positive core is surrounded by a non-ThioS-positive penumbra of Aβ, only the fibrillar core of the Aβ deposit is toxic. Computer simulations and mathematical modeling of the effect of ThioS-positive Aβ deposits on adjacent neurons is compatible with the hypothesis that these deposits are toxic to neurons, rather than acting as nontoxic space-occupying lesions that merely push neurons away.
Methods
Human Tissue Resources and Image Acquisition.
Confocal images were obtained on a Bio-Rad 1024MP system, visualizing plaques with ThioS and anti-Aβ (antibody R1282, visualized with Cy5). Neurons were immunostained with anti-Neu-N and visualized with Cy-3. Three scans per image were obtained by using single lines of the lasers to avoid optical bleed-through. Cortical areas to be studied were chosen on the basis of gross anatomical landmarks and included a region ≈1 cm from the crown of the superior temporal gyrus in the superior temporal sulcus (STS) cortical area. We studied the STS region at the level of the lateral geniculate (18). Layer III of the STS area was identified on Neu-N-immunostained sections based on standard cytoarchitectonic criteria by an observer unaware of the disease category of the case.
The STS region was selected for study because it is a cortical area with strong limbic and association connections known to be affected in AD. Quantitative analyses have been previously applied to the STS area (13, 18), and neuronal loss proceeds in parallel with duration of illness. The STS is advantageous also because previous experience suggests that neurofibrillary tangles, a wide morphologic variety of senile plaques, and neuronal loss all consistently occur in this region from mild to severe AD (13). Four control cases (ages 63–104 years, mean 92 years), without clinical history of dementia or neuropathological signs of AD, and five cases of AD (ages 69–92 years, mean 81 years) were examined. Controls were two females and two males, AD cases were two females and three males. Detailed clinical histories are not available. Postmortem intervals were less than 24 h in all cases.
Transgenic Mouse Resources and Image Acquisition.
Hemizygous transgenic mice expressing mutant human APPSw (19) line Tg2576 and hemizygous mice expressing mutant human PS1M146L (5) line 6.2 were crossed. Double transgenic mice (referred to as PSAPP) were compared with nontransgenic mice. All mice examined were 12 months of age. Four mice from each genotype were used in each experiment. Brains were fixed in 4% paraformaldehyde and cryoprotected with 10% glycerol, and coronal sections were prepared at 40-μm thickness on a freezing sledge microtome. Immunohistochemistry was performed by using a polyclonal antibody against Aβ (R1282, 1:200, courtesy of D. Selkoe, Brigham and Women's Hospital, Boston) and monoclonal neuronal marker anti-Neu-N, 1:500 (Chemicon). Detection used secondary antibodies linked to Cy-3 or Cy-5 (Jackson ImmunoResearch). Sections were counterstained with 0.1% ThioS (Sigma). Confocal images of cingulate cortex were obtained on a Bio-Rad 1024 scanning laser confocal microscope mounted on a Nikon TE300 inverted microscope. Sequential images were obtained by using 488, 568, and 647 lines of the laser to excite ThioS, Cy-3, and Cy-5, respectively. Identical settings were used in each imaging session. Images were collected from the crown of the cingulate cortex at the coronal level of the corpus callosum. Images were obtained at ×20 power, and 12–15 adjacent fields of 447 μm × 447 μm per animal were collected over the anatomical region of interest.
Mice do not have a cortical homologue of the STS area. The cingulate cortex was selected as a cortical area for study in mice because it also has strong limbic and cortical projections, and the cingulate area develops early Aβ deposits in these mice. Importantly, our preliminary studies suggested that the cingulate was an area that consistently developed both diffuse and compact ThioS-positive plaques at 12 months of age.
Position Registration of Neurons and Plaques.
We developed a computer algorithm to detect neurons, Aβ-immunoreactive deposits, and ThioS-reactive deposits and to coregister the locations of each structure in x and y coordinates. Wavelet filtering allowed for the enhancement and separation of individual neurons even when they were partially overlapping in regions of high neuronal density. The detection algorithm proved to be accurate to levels >95% in the case of the PSAPP mouse model as compared with manual detection with stereological methods. In the human autopsy tissue, autofluorescent lipofuscin was also present in the confocal images, including relatively large, round corpora amylacia and noncell-associated smaller fainter deposits. Both were deleted by visual inspection. In addition, a small number of neurons had overlapping processes, leading to the incorrect identification of a single cell; these were separated by manual correction. With this additional manual correction we achieved 95% accuracy in detection of (x, y) neuronal positions. The detection benefited from the clear signal, specificity, and high contrast that the fluorescence of ThioS, Neu-N, and R1282 antibodies provides. The result of the detection is a coregistered 2D map of the x−y coordinate locations of the neurons, Aβ-immunoreactive deposits, and ThioS deposits in the thin confocal image plane.
