

Abstract
We report the selective and real-time detection of label-free DNA using an electronic readout. Microfabricated silicon field-effect sensors were used to directly monitor the increase in surface charge when DNA hybridizes on the sensor surface. The electrostatic immobilization of probe DNA on a positively charged poly-l-lysine layer allows hybridization at low ionic strength where field-effect sensing is most sensitive. Nanomolar DNA concentrations can be detected within minutes, and a single base mismatch within 12-mer oligonucleotides can be distinguished by using a differential detection technique with two sensors in parallel. The sensors were fabricated by standard silicon microtechnology and show promise for future electronic DNA arrays and rapid characterization of nucleic acid samples. This approach demonstrates the most direct and simple translation of genetic information to microelectronics.
A wide range of techniques for detecting nucleic acids is based on their hybridization to DNA probes on a solid surface (ref. 1, entire issue, and ref. 2). In the methods used most routinely, the physical nature of the readout requires the attachment of reporter molecules such as fluorescent, chemiluminescent, redox, or radioactive labels (1, 3, 4). Although label-dependent methods achieve the highest sensitivities (5–7), eliminating the labeling steps has the advantage of simplifying the readout and increasing the speed and ease of nucleic acid assays, which is especially desirable for characterizing infectious agents, scoring sequence polymorphisms and genotypes, and measuring mRNA levels during expression profiling. The development of label-independent methods that can monitor hybridization in real time and that are simple and scalable is still in its infancy (8–11). Here we describe a label-free method for electronically detecting DNA by its intrinsic molecular charge using microfabricated field-effect sensors.
The field-effect sensor is based on an electrolyte-insulator-silicon (EIS) structure. Variations in the insulator-electrolyte surface potential, which arise from the binding of charged molecules (e.g., nucleic acids) to the insulator surface (Fig. 1 a and b), modify the charge distribution in the silicon below the electrolyte. Surface charge and surface potential at this interface are related according to the Grahame equation (12). The surface potential can be measured by changes in conductivity (13) or capacitance (14) in the silicon part of the EIS structure. We have chosen to measure the capacitance, because it requires only one electrical connection to the silicon. The measured capacitance between the silicon and counter electrode in the electrolyte solution is dominated by the insulator capacitance and the capacitance of the charge-depleted region in the silicon. These two capacitances appear in series, and only the silicon depletion capacitance is modulated by the insulator surface potential. The capacitance-versus-voltage dependence of EIS structures (15) is similar to that of metal-oxide-semiconductor (MOS) structures (16).
Fig 1.

Sensor schematics. (a and b) EIS interface of a n-type field-effect sensor. DNA exhibits one intrinsic negative charge per base at its sugar-phosphate backbone. Probe DNA is bound electrostatically to a layer of PLL (gray) on the surface. (a) Binding of negatively charged target DNA (green) to its complementary probe DNA (red) at the sensor surface (yellow) extends the depletion region (black arrow) in the silicon portion of the sensor compared with b, where no binding occurs to noncomplementary probe DNA (blue). (c) Optical micrograph of a device consisting of field-effect sensors at the terminus of two cantilevers. The cantilevers are 500 μm long, 75 μm wide, and 3 μm thick. (d) Cross section of a cantilever field-effect sensor. The sensing area at the terminus of the cantilever is connected electrically to a metal contact on the substrate by a layer of highly doped silicon inside the cantilever.
In biology, field-effect sensors have been used primarily to monitor enzymatic reactions (17) and cell metabolism (18), and to record electrical signals from muscle cells and neurons (19, 20). Although promising approaches to the detection of DNA with field-effect devices have been reported (21, 22), thus far they have not achieved the speed, sensitivity, and selectivity necessary for genetic analysis (4). We demonstrate that micrometer-scale field-effect sensors can detect and distinguish label-free 12-mer oligonucleotides with a single base mismatch within minutes. The field-effect readout is enabled by operating the sensor at low ionic strength where field-effect detection is most sensitive. Rapid hybridization at this ionic strength is enabled by a positively charged surface that compensates for electrostatic repulsion between complementary DNA strands and accelerates hybridization. The single base mismatch selectivity is enabled by a differential configuration that corrects for background signals.
Materials and Methods
Field-Effect Sensing System.
