

Abstract
Tat is a critical regulatory factor in HIV-1 gene expression. It mediates the transactivation of transcription from the HIV-1 LTR by binding to the transactivation response (TAR) element in a complex with cyclin T1. Because of its critical and early role in HIV gene expression, Tat and its interaction with the TAR element constitute important therapeutic targets for the treatment of HIV-1 infection. Based on the known nucleolar localization properties of Tat, we constructed a chimeric small nucleolar RNA-TAR decoy that localizes to the nucleoli of human cells and colocalizes in the nucleolus with a Tat-enhanced GFP fusion protein. When the chimeric RNA was stably expressed in human T lymphoblastoid CEM cells it potently inhibited HIV-1 replication. These results demonstrate that the nucleolar trafficking of Tat is critical for HIV-1 replication and suggests a role for the nucleolus in HIV-1 viral replication.
The Tat protein is a key regulator of HIV-1 replication (1). Its major function is to transactivate RNA polymerase (pol) II transcription from the viral LTR promoter. The binding of Tat to a transactivation response (TAR) element in the 5′ UTR of the viral RNAs stimulates the processivity of RNA pol II, greatly increasing HIV-1 RNA transcription (2–6). In the absence of Tat the transcription complex is able to initiate transcription from the LTR, but elongates very inefficiently (3). The transactivation activity of Tat is mediated by the interaction with a positive transcription elongation factor, P-TEFb (7–9), composed of CDK9 and cyclin T1 (10–16). The binding of Tat with cyclin T1 increases its affinity for the TAR element and induces the cooperative binding of the P-TEFb complex to TAR (13). CDK 9 can phosphorylate the carboxyl-terminal domain of the largest subunit of RNA pol II, stimulating its processivity (10, 16, 17). Because Tat affects one of the earliest stages of HIV-1 gene expression and may be involved in other critical steps in virus replication (reverse transcription for example, ref. 18), it has been considered a good candidate for therapeutic intervention against HIV-1. Among the different gene therapy strategies that have been tested to block Tat activity in human cells one of the most successful is the use of small RNA molecules that work as “decoys.” These RNAs mimic the specific RNA binding element for Tat, subsequently leading to its titration. TAR RNA or in vitro-evolved Tat binding aptamers have been previously used as decoys for Tat and have resulted in inhibitory effects on HIV-1 replication (19–23).
In addition to its known localization in the nucleoplasm, Tat has been shown to have nucleolar localization properties (24–28). The functional role of Tat nucleolar trafficking is unclear, but it may associate with cyclin T1 in this compartment (29). We have previously used strategies for localizing an anti-HIV ribozyme as well as a Rev binding element in the nucleolus and demonstrated that both of these strategies inhibit HIV-1 replication (30, 31). To test whether Tat nucleolar localization is functionally important, we have used a similar strategy to direct a TAR element into this compartment. We demonstrate here that human T-lymphoblastoid CEM cells stably expressing a nucleolar-localized TAR element are highly resistant to HIV-1 infection. Using in situ hybridization analyses we also show that a Tat-enhanced GFP (EGFP) fusion protein colocalizes with the nucleolar-localized TAR element. The present observations taken together with the nucleolar localization properties of HIV-1 Rev (32–34), as well as some HIV RNAs (30, 35, 36), represent an additional paradigm for the role of the nucleolus in HIV-1 replication. The potent inhibition of HIV-1 replication mediated by the nucleolar-localized TAR decoy also suggests a strategy for genetic therapy of HIV-1 infection.
Materials and Methods
Plasmid Constructs.
The U16TAR DNA was prepared synthetically by PCR (37) using the primers A, B, C, D, E, and F: A, 5′-CTTGCAATGATGTCGTAATTTGCGTCTTACCTCGTTCTC-3′; B, 5′ CCAGATCTGAGCCTGGGAGCTCTCTGGCTGTCAGTAAGCTGGTACAGAA-3′; C, 5′-GCTCCCAGGCTCAGATCTGGCTGTCGCTGAGAACAGAGTAAGACGCAA-3′; D, 5′-TTTCTTGCTCAGTAAGAATTTTCGTCAACCTTCTGTACCAGCTTACTGAC-3′; E, 5′- CCCCCCCCGTCGACCTTGCAATGATGTCGTAATTTG -3′; and F, 5′- CCCCTCTAGAAAAAATTTCTTGCTCAGTAAGAATTT-3′.
The TAR nucleotides are in bold. The PCR product was digested with SalI and XbaI restriction enzymes and subcloned in the corresponding sites of the pTz/U6+1 expression cassette (38), generating the U6+1/U16TAR construct. The BamHI and XbaI sites on the cleaved fragment from U6+1/U16TAR (containing the U6 promoter) were filled in, and the resulting fragment was inserted into the NheI site of the pBabe Puro retroviral vector (U3 region of the 3′ LTR), giving rise to pBabe Puro/U16TAR (see Fig. 3A). We used only those constructs in which the transcriptional unit was in the same orientation as the vector LTR.
Fig 3.