Aβ Deposit Classification.
We performed subgroup analysis on Aβ deposits by segregating them in terms of size and optical density (the latter depends on the intensity of staining and on the image contrast). All of the tissue sections, mouse or human, were immunostained in parallel and were analyzed at identical laser settings with the confocal microscope. We classified each Aβ deposit according to two properties: (i) effective radius R and (ii) average optical density. We defined the effective radius R as proportional to the radius of gyration Rg of the deposit. The definition that is given below is such that if a deposit was a disk of a radius rd and homogeneous optical density, its effective radius would be R = rd. The radius of gyration is defined by
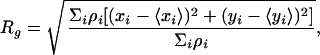 |
where ρi ∈ [0,255] is the optical density of pixel i, and i ranges over all of the pixels in the deposit. The effective radius is then calculated as R = 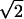 Rg. The average optical density is defined by averaging over the optical densities ρi of individual pixels in the Aβ deposit.
Rg. The average optical density is defined by averaging over the optical densities ρi of individual pixels in the Aβ deposit.
CCDM Method.
The method is a modified version of the density map method described and used in our recent publication (18) where it was applied to study local neuronal architecture. Here we adapt it to study the spatial relationship between two different populations: Aβ deposits and neurons. To apply the CCDM method, we needed spatial locations of the centers of mass of Aβ deposits and (x, y) coordinates of neurons. We labeled N deposits in the sample by i ∈ {1, 2, … N}. For each deposit we calculate the local neuronal density field gi(x, y), which is centered at the center of mass of the deposit and coarse grained by a square grid with an edge Δ. The quantity gi(x, y) is then calculated as a number of neurons in the grid cell of size Δ2, which is centered at (x, y). For the calculation to be meaningful Δ should be large enough compared with the size of neurons, and small enough to allow for spatial resolution (18). In our calculations 10 μm < Δ < 20 μm. After we calculate all of the local neuronal density fields, gi(x, y), for every deposit i, we then average over the local neuronal density fields, gi(x, y), to obtain the average local neuronal density g(x, y). Finally, radial neuronal density ρ(r), is calculated from g(x, y) by integrating g(x, y) over all angles.
Results
Characterization of Aβ Deposits in AD.
We used triple-labeled images of immunostained samples of five different AD cases with average neuronal densities of 1,563 mm−2, 763 mm−2, 1,087 mm−2, 1,597 mm−2, and 862 mm−2. Four control cases with average neuronal densities of 1,615 mm−2, 1,816 mm−2, 1,486 mm−2, and 1,632 mm−2 were also studied. We applied the CCDM method to study the local neuronal density around Aβ deposits within neuronal layer III of the STS area. The percentages of the areas covered by Aβ were, case by case, 13.6%, 7.5%, 5.7%, 9.1%, and 20.1%. The subclass of Aβ deposits that was marked as ThioS-positive covered 6.3%, 0.8%, 2.6%, 2.4%, and 6.0% of the total area of layer III.
Results of the CCDM Method in AD.
We examined the neuronal density within and in the immediate vicinity of the Aβ- and ThioS-positive Aβ deposits. Three-color confocal images were used to determine the degree of overlap and spatial relationships among Aβ immunoreactivity, ThioS deposits, and Neu-N-immunostained neurons. The color superposition of the three channels is shown in Fig. 1a. We applied the CCDM method to quantify the local neuronal density in the vicinity of Aβ deposits. As we were interested only in the neuronal density distribution and its dependence on the distance from the radius of the deposit, we integrated the averaged density map over all angles to obtain the radial neuron density, ρ(r), which describes the density of neurons as it varies over the radial distance r from the center of the deposit: ρ(r) at distance r from the origin measures the number of neurons within a ring centered at r, divided by the area of the ring as shown in Fig. 1a.
Fig 1.

(a) Color superposition of triple-labeled images of human brain tissue (AD case) from a dual photon microscope showing coregistration of neurons with Aβ-immunoreactive and ThioS-labeled deposits in layer III of the STS: Neu-N-stained neurons (red), Aβ-immunoreactive deposits (blue), and ThioS labeling (green), showing tangles in addition to ThioS-positive Aβ deposits. A schematic picture of concentric rings around one deposit illustrates the density map method used in the calculation. ThioS-positive Aβ channel (green) was used to classify the plaques into ThioS-positive and non-ThioS deposits. (b) Color superposition of confocal double-labeled immunostained images of transgenic mouse brain tissue showing coregistration of neurons (red) with Aβ-immunoreactive (blue) and ThioS-labeled (green) deposits. The arrows point to two ThioS deposits that coincide with two holes in the neuronal density.