The field-effect sensors used in this work are EIS capacitors microfabricated at the termini of silicon cantilevers according to a process described in detail elsewhere (ref. 23; Fig. 1c). The cantilever design enables functionalization of individual sensors in distinct reservoirs. Each sensor has a 50 × 50-μm2 sensing region doped with ≈1015 atoms per cm3 and is covered with an ≈2-nm layer of chemically grown oxide. Both the low doping level and extremely thin insulator thickness enhance the surface potential sensitivity of the sensors (16). A highly doped silicon layer inside the cantilever (1018 atoms per cm3) electrically connects the sensing region at the cantilever terminus to an aluminum contact on the substrate (Fig. 1d). This layer is covered with 1.1 μm of thermally diffused silicon dioxide. With the exception of the ion implant step, all fabrication was performed at the Massachusetts Institute of Technology Microsystems Technology Laboratories.
A device with two sensors in parallel is mounted in a fluid cell such that the cantilevers are inside the cell and the metal contacts are isolated from the fluids. Test solutions of ≈500-μl volume are injected with a pipette. An Ag/AgCl electrode is used to establish a voltage bias in solution and to deliver a 1-kHz, 150-mVpp ac voltage. The high-impedance EIS structure prevents faradaic processes on the sensor surface that could deteriorate the sensors or analytes. Sensors are operated at a bias voltage where their response is linear and most sensitive to surface potential changes, i.e., at the inflection point of their capacitance-versus-voltage curves (23). The resulting periodic charging current on the sensors is amplified and measured by using a lock-in amplifier with a 100-ms time constant. An offset is added to the lock-in output to zero the signal. Hence all figures show relative surface potential. The amplitude of the charging current measured between the electrode and silicon is typically 25 nA, which corresponds to a total capacitance of the EIS structure of ≈20 pF. Current and capacitance signals scale linearly with sensor area. The adsorption of negative charges on the surface of an n-doped sensor increases the depth of its depletion region, leading to a decrease in depletion capacitance and a decrease in charging current or measured surface potential. Before each experiment, a 10-mV step is applied to the bias voltage to calibrate the surface potential response of each sensor. Data are sampled at 5 Hz.
Sensor Surface Functionalization.
After cleaning with piranha solution (1:1 30% H2O2 in H2O:H2SO4), etching in hydrofluoric acid for 10 s (buffered oxide etch 7:1), and chemically growing oxide for 1 min in piranha solution, the sensors were equilibrated in buffer (5 mM sodium phosphate, pH 7.0/10 mM NaCl), functionalized with 0.2 mg/ml poly-L-lysine (PLL, MW 16,000–22,100, Sigma) inside the fluid cell for 15–60 min and then incubated individually with 15 μl of 4–40 μM probe oligonucleotides for 15–60 min before being placed back in the fluid cell. Probe 12-mer oligonucleotide sequences were A = CTATGTCAGCAC, Am = CTATGTAAGCAC, B = AGGTCTAGTGCA, and C = CCTCTTGGAGAA, and their corresponding complementary target DNA sequences were cA, cAm, and cB (HPLC-purified, Synthegen, Houston). Hybridization was carried out at room temperature.
Ellipsometry and Radiolabeling Experiments.
Experiments were done on 1-cm2 pieces of silicon that were prepared identically to the sensor surfaces. PLL was used at 0.2 mg/ml, probe oligonucleotides at 4 μM, and target oligonucleotides at 80 nM. The incubation time was always 15 min followed by 1 min of equilibration in buffer. For radiolabeling experiments, oligonucleotides were end-labeled with [γ-32P]ATP (Perkin–Elmer Life Sciences) by using T4 polynucleotide kinase (New England Biolabs). Measurements were done with a PhosphorImager (Molecular Dynamics). Ellipsometry measurements were done in air with a discrete wavelength ellipsometer (Sentech, Berlin).
Results and Discussion
To demonstrate that the field-effect sensors are sensitive specifically to the charge of adsorbed molecular layers as opposed to their thickness (24), we monitored the growth of polyelectrolyte multilayers consisting of positively charged PLL and negatively charged oligonucleotides on the sensor surface (Fig. 2). Such layers bind to each other primarily by electrostatic interactions and are known to overcompensate for the surface charge of the previously adsorbed layer, which leads to a linear growth in multilayer thickness as positively and negatively charged molecules are successively applied to the surface (25). Using ellipsometry, we determined the incremental thickness increase of PLL-oligonucleotide multilayers to be ≈0.4 nm per layer (Fig. 2a). In contrast, the field-effect sensor showed an alternating response, over five successive cycles of PLL and DNA adsorption, with an increase of ≈16 mV after the addition of PLL and a subsequent decrease of ≈14 mV after the addition of oligonucleotides (Fig. 2b). Thus, our device measures net charge rather than layer thickness.