Delivery and intracellular activity of the U16TAR RNA in a pool of transfected CEM cells. (A) Schematic representation of the pBabe Puro retroviral vector. The U6+1/U16TAR cassette was inserted within the U3 region of the 3′ LTR (pBabe Puro/U16TAR). SV40, simian virus 40. (B) Pooled transfected CEM cells expressing the U16TAR RNA. CEM cells were transduced with the pBabe/U16TAR construct, and a pooled population of puromycin-resistant cells was selected. Total RNA was isolated from these cells, electrophoresed in a 6% polyacrylamide-7 M urea gel, and blotted onto a nylon membrane. Hybridization with the U16 snoRNA-specific probe allowed simultaneous detection of the U16TAR RNA and the endogenous U16 snoRNAs. Hybridization to endogenous tRNA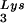 was used as a loading control. Lane 1 contains total RNA extracted from untransduced CEM cells. Lane 2 contains total RNA extracted from CEM cells transduced with pBabe/U16TAR. (C) U16TAR anti-HIV-1 activity. Parental CEM cells or the pooled transductants expressing U16TAR were challenged with HIV-1 NL4–3 at an moi of 0.01. At 7, 14, and 21 days postinfection supernatants were collected from the cell culture and analyzed by HIV reverse transcriptase (RT) assay.
was used as a loading control. Lane 1 contains total RNA extracted from untransduced CEM cells. Lane 2 contains total RNA extracted from CEM cells transduced with pBabe/U16TAR. (C) U16TAR anti-HIV-1 activity. Parental CEM cells or the pooled transductants expressing U16TAR were challenged with HIV-1 NL4–3 at an moi of 0.01. At 7, 14, and 21 days postinfection supernatants were collected from the cell culture and analyzed by HIV reverse transcriptase (RT) assay.
A cDNA from Tat was prepared by PCR using the HIV-1 GST Tat expression vector (National Institutes of Health repository no. 2346) as a substrate for the following primers: Tat A, 5′-AGCTCAAGCTTCGATGGAGCCAGTAGATCCTAGA-3′; and Tat B, 5′-TCCGGTGGATCCCTATTCCTTCGGGCCTGTCGG-3′.
The PCR product was digested with BamHI and HindIII restriction enzymes and inserted in-frame with EGFP in the corresponding sites of the pLEGFP-C1 plasmid (CLONTECH), giving rise to the Tat-EGFP fusion protein clone (pLEGFP-C1/Tat).
Cell Culture.
Human 293 and human CEM cells were maintained in culture as described (30). The PG13 packaging cell line (ATCC CRL-10686) was cultured in DMEM (Irvine Scientific) containing 10% FCS (Irvine Scientific), penicillin (10 units/ml, Irvine Scientific), and streptomycin (100 μg/ml, Irvine Scientific). Transient transfection of 293 cells was carried out by using cells plated at ≈1 × 106 per 100-mm dish 1 day before transfection with 2–10 μg of DNA by using the calcium phosphate DNA precipitation method (GIBCO/BRL, Invitrogen).
In Situ Hybridization.
The hybridizations and microscopic analyses were performed as described (30) with some variations. To reduce the loss of EGFP fluorescence caused by 70% ethanol treatment after fixation, the cells were rinsed in PBS 1× (Irvine Scientific) and then permeabilized with 0.5% Triton X-100 (Sigma-Genosys, The Woodlands, TX) in PBS 1× (Dulbecco's PBS solution) for 10 min at 4°C. Cells were next washed three times in PBS followed by in situ hybridization. The following amino-allyl T-modified oligonucleotides were used as probes: U3, 5′- GT*TCTCTCCCTCT*CACTCCCCAAT*ACGGAGAGAAGAACGAT*CATCAATGGCT*G-3′; and TAR, 5′-GT*GGTTCCCTAGT*TAGCCAGAGAGCT*CCCAGG- CTCAGAT*CTGTTCTAACCT*-3′. The symbol * indicates the allyl-T-modified nucleotides.
The U3 and TAR probes were chemically conjugated to specific fluorophores: either Cy3 (CyTM3 monofunctional reactive dye, Amersham Pharmacia) or Oregon green 488 (Molecular Probes).
Cell Extract Preparation and Immunoselection.
The 293 cells were sonicated in NET2 buffer (200 mM NaCl/40 mM Tris⋅HCl, pH 7.5/0.05% Nonidet P-40) three times for 13 sec each with a 13-sec cooling interval in ice by using a Branson sonicator (model 250/450) with a cup horn. The following settings were used: time on hold, duty cycle on constant, and output control on 3 (A. Ehsani and J.J.R., unpublished results). After sonication, the cell debris was pelleted by centrifugation at 14,000 rpm for 10 min at 4°C, and the supernatant was collected and used for the immunoselection assays. The assays were carried out with 30 μl of an antifibrillarin mAb (72B9, kindly provided by Michael Pollard, The Scripps Research Institute, La Jolla, CA) coupled to 500 μl preswollen Protein A-Sepharose (5 mg/ml, Sigma) in NET2 buffer and incubated either 1 h or overnight with gentle rocking at 4°C. The 293 cell extract was then added to the antibody-coupled Protein A-Sepharose and incubated for 1–2 h with gentle rocking at 4°C. Washing was performed in NET2 buffer, and the samples were digested with Proteinase K (0.5 mg/ml). The recovered RNA was resolved in a 6% polyacrylamide-7 M urea gel.
Packaging Cell Line.
PG13 was used as the packaging cell line for producing the pBabe Puro/U16TAR construct (30).
RNA Preparation and Northern Blot Analyses.
Total RNA was prepared by using the RNA-STAT 60 reagent (Tel-Test, Friendswood, TX) according to the manufacturer's protocol. The RNA was electrophoresed in a 1.2% agarose-6.8% formaldehyde gel or a 6% polyacrylamide-7 M urea gel and blotted onto a nylon membrane.
HIV-1 Titers.