Radial neuron densities, ρ(r), plotted in Fig. 2a, are calculated for each of five AD cases separately, normalized by the average neuronal density per case, and then averaged over the cases. Fig. 2a shows ρ(r) for all Aβ deposits (n = 1287), non-ThioS (n = 996), and ThioS-positive (n = 223) Aβ deposits. In addition, as two kind of controls, we calculated ρ(r) for Aβ deposits, superimposed at random positions over the neuronal map of layer III of the control cases and for Aβ deposits, superimposed at random positions over the neuronal map of layer III of the AD cases. As expected, both control radial neuron density curves with randomly scattered phantom Aβ deposits over the neuronal layer III of human control and AD cases are flat with no evidence of local neuronal density change.
Fig 2.

(a) Normalized neuronal density ρN(r) averaged over five AD cases with average neuron densities of 1,563, 763, 1,087, 1,597, and 862 neurons per mm2 for (i) all Aβ deposits with n = 1,287 (black), (ii) non-ThioS Aβ deposits with n = 996 (cyan), (iii) ThioS-positive Aβ deposits with n = 223 (red), and (iv) randomly shuffled Aβ deposits with n = 1,287 (gray). (Inset) Normalized neuronal density ρN(r) for different size classes of Aβ deposits with respect to the radius R of the deposit, averaged over five different AD cases: (i) R < 7 μm (red), (ii) 7 < R < 10 μm (orange), (iii) 10 < R < 15 μm (green), and (iv) R > 15 μm (blue). (b) Neuronal density ρ(r) in transgenic mice with an average neuronal density of 2,468 per mm2 with respect to the center of mass of the Aβ deposits. We plot ρ(r) for all Aβ deposits with n = 982 (black), non-ThioS (cyan) with n = 819, and ThioS-positive (red) Aβ deposits with n = 166. (Inset) Neuronal densities ρ(r) around ThioS-positive Aβ deposits of different effective radia R: (i) R < 12.3 μm (red) with n = 31, (ii) 12.3 < R < 24.8 μm (orange) with n = 78, and (iii) R > 24.8 μm (green) with n = 58.
Fig. 2a shows that there is diminished neuronal density within the boundaries of Aβ deposits compared with its immediate surroundings. The main feature of the graphs in Fig. 2a is a significantly bigger decrease of neuronal density associated with ThioS-positive Aβ deposits compared with non-ThioS deposits. We performed a Kolmogorov-Smirnov test to determine the statistical significance of differences between the radial neuron density curves of ThioS, non-ThioS, and randomly placed deposits (a control curve). The results showed that for distances r, r < 15 μm, the ThioS and non-ThioS curves are significantly different (P < 0.05). We confirmed also that for r < 15 μm the non-ThioS radial neuron density curve significantly differs from the control curve (P < 0.05).
Further, we examined all immunofluorescent Aβ deposits. We separated all immunostained Aβ deposits into four size classes with respect to the effective radius of a deposit. We found the largest decrease of neuronal density for Aβ deposits of the radia R, 10 μm < R < 15 μm (see Fig. 2a Inset). Surprisingly, the largest deposits with R > 15 μm did not correlate with the strongest decrease in neuronal density at the center. We also found that optically denser deposits are associated with a larger decrease in neuronal density at the center of the deposit.
The results from control and AD cases indicate that there is a local decrease of neuronal density at the center of Aβ deposits that could be caused by either deposits being space-occupying objects that tend to push neurons away or toxicity. We found that three subclasses of Aβ deposits show a significantly stronger decrease of local neuronal density: (i) ThioS-positive deposits that contain β-pleated sheet conformation of amyloid, (ii) larger deposits with radius 10 μm < R < 15 μm, and (iii) optically denser deposits that presumably indicated higher amyloid density. To further test which of these classes has the largest impact on local neuronal decrease, we repeated the above study on a PSAPP transgenic mouse model.
Characterization of Aβ Deposits in PSAPP Mice.
R1282 immunostaining demonstrated Aβ deposition in cortical and hippocampal regions of PSAPP mice, as published (17). Fig. 1b shows a coexistence of Neu-N-stained neuronal bodies (red), amyloid deposits (blue), and ThioS-positive labeling (green). We find “holes” within the cortex that are neuron free and contain amyloid as observed by others (20–22). In the four PSAPP mice used in this study, the percentage of the crown of the cingulate covered with Aβ immunoreactivity using the protocol published in ref. 17 was 24.96 ± 8.1%. ThioS-staining deposits covered 1.83 ± 0.42% of the cingulate cortex. The distribution of the sizes of the Aβ-immunoreactive deposits as well as the ThioS-staining deposits followed a peak distribution (23) that indicates a decrease in their number with increasing deposit size. The average effective radius of all Aβ-immunoreactive deposits was 7.65 ± 6.03 μm and ThioS deposits was 4.26 ± 3.74 μm. The neuronal density by Neu-N-staining was on average 2,400 neurons/mm2.