Fig 2.

PLL/oligonucleotide multilayer growth. (a) Thickness of PLL/oligonucleotide multilayer from ellipsometry measurements on silicon substrates that were prepared identically to the field-effect sensor surfaces. The thickness increases linearly. (b) Surface potential signal of multilayer growth in solution measured with a field-effect sensor. The signal alternates according to the charge of the adsorbed layer (positive for PLL and negative for oligonucleotides). The same PLL and oligonucleotide concentrations as described for a were used. Each solution was injected twice and followed by an injection of buffer before the next layer was adsorbed. Blue arrows indicate PLL injections, and red arrows indicate oligonucleotide injections.
To explore the utility of our field-effect sensor for detecting DNA in solution, two sensors were first functionalized with a PLL layer. Next, the sensing area of one sensor was functionalized with the 12-mer oligonucleotide A (sensor 1), and the adjacent sensor was functionalized with the unrelated 12-mer oligonucleotide B (sensor 2). The sensors were then mounted in a fluid cell. Solutions containing various target DNA oligonucleotides were injected in succession, and the surface potential of the sensors was measured. The addition of control solutions such as buffer or oligonucleotide B generated similar signals from both sensors (Fig. 3a). These signals arose because the surface potential is sensitive to thermal fluctuations, drifts, nonspecific binding, and changes in electrolyte composition. However, because these unwanted signals are similar for both sensors, they can be eliminated by taking the differential response from the two sensors (sensor 1-sensor 2). When oligonucleotide cA, complementary to A, was injected, the surface potentials of sensor 1 and sensor 2 diverged. The sensors showed a differential response of −3 mV for oligonucleotide cA and of +3 mV for the subsequent addition of cB (Fig. 3b). These observations demonstrate that a differential field-effect sensor configuration is able to measure the sequence-specific formation of A–cA and B–cB hybrids.
Fig 3.

Field-effect detection of DNA hybridization. (a) Surface potential response from sensor 1 (blue) functionalized with probe oligonucleotide A and sensor 2 (red) functionalized with probe oligonucleotide B during a hybridization experiment. Downward arrows indicate injections of oligonucleotides, and upward arrows indicate injections of buffer into the fluid cell. (b) Differential signal obtained by subtracting the two sensor signals shown in a (sensor 1-sensor 2). The order of injections was: buffer, B (80 nM), buffer, cA (80 nM), cA (200 nM), buffer, cB (80 nM), cB (200 nM), and buffer. The second injection of either cA or cB did not result in a change in hybridization signal, indicating that saturation was reached.
We observed that sensors can be reused more than 10 times by regenerating their surface with a piranha-hydrofluoric acid-piranha cleaning. After a new round of functionalization with PLL and oligonucleotides, the difference in signal strengths between the original and regenerated surfaces was 10–20% for hybridizing oligonucleotides at a concentration of 80 nM.
The ionic strength of the buffer used in our experiments (23 mM) is much lower than that commonly used for DNA hybridization (e.g., 825 mM for a 5× saline sodium citrate buffer). We chose such a low ionic strength because field-effect detection is most sensitive when counterion screening of the charged molecules is minimized (12, 26). However, at low ionic strength, the electrostatic repulsion between two complementary DNA strands strongly reduces their probability of hybridization and extends the time required for annealing (27, 28). Still, the data shown in Fig. 3 indicate that saturation is reached within a few minutes of the addition of target oligonucleotides to the fluid cell, which is unusually rapid at this ionic strength. The positively charged PLL layer seems to compensate the negative charge on the probe DNA and reduce the electrostatic repulsion between target and probe DNA. In addition, a positively charged surface may increase the local concentration of target oligonucleotides near the surface (29). The method of hybridizing nucleic acids on a charge-compensated surface at low ionic strength has been investigated recently for use in rapid microarray hybridization (29). It might also be used for other field-effect sensing devices such as silicon nanowires (30), which could lead to a dramatic size reduction of nucleic acid sensors.