The HIV-1 titers were accomplished by using a modified version of the endpoint dilution method described by Ho et al. (39). A total of 5 × 105 H9 cells were suspended in 0.9 ml RPMI medium with glutamine, penicillin/streptomycin, and 10% FBS (heat-inactivated). A total of 0.1 ml of 10-fold serial dilutions of HIV-1 NL4–3 was added to cultures in quadruplicate. After overnight incubation at 37°C, the cultures were washed three times to remove free virus. On day 4 postinfection, half of the medium was changed, and on day 7, the culture was measured for supernatant reverse transcriptase activity.
HIV-1 Inhibition Assays.
pBabe-Puro-transduced constructs were analyzed for viability and growth kinetics before challenges with HIV-1. The challenges were carried out by using the HIV-1 NL4–3 strain. We used a modification of a standard method described by Ho et al. (40) for virus neutralization. HIV-1 was inoculated into 5 × 105 transduced cells with a multiplicity of infection (moi) of 0.01 with overnight incubation at 37°C, followed by three washings with PBS. Cells were fed twice weekly by removing half of the medium. On days 7 (or 8), 14, and 28 each culture supernatant was assayed for reverse transcriptase activity by using a modification of the assay described by Goldstein et al. (41). This assay is based on the incorporation of 32P-labeled thymidine triphosphate in a reverse transcription reaction using a poly(A) template to initiate transcription. Fifteen microliters of cell-free supernatants was aliquoted into 96-well plates in duplicate, and each sample was mixed with 50 μl 32P-TTP and a mixture containing 50 mM Tris⋅HCl (pH 7.8), 75 mM KCl, 2 mM DTT, 5 mM MgCl2, 5 μg/ml poly(A) template, 6.2 μg/ml oligo(dT) 12–18 primer, 0.05% Nonidet P-40, 0.5 mM EGTA, and 10 μCi/ml 32P. The mixture was incubated under a sealed lid in a humidified chamber at 37°C for 1.5–2 h. Ten microliters from each well was dotted onto CD81 filter paper and air-dried for 30 min. The filter was then washed five times with 1 × SSC (150 mM NaCl, 15 mM Na citrate, pH 7.0) and once with ethanol. Reverse transcriptase-incorporated 32P was quantified by using a PhosphorImager (Molecular Dynamics, Amersham Pharmacia).
Results
The HIV-1 TAR element was localized in the nucleolus by expressing it in the context of the U16 small nucleolar RNA (snoRNA) backbone. U16 is a member of the C/D box snoRNA family that is primarily involved in the 2′ O-methylation of specific rRNA residues (42). We chose the U16 snoRNA because it has been extensively studied (43) and has been successfully used for the nucleolar localization of a ribozyme (30) and a Rev binding element decoy (31, 44). To insert the TAR element in the U16, we replaced the U16 apical loop with a minimal, functional TAR sequence (Fig. 1A Right).
Fig 1.

U16TAR RNA design and intracellular expression. (A) Schematic representation of the U16 snoRNA (Left) and U16TAR RNA (Right). The apical loop of the U16 snoRNA was replaced by a minimal, functional TAR sequence. (B) Intracellular U16TAR expression. The U16TAR sequence was inserted within a U6 snoRNA expression cassette (pTZ/U6+1) generating the transcription unit U6+1/U16TAR (Upper). The U6 expression cassette allows intracellular expression mediated by RNA pol III. Six thymidines were added immediately downstream of the U16TAR sequence to function as an RNA pol III termination signal. The 293 cells were transiently transfected with 10 μg of the U6+1/U16TAR plasmid. After 48 h total RNA was isolated, electrophoresed in a 6% polyacrylamide-7 M urea gel, and blotted onto a nylon membrane (Lower). The U16 snoRNA-specific probe was used to simultaneously detect the U16TAR and the endogenous U16 snoRNAs. An additional probe was used to detect endogenous tRNA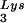 . Lane 1 contains total RNA isolated from untransfected 293 cells. Lane 2 contains total RNA extracted from 293 cells transfected with U6+1/U16TAR. (C) U16TAR stable association with fibrillarin. The 293 cells were transiently transfected with 10 μg of the U6+1/U16TAR plasmid and/or 4 μg of the pLEGFP-C1/Tat plasmid. After 48 h total cell extract was prepared and immunoselected with antifibrillarin antibody. Total RNAs were isolated from the input cell extract and the pellet and electrophoresed in a 6% polyacrylamide-7 M urea gel followed by blotting onto a nylon membrane. A 32P-labeled probe was used to detect U16TAR. A probe complementary to the endogenous U1 snoRNA was used to detect this snoRNA, which is not associated with fibrillarin. A probe complementary to endogenous U3 snoRNA, which is fibrillarin-associated, was used to detect this RNA. Lane 1 contains total RNA extracted from the untransfected 293 cells. Lane 2 contains total RNA extracted from 293 cells transiently transfected with the pLEGFP-C1/Tat plasmid. Lanes 3 and 4 contain total RNAs extracted from 293 cells transfected with U6+1/U16TAR without or with the pLEGFP-C1/Tat plasmid. Lane 5 contains total RNA extracted from the immunoselected pellet of untransfected 293 cells. Lane 6 contains total RNA extracted from the immunoselected pellet of 293 cells transiently transfected with pLEGFP-C1/Tat plasmid. Lanes 7 and 8 contain total RNAs extracted from immunoselected pellets of 293 cells transfected with U6+1/U16TAR without or with the pLEGFP-C1/Tat plasmid.