Results of the CCDM Method in the PSAPP Mouse Model.
We performed the CCDM analysis on n = 989 Aβ- immunoreactive deposits from four 12-month-old double transgenic PSAPP mice (176.4 ± 76.5 deposits/mm2 per mouse). We applied the CCDM method to all Aβ deposits and separately to non-ThioS and ThioS-positive Aβ deposits. The corresponding radial neuronal densities, ρ(r), are plotted in Fig. 2b. ρ(r) is only slightly decreased at the center of the deposit if all Aβ deposits are included in the CCDM calculation. Although non-ThioS deposits do not seem to affect ρ(r) at all, there is a profound decrease in ρ(r) at a center of a typical ThioS-immunostained Aβ deposit, at r = 0. Randomly shuffling the position of ThioS plaques over a field of neurons, used as a negative control, did not show any correlations.
In a further search for a subclass of Aβ deposits that is correlated with the largest decrease of local neuronal density, we subdivided Aβ deposits separately into groups by effective radius R and optical density. There was an ≈20% reduction in ρ(r) within the class of Aβ deposits with the largest effective radius R > 12 μm (see Fig. 2b Inset). We also found optically denser Aβ deposits to be associated with larger decrease in ρ(r). We found that deposits with the largest effective radius and largest optical density coincide with ThioS-positive-labeled deposits in 95% of the cases. The majority of Aβ deposits were non-ThioS-positive: examination of this group, in contrast to the ThioS-positive plaques, showed no change in the local neuronal density. Our results are thus consistent with previous observations that, taken as a whole, Aβ deposits were not associated with measurable neuronal loss in PSAPP mice (17).
Our findings described above suggested that the larger and more dense Aβ deposits were associated with a decrease in ρ because they contained fibrillar rather than diffuse Aβ. This conclusion was possible in large part due to the CCDM method, which proved to be a powerful tool for testing predictions regarding the local effects of Aβ on neurons, specifically by quantifying disruption in the neuronal distribution in the vicinity of each Aβ deposit. This analysis allowed us to test four models of the effect of Aβ deposition (Fig. 3):
Fig 3.

Schematic pictures and diagrams for possible biological scenarios of Aβ deposit effects on neuronal landscape. (A) Nontoxic diffuse deposit. (B) Nontoxic deposit displacing neurons. (C) Focally toxic deposit. (D) Focally toxic deposit with a toxic penumbra.
A: Nontoxic diffuse deposit.
The neuronal density is constant both within the Aβ deposit and in the vicinity, resulting in a horizontal line in ρ(r) independent of r (Fig. 3A).
B: Nontoxic deposit.
The Aβ deposit displaces neurons, resulting in a reduced ρ(r) within the Aβ deposit, a local increase in ρ(r) representing neurons pushed to the periphery, followed by a constant ρ(r) thereafter (Fig. 3B).
C: Focally toxic deposit.
The Aβ deposit kills neurons within the Aβ deposit, resulting in a reduced ρ(r) inside the deposit, followed by a constant ρ(r) afterward (Fig. 3C).
D: Toxic deposit with toxic penumbra.
The Aβ deposit kills neurons, resulting in a reduced ρ(r) both within and in the vicinity of the Aβ deposit (Fig. 3D).
The results of our CCDM analysis on PSAPP mouse brain showed that non-ThioS Aβ deposits behave according to model A; the local neuronal density ρ is not affected by the presence of non-ThioS Aβ deposits. The challenge was then to understand the decrease in ρ within ThioS-positive Aβ deposits and figure out which of the three models (B, C, or D) best fit the observed behavior of ρ(r), in Fig. 2.
Modeling Spatial Relationship Between Neurons and ThioS Aβ Deposits.
We explored two plausible physical mechanisms for the decreased neuronal density within a ThioS-staining deposit: (i) compact Aβ behaves as a nontoxic space-occupying deposit that simply pushes neurons away (model B), or (ii) compact Aβ kills neurons as it grows (models C and D). To clarify the biophysical nature of ρ(r) from the ThioS-staining deposits (e.g., dependence on size, density, and β-pleated sheet conformation of Aβ), we investigated the effect of the growth of a dense object, the ThioS-positive Aβ deposit, on adjacent neurons by using a molecular dynamics algorithm (24) typically used in statistical physics to simulate many-body interactions between objects. The molecular dynamics algorithm consists of determining the speed and the direction of motion of objects based on the total force resulting on them. In our case, however, we were interested only in the final distribution of objects (by which we modeled neurons), thus we considered only the direction of motion of the surrounding objects due to a slow growth of Aβ deposit, discarding all information regarding velocities and magnitudes of forces. To be able to compare simulations to the experimental data, we used for the simulations the same ThioS-staining deposit size distribution.