We used autoradiography to verify selective hybridization with radiolabeled A, cA, and cB oligonucleotides. We found that the surface coverage after functionalization was approximately 1 probe oligonucleotide per 2 nm2. Specific target oligonucleotides hybridized with an efficiency of 5% at 80 nM, whereas binding by noncomplementary oligonucleotides was less than 0.6%. These experiments with radiolabeled DNA showed that a change in surface potential of ≈3 mV corresponds to the binding of ≈3 × 104 12-mer oligonucleotides per μm2 or 15 ng of DNA per cm2. The expected change in surface potential Ψ0 for a given change in surface charge density σ0 can be calculated by using the Grahame equation (12),
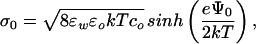 |
where k is the Boltzmann constant, T is absolute temperature, e is elementary charge, ɛo is the permittivity of free space, ɛw is the dielectric constant of water, and co is the buffer ionic strength. Starting with a surface charge density of silicon dioxide of 0.8 C/m2 (31), we calculated that a change of the surface charge density from the 3 × 104 hybridized 12-mer oligonucleotides per μm2 leads to a change in surface potential of ≈3 mV, which is in good agreement with the observed value. Here we assumed an upper value of 12 charges per oligonucleotide, although their effective charge may be less than that, which would reduce the calculated change in surface potential. The surface charge density of 0.8 C/m2 is an upper limit for silicon dioxide, and using a lower effective surface charge density would increase the calculated change in surface potential.
Fig. 4 shows the dependence of the hybridization signal on target oligonucleotide concentration and demonstrates that a 2 nM concentration of 12-mer oligonucleotides or 8 ng/ml DNA can be detected easily. Although our detection method is less sensitive than state-of-the-art label-dependent methods (5–7) or typical microarray applications that have sensitivities in the tens of picomolar range, our detection limit of 2 nM is one of the lowest values reported thus far for label-independent methods (8–11). Because the sensor response depends on the change in surface charge during hybridization, the higher charge associated with longer DNA molecules will create a stronger signal per molecule. Whether this will improve the detection limit remains unclear, because the total number of duplexes created during hybridization will also depend on the surface coverage of longer probe oligonucleotides as well as the affinity and unspecific binding of longer target oligonucleotides.
Fig 4.

Concentration dependence and detection limit. Shown is the differential surface potential response for the hybridization of target oligonucleotides at concentrations of 2, 5, 20, and 80 nM.
To demonstrate the specificity of our sensors, we investigated two cases where unspecific binding of oligonucleotides to the sensor surface can affect sensor response. First we successfully detected a specific oligonucleotide within a high concentration of other unrelated oligonucleotides. Fig. 5 shows the binding of 20 nM cA and cB to A and B, respectively, within a 10-times-higher concentration (200 nM) of unrelated sequences. Comparing this signal with a signal from a pure oligonucleotide (Fig. 4) shows that the high unspecific background reduces the hybridization signal by a factor of 3. Although conventional microarrays are operated at an even higher background-to-target ratio (1), we anticipate that further improvement of surface functionalization and hybridization conditions will improve the specificity as well as the sensitivity of our sensor (32). Second, Fig. 6 shows that our field-effect device can identify a single base mismatch in 12-mer oligonucleotides, i.e., it can differentiate between a specific sequence and a sequence that differs only by one base. This is particularly important, because a potential application of DNA sensors is to detect DNA point mutations associated with disease. A pair of sensors was functionalized with oligonucleotides A and Am, a sequence that differs from A at a single base. When cA was injected, the differential signal decreased, showing specific hybridization to A. When cAm was injected, the differential signal increased, showing hybridization to Am. The smaller differential signal from binding of cAm to Am compared with that of cA to A could result from a difference in surface functionalization of the two sensors with probe oligonucleotides. However, this trend was subsequently verified by radiolabeling experiments, suggesting that the difference could also result from the several degrees lower melting temperature of the Am–cAm duplex relative to the A–cA duplex. The smaller differential signal from cA in Fig. 6b compared with that from cA in Fig. 3b suggests that cA also binds unspecifically, to some extent, to Am, and this reduces the differential signal. Nevertheless, the data in Fig. 6 demonstrate that our device can distinguish between complementary and mismatched DNA sequences.
Fig 5.

Field-effect detection within a complex sample. Absolute surface potential (a) and differential surface potential (b) for the hybridization of 20 nM oligonucleotides cA to A and cB to B within a 10-times-higher concentration of unrelated oligonucleotides. All injections were made at a constant 200 nM concentration. Arrows indicate injections of 200 nM C, then 20 nM cA + 180 nM C, and then 20 nM cB + 20 nM cA + 160 nM C.