. Lane 1 contains total RNA isolated from untransfected 293 cells. Lane 2 contains total RNA extracted from 293 cells transfected with U6+1/U16TAR. (C) U16TAR stable association with fibrillarin. The 293 cells were transiently transfected with 10 μg of the U6+1/U16TAR plasmid and/or 4 μg of the pLEGFP-C1/Tat plasmid. After 48 h total cell extract was prepared and immunoselected with antifibrillarin antibody. Total RNAs were isolated from the input cell extract and the pellet and electrophoresed in a 6% polyacrylamide-7 M urea gel followed by blotting onto a nylon membrane. A 32P-labeled probe was used to detect U16TAR. A probe complementary to the endogenous U1 snoRNA was used to detect this snoRNA, which is not associated with fibrillarin. A probe complementary to endogenous U3 snoRNA, which is fibrillarin-associated, was used to detect this RNA. Lane 1 contains total RNA extracted from the untransfected 293 cells. Lane 2 contains total RNA extracted from 293 cells transiently transfected with the pLEGFP-C1/Tat plasmid. Lanes 3 and 4 contain total RNAs extracted from 293 cells transfected with U6+1/U16TAR without or with the pLEGFP-C1/Tat plasmid. Lane 5 contains total RNA extracted from the immunoselected pellet of untransfected 293 cells. Lane 6 contains total RNA extracted from the immunoselected pellet of 293 cells transiently transfected with pLEGFP-C1/Tat plasmid. Lanes 7 and 8 contain total RNAs extracted from immunoselected pellets of 293 cells transfected with U6+1/U16TAR without or with the pLEGFP-C1/Tat plasmid.
Intracellular Expression of U16TAR.
The chimeric U16TAR RNA encoding sequence was inserted downstream of the human U6(+1) snRNA promoter (38). To facilitate RNA pol III transcriptional termination, six thimidines were inserted downstream of the U16TAR gene sequence. The U6+1/U16TAR plasmid was transiently transfected into human 293 cells, and total RNA was isolated 48 h later. Northern blot analyses demonstrated a high level of expression of U16TAR sequence (Fig. 1B). In the experiment shown, precursors or alternative structures are visible, most likely a result of the overexpression of the fusion transcript.
U16TAR Is Associated with Fibrillarin.
The 293 cells were transiently transfected with a Tat-EGFP expression plasmid (pLEGFP-C1/Tat) and/or the U16TAR construct, and total cell extracts were prepared for immunoselection experiments by using the 72B9 mAb raised against fibrillarin. The fibrillarin protein is a component of C/D box snoRNA complexes that are involved in ribosomal RNA processing (42, 45). Total RNA was isolated from the immunoselected fraction and analyzed by Northern blotting. The RNA analyses showed that U16TAR was associated with fibrillarin in both the presence and absence of the Tat-EGFP fusion protein (Fig. 1C). These results demonstrated that the RNA decoy is assembled in a C/D box snoRNA ribonucleoprotein-like complex.
Intracellular Localization of U16TAR.
To further examine the intracellular localization of U16TAR, in situ hybridization was performed by using fluorescent probes specific either for U16TAR RNA (red fluorescence) or endogenous U3 snoRNA (green fluorescence) on transiently transfected 293 cells with the U6+1/U16TAR plasmid. U3 is an abundant snoRNA that serves as a nucleolar control. The overlapping of the two fluorescent signals confirmed the nucleolar localization of the U16TAR RNA (Fig. 2A Lower Left). In some transfected cells, U16TAR speckles that were not colocalized with U3 snoRNA were also observed (white arrow in Fig. 2A Lower Left). This non-nucleolar localization is a consequence of overexpressing the U16TAR RNA in a transient transfection.
Fig 2.

Intracellular localization of U16TAR and colocalization with Tat-EGFP. The 293 cells were grown on coverslips and transiently transfected with 10 μg of the U6+1/U16TAR plasmid and/or 4 μg of pLEGFP-C1/Tat plasmid. After 48 h the cells were fixed in 4% para-formaldehyde dissolved in 1× PBS and in situ hybridizations were carried out. (A) The 293 cells transiently transfected with the U6+1/U16TAR plasmid. Hybridization was performed by using a U16TAR RNA-specific probe conjugated with the CY3 fluorophore (red fluorescence, Upper Left) and a U3 snoRNA-specific probe (for a nucleolar control), conjugated with Oregon green fluorophore (green fluorescence, Upper Right). The yellow signal depicts overlapping of the two hybridization signals and confirms the U16TAR nucleolar localization (Lower Left). The blue staining nuclei (4′-6′-diamino-2-phenyindole, DAPI) are indicated (Lower Right). In some of the 293 cells a fraction of the U16TAR signal (red) does not overlap with the U3 snoRNA (green) signal (white arrow, Lower Left), suggesting a small amount of localization at a site other than the nucleolus when U16TAR is overexpressed (data not shown). (B) The 293 cells transiently transfected with the pLEGFP-C1/Tat plasmid. The Tat EGFP green fluorescence was detected in the nucleus of 293 cells (Left) (confirmed by DAPI staining, Right). (C) The 293 cells transiently transfected with 4 μg of pLEGFP-C1/Tat plasmid. Hybridization was performed by using a U3 snoRNA-specific probe conjugated with the CY3 fluorophore (red fluorescence, Upper Right). Tat EGFP is detectable via the green fluorescence (Upper Left). The yellow signal depicts overlapping signals of the green fluorescence from the Tat EGFP fusion protein and the red fluorescence of the U3 snoRNA hybridization (Lower Left). (D) The 293 cells transiently transfected with pLEGFP-C1/Tat and U6+1/U16TAR plasmids. Hybridization was performed by using a U16TAR-specific probe conjugated with CY3 (red fluorescence, Upper Right). Tat EGFP is detected by the green fluorescence (Upper Left). The yellow signal in the nucleoli indicates an overlap between the red fluorescence of the U16TAR probe and the green fluorescence of the Tat EGFP fusion protein (Lower Left).