In Fig. 4a we show ρ(r) for the model assuming either a nontoxic or a toxic deposit. Within 30 μm of a nontoxic deposit, there were two peaks of increased neuronal density, corresponding to the nearest and next-nearest neurons that surround the deposit. The increase in value of the first peak, as compared with the average neuronal density beyond 30 μm, indicated the increased density of neurons that had been pushed and compressed together in the immediate periphery of the deposit. The radial neuronal density ρ(r) around a toxic deposit was flat and featureless, indicating an undisturbed neuronal neighborhood outward from the periphery of the deposit.
Fig 4.

(a) Neuronal density ρ(r) from the molecular dynamics simulations for the nontoxic and toxic model. In each case, ρ(r) is averaged over a set of sizes obtained from the ThioS size distribution with n = 254. Neuronal density ρ(r) for the nontoxic model shows a rather prominent peak of neuronal density that would correspond to a denser ring of neurons around the deposit. Neuronal density ρ(r) for the toxic model displays no such peak, indicating that neurons are not merely pushed away as the deposit grows but rather destroyed. The Insets correspond to typical computer realizations for each model, where the initial position for the nonoverlapping small circles (model neurons) is random and the total density and radius [Nneu = 1,850 in a 512 × 512 pixel2 box, R = 4 pixels (3.5 μm)] is taken to be equivalent to the average density and radius of neurons in the PSAPP transgenic mice. The central deposit (only the hole shown) in each Inset has a radius of 18 pixels (15.7 μm). (b) Neuronal density ρ(r) in dependence on the distance from the surface of a ThioS core in transgenic mice. For each deposit ρ(r) is shifted by the radius of the ThioS core. Individual functions ρ(r) are then averaged over all 281 ThioS cores. (Inset) A confocal image of Neu-N-stained PsAPP mouse brain showing a hole in the otherwise rather homogeneous distribution of neurons.
The result for ρ(r) in Fig. 4a remarkably approximates the radial neuronal density for ThioS-staining deposits shown in Fig. 2, implying that ThioS-staining deposits behaved as toxic deposits, following either model C or D. Aβ deposits that were ThioS-positive frequently contain a core of ThioS staining surrounded by a region of more diffuse, nonfibrillar Aβ. To distinguish between models C and D, we next asked whether the toxicity of ThioS-staining deposits extended beyond the ThioS-positive core—whether there was a “penumbra” of toxicity. ρ(r) was subjected to a linear transformation so that for each ThioS deposit ρ(r) was shifted by the radius of the ThioS core itself. Thus, in the transformed x axis, R = 0 represented the ThioS core surface, positive R represented the distance from the ThioS core surface, and negative R represented the distance from the ThioS core surface to an interior region of the ThioS deposit. Fig. 4b shows (i) that there was no dense ring around the neuronal “exclusion” area, even for deposits with the largest effective radius, and (ii) that the neuronal density plummeted within the ThioS core, but was normal within 10 μm of its surface, independent of size, suggesting no detectable penumbra effect beyond the ThioS core region. These two features together indicate that model C, the focal toxic model, fit our results better. In AD cases that we analyzed, the border of the ThioS core was not as well defined as in the mouse model. However, we repeated a similar analysis by shifting ρ(r) for each individual ThioS-positive Aβ deposit by its effective radius (rather than the radius of the core). The resulting ρ(r) looked similar to the curve in Fig. 4b except that it was shifted to negative distances r by about 16 μ m, indicating that the main drop in neuronal density occurs within the relatively small region at the center of the ThioS deposit.
Discussion
Our study addresses one of the paradoxes of AD neuropathology: the fact that Aβ metabolism is so strongly implicated in the pathogenesis of AD by genetic studies, but that the amount of Aβ deposits observed in the brain does not correlate well with the degree of dementia or extent of neuronal loss. We used a CCDM method to examine the possibility that there might be neuronal loss associated with only a subset of plaques. Aβ neurotoxicity has been demonstrated in cell culture (25–31) where the extent of toxicity is clearly influenced by the aggregation state of Aβ, with fibrillar Aβ being far more toxic. We show evidence for a specific association between ThioS Aβ plaques and neuronal loss in both human brain and a transgenic model of AD. Based on the form of ρ(r), we found that: (i) larger and optically denser Aβ-immunoreactive deposits are associated with neuronal loss, (ii) ThioS-positive Aβ deposits were almost entirely associated with neuronal loss, and (iii) there is no penumbra of toxicity beyond the ThioS-staining deposit. Modeling the Aβ deposit interaction with neurons as a biophysical many-body interaction problem by computer simulations suggested that ThioS-staining deposits behave as toxic lesions.