Nucleolar Colocalization of the U16TAR RNA Decoy with the Tat-EGFP Protein.
The 293 cells were transiently transfected with the U16TAR expression plasmid (U6+1/U16TAR) and/or with the pLEGFP-C1/TAT plasmid. Forty eight hours posttransfection, in situ hybridization analyses were carried out to detect the U16TAR and/or U3 snoRNAs (Fig. 2 C and D). Tat EGFP localization was analyzed by EGFP fluorescence. This protein showed both nuclear and nucleolar localization (green fluorescence in Fig. 2 B–D, Tat EGFP). The green fluorescence in the nucleolus overlapped with the U3 snoRNA red fluorescence (Fig. 2C Lower Left). When 293 cells were transiently cotransfected with both the U16TAR and the Tat EGFP expression plasmids, nucleolar colocalization of the U16TAR RNA with the Tat EGFP protein was observed (yellow fluorescence in Fig. 2D Lower Left).
U16TAR Anti-HIV Activity.
The U6 expression cassette (U6+1/U16TAR) was inserted within the U3 region (3′ LTR) of the pBabe Puro retroviral vector (Fig. 3A Lower). This construct was used to transduce human CEM T-lymphoblastoid cells followed by selection for puromycin resistance.
Northern blot analyses were performed on total RNA isolated from pools of transduced cells demonstrating high level of expression of the U16TAR RNA (Fig. 3B). The pool of U16TAR-transduced cells was challenged with HIV-1 NL4–3 at an moi of 0.01. HIV-1 replication in these experiments was assayed by monitoring HIV-1-reverse transcriptase at 7, 14, and 21 days postinfection. The results demonstrated that the pooled population of cells expressing the U16TAR chimeric RNA is highly resistant to HIV-1 infection (Fig. 3C). Using limiting dilution of the puromycin-resistant pool of cells, we selected single stable clones. Northern blot analyses were performed on total RNAs isolated from these clones, again demonstrating high U16TAR expression (Fig. 4A).
Fig 4.

Selection of stably transduced clones expressing U16TAR RNA and HIV-1 challenge assays. Individual clones stably expressing the various constructs were isolated by limiting dilution of the pool of transduced CEM cells. Northern blot analyses were performed after electrophoresis of total RNAs in a 6% polyacrylamide-7 M urea gel (A and B) or in a 1.2% agarose-formaldehyde gel (C). (A) Hybridizations were carried out by using a specific U16 snoRNA probe to detect the U16TAR RNAs and the endogenous U16 snoRNA. Probing for endogenous tRNA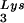 serves as a loading control. Lane 1 contains RNA extracted from untransduced CEM cells. Lanes 2–9 contain total RNAs extracted from clones of transduced CEM clones expressing U16TAR. (B) Hybridizations were carried out under the same condition used for A. Lane 1 contains RNA extracted from untransduced CEM cells. Lanes 2 and 3 contain RNAs extracted from individual CEM clones expressing the U16Rz WT and mutant, respectively (30). Lanes 4 and 5 contain RNAs extracted from individual CEM clones expressing U16TAR (clones 1 and 2). (C) Hybridizations were carried out by using a U16TAR-specific probe and a probe for glyceraldehyde phosphate dehydrogenase (GAPDH) mRNA (loading control). Lane 1 contains RNA extracted from untransduced CEM cells. Lanes 2–9 contain total RNAs extracted from individual CEM clones expressing U16TAR. SV40, simian virus 40. (D) Individual CEM clones expressing the U16TAR RNA are resistant to HIV-1 infection. Cells expressing U16TAR were challenged with HIV-1 NL4–3 at an moi of 0.01. At 8, 14, and 21 days postinfection supernatants were collected from the cultures and analyzed by HIV-1 reverse transcriptase assays. CEM clones expressing a nucleolar-localized hammerhead ribozyme (WT or a nonfunctional mutant) (U16Rz WT and mutant, ref. 30) were used as controls. The data presented represent an average of four independent challenge experiments, and the standard errors for each point are around 5% maximum. (E) Total RNAs extracted from challenged cells 21 days postinfection (see D) were electrophoresed in a 1% agarose-formaldehyde gel, blotted onto a nylon membrane, and hybridized with an HIV-1 probe or a probe for GAPDH mRNA. Lane 1 contains RNA from uninfected CEM cells. Lanes 2 and 3 contain total RNAs extracted from HIV-1-infected CEM cells expressing the U16Rz mutant or U16Rz WT RNAs, respectively. Lanes 4–6 contain RNAs extracted from HIV-1-infected CEM cells expressing U16TAR RNA.