The presence of a small amount of neuronal loss in CA1 of APP23 mice, contrasting with the absence of neuronal loss in the cortex of APP23 mice (16) or in hippocampus or cortex of other mouse models of Aβ deposition (14–17), raised the possibility that the CA1-specific loss identified in APP23 mice was due to transgene or mouse-specific effects such as concurrent vascular pathology. However, our current analysis provides strong evidence for focal Aβ-associated neuronal toxicity. The statistical power of stereological techniques in our prior studies, although sufficient to detect overall neuronal losses in the order of several percent, was insufficient to detect focal losses. The current analysis shows that the vast majority of Aβ deposits do not alter the neuronal landscape; in fact, only the densest and largest Aβ deposits (<3% area coverage) are associated with neurotoxicity. For example, ThioS cores occupy only 1.8% of the cortical surface and are distributed widely throughout cortical areas. Even if all neurons within ThioS cores were lost, this difference in total neuron numbers would be difficult to detect. As an example, if the animal-to-animal variation in cortical neuron counts is 5–10%, we estimate that 175 animals would be required to detect the loss of 1.5% of the neurons. Our present method of focusing on the microenvironment around individual Aβ deposits substantially increases the sensitivity of our analysis to detect neuronal toxicity. It complements stereological methods, which provide a powerful approach to measuring total neuronal number, but which are less sensitive to local microenvironments and microarchitecture in the brain. PSAPP mice do not develop neurofibrillary changes nor widespread neuronal loss, and so can be used as a model to determine the unique effect of overproduction and deposition of Aβ in its various forms on the brain. Changes associated with Aβ deposits in both AD and transgenic mice include astrocytosis, microgliosis, and neuritic dystrophy; to this list we now add focal, discrete, neuronal loss.
In AD, neuronal loss exceeds that which can be accounted for by ThioS-positive plaques alone (only 4%). Yet AD is a complex pathophysiology, with neuronal loss also no doubt related to neurofibrillary tangle formation (13) and potentially even retrograde degeneration phenomenon. Frequently, however, ThioS-positive plaques are associated with phosphorylated tau-positive dystrophic neurites, suggesting a link between ThioS plaques and alterations in tau metabolism, and further suggesting an impact of ThioS on the normal morphology of neurites in the neuropil (32). Recent reports show that in transgenic mouse models Aβ deposits influence the formation of tau tangles in areas of brain known to be affected in AD (33, 34). These recent studies in combination with our results support the hypothesis that Aβ is indeed a causative pathogenic factor in AD development; other factors present in AD that may yet prove to contribute to neuronal loss and dysfunction may include oxidative damage and local inflammation, as well as the potentially toxic effect of oligomers of Aβ and activation of apoptotic mechanisms.
In conclusion, the neurotoxic effects of Aβ in the brains of PSAPP mice are limited to the largest and densest deposits that have a β-pleated sheet conformation. Although a small percentage of all Aβ deposits, these Aβ deposits have a marked effect on neurons in their immediate environment. Although an alternative possibility, that neuronal death precedes and is a prerequisite for ThioS plaque formation, cannot be excluded, our results are consistent with in vitro and in vivo studies demonstrating that the toxicity of Aβ is associated with the conformational state of the peptide. Aggregated fibrillar Aβ, but not monomeric or amorphous polymeric Aβ, is toxic to neurons in culture (27, 29, 35). In neuropathological studies of human AD brain, ThioS plaques but not non-ThioS plaques are associated with dramatic alterations in neuritic geometry (36, 37), which, we have argued, may underlie functional deficits in neural systems in AD. The current results are also consistent with the idea that the β-pleated sheet conformation of Aβ is important for toxicity in vivo, perhaps by inducing cross linking or conformational changes of cell surface proteins (38). In vitro data demonstrate that fibrillar Aβ can affect calcium homeostasis, promote free-radical and oxidative injury, and potentiate excitotoxic and apoptotic insult. Within PSAPP transgenic mice, oxidative stress markers have been associated with neurites around fibrillar but not diffuse deposits (39). Within APP transgenic mice, Aβ deposition can promote inflammatory responses as well, which may lead to bystander injury of neurons. Taken together, our data show that fibrillar Aβ toxicity is one of the mechanisms of neuronal death active in both AD and APP transgenic mice, emphasizing the potential importance of therapeutic attempts focusing on prevention or clearance of fibrillar Aβ deposits.
Acknowledgments
We thank S. V. Buldyrev and S. Peng for helpful discussions. This work was supported by National Institute of Aging Grants AG08487, AG00793, AG15453, AG15379, and AG17216 and National Institutes of Health/National Center for Research Resources Grant P41 RR13622. We acknowledge additional support by the National Science Foundation, the Adler Foundation, and the Memory Ride Foundation.
Abbreviations
AD, Alzheimer's disease
Aβ, amyloid-β
APP, amyloid precursor protein
ThioS, thioflavin S
CCDM, cross-correlation density map
STS, superior temporal sulcus
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Selkoe D. (1994) J. Neuropathol. Exp. Neurol. 53, 438-447. [DOI] [PubMed] [Google Scholar]
- 2.Goate A., Chartier-Harlin, M.-C., Mullan, M., Brown, J., Crawford, F., Fidani, L., Giuffra, L., Haynes, A., Irving, N., James, L., et al. (1991) Nature 349, 704-706. [DOI] [PubMed] [Google Scholar]
- 3.Citron M., Oltersdorf, T., Haass, C., McConlogue, L., Hung, A. Y., Seubert, P., Vigopelfrey, C., Lieberburg, I. & Selkoe, D. J. (1992) Nature 360, 672-674. [DOI] [PubMed] [Google Scholar]
- 4.Cai X.-D., Golde, T. E. & Younkin, S. G. (1993) Science 259, 514-516. [DOI] [PubMed] [Google Scholar]
- 5.Duff K., Eckman, C., Zehr, C., Yu, X., Prada, C. M., Perez-tur, J., Hutton, M., Buee, L., Harigaya, Y., Yager, D., et al. (1996) Nature 383, 710-713. [DOI] [PubMed] [Google Scholar]
- 6.Gomez-Isla T., West, H. L., Rebeck, G. W., Harr, S. D., Growdon, J. H., Locasio, J. T., Perls, T. T., Lipsitz, L. A. & Hyman, B. T. (1996) Ann. Neurol. 39, 62-70. [DOI] [PubMed] [Google Scholar]
- 7.Scheuner D., Eckman, C., Jensen, M., Song, X., Citron, M., Suzuki, N., Bird, T. D., Hardy, J., Hutton, M., Kukull, W., et al. (1996) Nat. Med. 2, 864-870. [DOI] [PubMed] [Google Scholar]
- 8.Borchelt D. R., Ratovitski, T., van Lare, J., Lee, M. K., Gonzales, V., Jenkins, N. A., Copeland, N. G., Price, D. L. & Sisodia, S. S. (1997) Neuron 19, 939-945. [DOI] [PubMed] [Google Scholar]
- 9.Mann D. M. A., Iwatsubo, T., Nochlin, D., Sumi, S. M., Levy-Lahad, E. & Bird, T. D. (1997) Ann. Neurol. 41, 52-57. [DOI] [PubMed] [Google Scholar]
- 10.Gomez-Isla T., Growdon, W. B., McNamara, M. J., Nochlin, D., Bird, T. D., Arango, J. C., Lopera, F., Kosik, K. S., Lantos, P. L., Cairns, N. J. & Hyman, B. T. (1999) Brain 122, 1709-1719. [DOI] [PubMed] [Google Scholar]
- 11.West M. J., Coleman, P. D., Flood, D. G. & Troncoso, J. C. (1994) Lancet 344, 769-772. [DOI] [PubMed] [Google Scholar]
- 12.Gomez-Isla T., Price, J. L., McKeel, D. W., Morris, J. C., Growdon, J. H. & Hyman, B. T. (1996) J. Neurosci. 16, 4491-4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomez-Isla T., Hollister, R., West, H., Mui, S., Growdon, J. H., Peterson, R. C., Parisi, J. E. & Hyman, B. T. (1997) Ann. Neurol. 41, 17-24. [DOI] [PubMed] [Google Scholar]
- 14.Irizarry M. C., Soriano, F., McNamara, M., Page, K. J., Schenk, D., Games, D. & Hyman, B. T. (1997) J. Neurosci. 17, 7053-7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irizarry M. C., McNamara, M., Fedorchak, K., Hsiao, K. & Hyman, B. T. (1997) J. Neuropathol. Exp. Neurol. 56, 965-973. [DOI] [PubMed] [Google Scholar]
- 16.Calhoun M. E., Wiederhold, K.-H., Abramowski, D., Phinney, A. L., Probst, A., Sturchler-Pierrat, C., Staufenbiel, M., Sommer, B. & Jucker, M. (1998) Nature 395, 755-756. [DOI] [PubMed] [Google Scholar]