serves as a loading control. Lane 1 contains RNA extracted from untransduced CEM cells. Lanes 2–9 contain total RNAs extracted from clones of transduced CEM clones expressing U16TAR. (B) Hybridizations were carried out under the same condition used for A. Lane 1 contains RNA extracted from untransduced CEM cells. Lanes 2 and 3 contain RNAs extracted from individual CEM clones expressing the U16Rz WT and mutant, respectively (30). Lanes 4 and 5 contain RNAs extracted from individual CEM clones expressing U16TAR (clones 1 and 2). (C) Hybridizations were carried out by using a U16TAR-specific probe and a probe for glyceraldehyde phosphate dehydrogenase (GAPDH) mRNA (loading control). Lane 1 contains RNA extracted from untransduced CEM cells. Lanes 2–9 contain total RNAs extracted from individual CEM clones expressing U16TAR. SV40, simian virus 40. (D) Individual CEM clones expressing the U16TAR RNA are resistant to HIV-1 infection. Cells expressing U16TAR were challenged with HIV-1 NL4–3 at an moi of 0.01. At 8, 14, and 21 days postinfection supernatants were collected from the cultures and analyzed by HIV-1 reverse transcriptase assays. CEM clones expressing a nucleolar-localized hammerhead ribozyme (WT or a nonfunctional mutant) (U16Rz WT and mutant, ref. 30) were used as controls. The data presented represent an average of four independent challenge experiments, and the standard errors for each point are around 5% maximum. (E) Total RNAs extracted from challenged cells 21 days postinfection (see D) were electrophoresed in a 1% agarose-formaldehyde gel, blotted onto a nylon membrane, and hybridized with an HIV-1 probe or a probe for GAPDH mRNA. Lane 1 contains RNA from uninfected CEM cells. Lanes 2 and 3 contain total RNAs extracted from HIV-1-infected CEM cells expressing the U16Rz mutant or U16Rz WT RNAs, respectively. Lanes 4–6 contain RNAs extracted from HIV-1-infected CEM cells expressing U16TAR RNA.
Because, as illustrated in Fig. 3A, the U6 expression cassette containing U16TAR was inserted within the U3 region of the 3′ LTR, the U16TAR RNA could potentially be generated by transcription initiating from the LTR and/or simian virus 40 pol II promoters as well as from the U6 small nuclear RNA pol III promoter. However, Northern blot analyses carried out on RNA isolated from the selected clones and electrophoresed in a 1.2% agarose-formaldehyde gel showed that more than 95% of the U16TAR RNA was of the size predicted from U6 promoter (Fig. 4C), although some processing of the longer pol II transcripts to the smaller size could not be completely ruled out. Several U16TAR-expressing CEM cell lines were challenged with HIV-1 NL4–3 at an moi of 0.01. As controls, we used cells transduced with either the vector harboring an anti-HIV-1 nucleolar ribozyme (U16Rz WT-positive control) or a nonfunctional mutant ribozyme (U16Rz mutant-negative control) (Fig. 4B) (30). After HIV-1 challenge, supernatants were collected at 8, 14, and 21 days postinfection and analyzed for HIV-1 reverse transcriptase. As shown in Fig. 4D, each of the single clones expressing the U16TAR (as well as the clone expressing the U16Rz WT), were highly resistant to HIV-1 infection, whereas the control cells were readily infected. Northern blot analyses using total RNAs isolated from the stable clones infected with HIV-1 (21 days postinfection, Fig. 4D) were carried out. Using a probe complementary to the Rev sequence of HIV-1, HIV-1 RNA was detectable only in the CEM cells expressing the U16Rz mutant RNA (Fig. 4E).
Discussion
We have used a C/D box snoRNA (U16 snoRNA) (43) as an RNA vector to deliver a TAR decoy into the nucleoli of human cells. Based on previous observations that demonstrated Tat has nucleolar localization properties (24–28), our goal was to test whether or not a nucleolar-localized TAR decoy could effectively block viral replication, thereby providing further evidence that the nucleolus plays a critical role in HIV-1 replication (30). We have shown that U16TAR is stably expressed and associated with a basic component of snoRNP complexes, fibrillarin (Fig. 1 B and C). Our results also demonstrate that U16TAR RNA is exclusively localized within the nucleolus (Fig. 2A) and colocalizes with the target Tat-EGFP (Fig. 2D). Human T lymphoblastoid CEM cells stably expressing this chimeric RNA or the U16Rz WT are highly resistant to the HIV-1 infection (Figs. 3C and 4D). Northern blot analyses performed on RNAs isolated from several CEM clonal cell lines infected by HIV-1 (day 21 postinfection, Fig. 4E) showed no HIV-1-specific RNAs from cells expressing U16TAR. Interestingly, our probe detected a small amount of the 2-kb fully spliced HIV-1 RNA only in the clone expressing the U16Rz WT. This result potentially reflects the different modes of action of these two chimeric RNAs. By sequestering Tat, the U16TAR RNA is expected to block all transcript production whereas the ribozyme may be functioning to cleave only singly spliced and unspliced HIV-1 transcripts (30).
To investigate the role of nucleolar localization of U16TAR on its anti-HIV activity, we performed a comparative analysis between the nucleolar U16TAR decoy versus a nucleoplasmic-localizing TAR RNA decoy (see Supporting Materials and Methods, Results, and Discussion and Figs. 5 and 6, which are published as supporting information on the PNAS web site, www.pnas.org). The U16TAR completely blocked HIV-1 replication whereas the nuclear TAR element provided only partial inhibition (Fig. 6). From this analysis, we can conclude that the nucleolar-localized U16TAR decoy is a more potent inhibitor of HIV-1 replication than the nuclear-localized TAR decoy under otherwise identical conditions of transfection and viral replication (see Supporting Materials and Methods, Results, and Discussion, and Fig. 6).
Tat is one of the earliest viral proteins produced during HIV-1 infection and plays a critical role in HIV-1 transcription and replication (1). Recently Wu and Marsh (46) demonstrated that in resting T cells Tat and Nef are selectively transcribed before HIV-1 genome integration. The expression of these proteins may enhance T cell activation and promote infection. Tat accumulates in the nucleolus of human cells (see Fig. 2) and colocalizes with the U16TAR RNA. Titration of Tat in the nucleolus at an early stage of the infection (possibly even before HIV-1 genome integration) could substantially impair HIV-1 replication. Functional nucleolar localization of Tat is supported by the work of Marcello et al. (29) who used fluorescence resonance energy transfer analyses to demonstrate that Tat and cyclin T1 come into close juxtaposition in the nucleolus. Cyclin T1, a component of the P-TEFb complex, plays a crucial role in Tat function (7–9). Cyclin T1 binding to Tat enhances its affinity for the TAR element, and more interestingly, this interaction confers binding specificity of the complex for the loop sequence of TAR (13). Taken together, these results strongly suggest that the nucleolus serves a critical role in the regulation of the Tat activity and consequently in HIV-1 replication. We believe that the potential importance of this role has been underestimated to date.
Recent experiments clearly indicate that the nucleolus is not only the site of rRNA synthesis and ribosome biogenesis, but also the locale for a number of other important processes (47). In addition to ribosomal proteins, viral proteins including HIV-1 Rev (32–34) and Tat (24–28), HTLV-1 Rex (48), the coronavirus nucleoproteins (49), adenovirus protein V (50), herpes simplex virus type 2 UL3 protein (51), and Borna disease virus gp18 BDV (52) have nucleolar localization properties. Therefore many viruses may usurp nucleolar functions for optimizing gene function and replication efficiency.
Supplementary Material
Acknowledgments
We thank Dr. Michael Pollard (The Scripps Research Institute, La Jolla, CA) for the 72B9 mAb raised against fibrillarin and Dr. Daniela Castanotto (Beckman Research Institute of the City of Hope) for the critical reading of the paper. This research was supported by National Institutes of Health Grants AI29329 and AI46030.
Abbreviations
pol, polymerase
TAR, transactivation response
EGFP, enhanced GFP
moi, multiplicity of infection
snoRNA, small nucleolar RNA
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Jeang K. T., Xiao, H. & Rich, E. A. (1999) J. Biol. Chem. 274, 28837-28840. [DOI] [PubMed] [Google Scholar]
- 2.Garber M. E. & Jones, K. A. (1999) Curr. Opin. Immunol. 11, 460-465. [DOI] [PubMed] [Google Scholar]
- 3.Kao S. Y., Calman, A. F., Rich, E. A. & Peterlin, B. M. (1987) Nature 330, 489-493. [DOI] [PubMed] [Google Scholar]
- 4.Laspia M. F., Wendel, P. & Mathews, M. B. (1993) J. Mol. Biol. 232, 732-746. [DOI] [PubMed] [Google Scholar]
- 5.Marciniak R. A. & Sharp, P. A. (1991) EMBO J. 10, 4189-4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toohey M. G. & Jones, K. A. (1989) Genes Dev. 3, 265-282. [DOI] [PubMed] [Google Scholar]
- 7.Marshall N. F., Peng, J., Xie, Z. & Price, D. H. (1996) J. Biol. Chem. 271, 27176-27183. [DOI] [PubMed] [Google Scholar]
- 8.Marshall N. F. & Price, D. H. (1995) J. Biol. Chem. 270, 12335-12338. [DOI] [PubMed] [Google Scholar]
- 9.Price D. H. (2000) Mol. Cell. Biol. 20, 2629-2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mancebo H. S., Lee, G., Flygare, J., Tomassini, J., Luu, P., Zhu, Y., Peng, J., Blau, C., Hazuda, D., Price, D. & Flores, O. (1997) Genes Dev. 11, 2633-2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gold M. O., Yang, X., Herrmann, C. H. & Rice, A. P. (1998) J. Virol. 72, 4448-4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peng J., Marshall, N. F. & Price, D. H. (1998) J. Biol. Chem. 273, 13855-13860. [DOI] [PubMed] [Google Scholar]
- 13.Wei P., Garber, M. E., Fang, S. M., Fischer, W. H. & Jones, K. A. (1998) Cell 92, 451-462. [DOI] [PubMed] [Google Scholar]
- 14.Yang X., Gold, M. O., Tang, D. N., Lewis, D. E., Aguilar-Cordova, E., Rice, A. P. & Herrmann, C. H. (1997) Proc. Natl. Acad. Sci. USA 94, 12331-12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Q., Chen, D., Pierstorff, E. & Luo, K. (1998) EMBO J. 17, 3681-3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu Y., Pe'ery, T., Peng, J., Ramanathan, Y., Marshall, N., Marshall, T., Amendt, B., Mathews, M. B. & Price, D. H. (1997) Genes Dev. 11, 2622-2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dahmus M. E. (1996) J. Biol. Chem. 271, 19009-19012. [DOI] [PubMed] [Google Scholar]