- 17.Takeuchi A., Irizarry, M. C., Duff, K., Saido, T. C., Hsiao Ashe, K., Hasegawa, M., Mann, D. M. A., Hyman, B. T. & Iwatsubo, T. (2000) Am. J. Pathol. 157, 331-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buldyrev S. V., Cruz, L., Gomez-Isla, T., Gomez-Tortosa, E., Le, R., Stanley, H. E., Urbanc, B. & Hyman, B. T. (2000) Proc. Natl. Acad. Sci. USA 97, 5039-5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsiao K., Chapman, P., Nilsen, S., Eckman, C., Harigaya, Y., Younkin, S., Yang, F. & Cole, G. (1996) Science 274, 99-102. [DOI] [PubMed] [Google Scholar]
- 20.Wong T. P., Debeir, T., Duff, K. & Cuello, A. C. (1999) J. Neurosci. 19, 2706-2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaffar S., Counts, S. E., Ma, S. Y., Dadko, E., Gordon, M. N., Morgan, D. & Mufson, E. J. (2000) Exp. Neurol. 170, 227-243. [DOI] [PubMed] [Google Scholar]
- 22.Boncristiano S., Calhoun, M. E., Kelly, P. H., Pfeifer, M., Bondolfi, L., Stadler, M., Phinney, A. L., Abramowski, D., Sturchler-Pierrat, C., Enz, A., et al. (2002) J. Neurosci. 22, 3234-3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hyman B. T., West, H. L., Rebeck, G. W., Buldyrev, S. V., Mantegna, R. N., Ukleja, M., Havlin, S. & Stanley, H. E. (1995) Proc. Natl. Acad. Sci. USA 92, 3586-3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allen M. P. & Tildesley, D. J., (1989) Computer Simulation of Liquids (Oxford Univ. Press, Oxford).
- 25.Yankner B. A., Duffy, L. K. & Kirschner, D. A. (1990) Science 250, 279-282. [DOI] [PubMed] [Google Scholar]
- 26.Pike C. J., Walencewicz, A. J., Glabe, C. G. & Cotman, C. W. (1991) Brain Res. 563, 311-314. [DOI] [PubMed] [Google Scholar]
- 27.Busciglio J., Lorenzo, A. & Yankner, B. A. (1992) Neurobiol. Aging 13, 609-612. [DOI] [PubMed] [Google Scholar]
- 28.Mattson M. P., Tomaselli, K. J. & Rydel, R. E. (1993) Brain Res. 621, 35-49. [DOI] [PubMed] [Google Scholar]
- 29.Pike C. J., Burdick, D., Walencewicz, A. J., Glabe, C. G. & Cotman, C. W. (1993) J. Neurosci. 13, 1676-1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lorenzo A. & Yankner, B. A. (1994) Proc. Natl. Acad. Sci. USA 91, 12243-12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Howlett D. R., Jennings, K. H., Lee, D. C., Clark, M. S. G., Brown, F., Wetzel, R., Wood, S. J., Camilleri, P. & Roberts, G. W. (1995) Neurodegeneration 4, 23-32. [DOI] [PubMed] [Google Scholar]
- 32.Knowles R. B., Gomez-Isla, T. & Hyman, B. T. (1998) J. Neuropathol. Exp. Neurol. 57, 1122-1130. [DOI] [PubMed] [Google Scholar]
- 33.Dickson D. W., Lin, W.-L., Chisholm, L., Corral, A., Jones, G., Yen, S.-H., Sahara, N., Skipper, L., Yager, D., Eckman, C., et al. (2001) Science 293, 1487-1491. [DOI] [PubMed] [Google Scholar]
- 34.Götz J., Chen, F., van Dorpe, J. & Nitsch, R. M. (2001) Science 293, 1491-1495. [DOI] [PubMed] [Google Scholar]
- 35.Yankner B. A. (1996) Neuron 16, 921-932. [DOI] [PubMed] [Google Scholar]
- 36.Knowles R. B., Wyart, C., Buldyrev, S. V., Cruz, L., Urbanc, B., Hasselmo, M. E., Stanley, H. E. & Hyman, B. T. (1999) Proc. Natl. Acad. Sci. USA 96, 5274-5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le R., Cruz, L., Urbanc, B., Knowles, R. B., Hsiao-Ashe, K., Duff, K., Irizarry, M., Stanley, H. E. & Hyman, B. T. (2001) J. Neuropathol. Exp. Neurol. 60, 763-758. [DOI] [PubMed] [Google Scholar]
- 38.Lorenzo A., Yuan, M., Zhang, Z., Paganetti, P. A., Sturchler-Pierrat, C., Staufenbiel, M., Mautino, J., Sol Vigo, F., Sommer, B. & Yankner, B. A. (2000) Nat. Neurosci. 3, 460-464. [DOI] [PubMed] [Google Scholar]
- 39.Matsuoka Y., Malester, B., Picciano, M. & Duff, K. (2001) Neuroscience 104, 609-613. [DOI] [PubMed] [Google Scholar]