- 18.Harrich D., Ulich, C. & Gaynor, R. B. (1996) J. Virol. 70, 4017-4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sullenger B. A., Gallardo, H. F., Ungers, G. E. & Gilboa, E. (1990) Cell 63, 601-608. [DOI] [PubMed] [Google Scholar]
- 20.Sullenger B. A., Gallardo, H. F., Ungers, G. E. & Gilboa, E. (1991) J. Virol. 65, 6811-6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lisziewicz J., Sun, D., Smythe, J., Lusso, P., Lori, F., Louie, A., Markham, P., Reitz, M. & Gallo, R. C. (1993) Proc. Natl. Acad. Sci. USA 90, 8000-8004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee S. W., Gallardo, H. F., Gaspar, O., Smith, C. & Gilboa, E. (1995) Gene Ther. 2, 377-384. [PubMed] [Google Scholar]
- 23.Browning C. M., Cagnon, L., Good, P. D., Rossi, J., Engelke, D. R. & Markovitz, D. M. (1999) J. Virol. 73, 5191-5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruben S., Perkins, A., Purcell, R., Joung, K., Sia, R., Burghoff, R., Haseltine, W. A. & Rosen, C. A. (1989) J. Virol. 63, 1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siomi H., Shida, H., Maki, M. & Hatanaka, M. (1990) J. Virol. 64, 1803-1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luznik L., Martone, M. E., Kraus, G., Zhang, Y., Xu, Y., Ellisman, M. H. & Wong-Staal, F. (1995) AIDS Res. Hum. Retroviruses 11, 795-804. [DOI] [PubMed] [Google Scholar]
- 27.Li Y. P. (1997) J. Virol. 71, 4098-4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stauber R. H. & Pavlakis, G. N. (1998) Virology 252, 126-136. [DOI] [PubMed] [Google Scholar]
- 29.Marcello A., Cinelli, R. A., Ferrari, A., Signorelli, A., Tyagi, M., Pellegrini, V., Beltram, F. & Giacca, M. (2001) J. Biol. Chem. 276, 39220-39225. [DOI] [PubMed] [Google Scholar]
- 30.Michienzi A., Cagnon, L., Bahner, I. & Rossi, J. J. (2000) Proc. Natl. Acad. Sci. USA 97, 8955-8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michienzi A., Cagnon, L., Bozzoni, I. & Rossi, J. J. (1999) Nucleic Acids Symp. Res. Ser. 41, 211-214. [Google Scholar]
- 32.Cullen B. R., Hauber, J., Campbell, K., Sodroski, J. G., Haseltine, W. A. & Rosen, C. A. (1988) J. Virol. 62, 2498-2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dundr M., Leno, G. H., Hammarskjold, M. L., Rekosh, D., Helga-Maria, C. & Olson, M. O. (1995) J. Cell Sci. 108, 2811-2823. [DOI] [PubMed] [Google Scholar]
- 34.Stauber R., Gaitanaris, G. A. & Pavlakis, G. N. (1995) Virology 213, 439-449. [DOI] [PubMed] [Google Scholar]
- 35.Romanov V. I., Zolotukhin, A. S., Aleksandroff, N. N., Pinto da Silva, P. & Felber, B. K. (1997) Virology 228, 360-370. [DOI] [PubMed] [Google Scholar]
- 36.Canto-Nogues C., Hockley, D., Grief, C., Ranjbar, S., Bootman, J., Almond, N. & Herrera, I. (2001) Micron 32, 579-589. [DOI] [PubMed] [Google Scholar]
- 37.Dillon P. J. & Rosen, C. A. (1990) BioTechniques 9, 298-300. [PubMed] [Google Scholar]
- 38.Good P. D., Krikos, A. J., Li, S. X., Bertrand, E., Lee, N. S., Giver, L., Ellington, A., Zaia, J. A., Rossi, J. J. & Engelke, D. R. (1997) Gene Ther. 4, 45-54. [DOI] [PubMed] [Google Scholar]
- 39.Ho D. D., McKeating, J. A., Li, X. L., Moudgil, T., Daar, E. S., San, N. C. & Robinson, J. E. (1991) J. Virol. 65, 489-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ho D. D., Moudgil, T. & Alam, M. (1989) N. Engl. J. Med. 321, 1621-1625. [DOI] [PubMed] [Google Scholar]
- 41.Goldstein S., Engle, R., Olmsted, R. A., Hirsch, V. M. & Johnson, P. R. (1990) J. Acquired Immune Defic. Syndr. 3, 98-102. [PubMed] [Google Scholar]
- 42.Weinstein L. B. & Steitz, J. A. (1999) Curr. Opin. Cell Biol. 11, 378-384. [DOI] [PubMed] [Google Scholar]
- 43.Fragapane P., Prislei, S., Michienzi, A., Caffarelli, E. & Bozzoni, I. (1993) EMBO J. 12, 2921-2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buonomo S. B., Michienzi, A., De Angelis, F. G. & Bozzoni, I. (1999) RNA 5, 993-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Venema J. & Tollervey, D. (1999) Annu. Rev. Genet. 33, 261-311. [DOI] [PubMed] [Google Scholar]
- 46.Wu Y. & Marsh, J. W. (2001) Science 293, 1503-1506. [DOI] [PubMed] [Google Scholar]
- 47.Pederson T. (1998) Nucleic Acids Res. 26, 3871-3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Siomi H., Shida, H., Nam, S. H., Nosaka, T., Maki, M. & Hatanaka, M. (1988) Cell 55, 197-209. [DOI] [PubMed] [Google Scholar]
- 49.Wurm T., Chen, H., Hodgson, T., Britton, P., Brooks, G. & Hiscox, J. A. (2001) J. Virol. 75, 9345-9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matthews D. A. & Russell, W. C. (1998) J. Gen. Virol. 79, 1671-1675. [DOI] [PubMed] [Google Scholar]
- 51.Yamada H. Y., Jiang, M., Zhu, H. Y., Inagaki-Ohara, K. & Nishiyama, Y. (1999) J. Gen. Virol. 80, 2157-2164. [DOI] [PubMed] [Google Scholar]
- 52.Pyper J. M., Clements, J. E. & Zink, M. C. (1998) J. Virol. 72, 7697-7702. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.