Fig 6.

Field-effect detection of a single base mismatch. Surface potentials from two sensors that were functionalized with probe oligonucleotides A and Am, which differ only in a single base. Control solutions with noncomplementary target oligonucleotide cB show no differential signal, whereas injection of 80 nM of complementary sequences cA and cAm both show a distinct hybridization signal. (a) Absolute surface potential. (b) Differential surface potential.
Conclusions
In this report we have introduced an electronic method for the direct detection of unlabeled nucleic acids. We have presented its operating principle and investigated its concentration sensitivity and its specificity. We focused on real-time and label-free DNA detection with sensors manufacturable by conventional, high-yield fabrication processes that can produce hundreds of sensors in parallel. In doing so, we target applications where rapid, parallel DNA analysis is needed, e.g., for characterizing pathogens, measuring mRNA levels during expression profiling, or point-of-care applications. To approach these goals, we anticipate further research toward improvement of concentration sensitivity and of specificity through optimization of experimental conditions, improved surface functionalization, and integration in small-volume fluidic handling systems. The active area of our sensor can be further miniaturized to approximately 1 μm2 by using standard photolithography. The geometry and sensitivity of the sensor also implies the potential for novel application to real-time, single-cell measurements. Given that an individual gene can be expressed at concentrations greater than 1 nM inside a mammalian cell, a sensor at the terminus of a silicon cantilever, similar to that shown in Fig. 1, might be able to detect gene expression from a single cell.
Acknowledgments
Devices were fabricated in the Massachusetts Institute of Technology Microsystems Technology Laboratories. We are thankful to M. Trulson for a critical reading of the manuscript. This work was supported by the Defense Advanced Research Projects Agency, the Air Force Office of Scientific Research, and the Media Laboratory's Things That Think consortium. E.B.C. acknowledges support from a National Science Foundation Graduate Research fellowship.
Abbreviations
EIS, electrolyte-insulator-silicon
PLL, poly-l-lysine
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Lander E. S. (1999) Nat. Genet. 21, Suppl. 1, 3-4. [DOI] [PubMed] [Google Scholar]
- 2.Wang J. (2000) Nucleic Acids Res. 28, 3011-3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kricka L. J. (1999) Clin. Chem. 45, 453-458. [PubMed] [Google Scholar]
- 4.Wang J. (1999) Chem. Eur. J. 5, 1681-1685. [Google Scholar]
- 5.Castro A. & Williams, J. G. K. (1997) Anal. Chem. 69, 3915-3920. [DOI] [PubMed] [Google Scholar]
- 6.Budach W., Abel, A. P., Bruno, A. E. & Neuschäfer, D. (1999) Anal. Chem. 71, 3347-3355. [Google Scholar]
- 7.Taton T. A., Mirkin, C. A. & Letsinger, R. L. (2000) Science 289, 1757-1760. [DOI] [PubMed] [Google Scholar]
- 8.Nelson B. P., Grimsrud, T. E., Liles, M. R., Goodman, R. M. & Corn, R. M. (2001) Anal. Chem. 73, 1-7. [DOI] [PubMed] [Google Scholar]
- 9.Okahata Y., Kawase, M., Niikura, K., Ohtake, F., Furusawa, H. & Ebara, Y. (1998) Anal. Chem. 70, 1288-1296. [DOI] [PubMed] [Google Scholar]
- 10.Fritz J., Baller, M. K., Lang, H. P., Rothuizen, H., Vettiger, P., Meyer, E., Güntherodt, H.-J., Gerber, C. & Gimzewski, J. K. (2000) Science 288, 316-318. [DOI] [PubMed] [Google Scholar]
- 11.Howorka S., Cheley, S. & Bayley, H. (2001) Nat. Biotechnol. 19, 636-639. [DOI] [PubMed] [Google Scholar]
- 12.Israelachvili J., (1992) Intermolecular and Surface Forces (Academic, London).
- 13.Bergveld P. (1972) IEEE Trans. Biomed. Eng. 19, 342-351. [DOI] [PubMed] [Google Scholar]
- 14.Bousse L. & Bergveld, P. (1983) J. Electroanal. Chem. 152, 25-39. [Google Scholar]
- 15.Bousse L. (1982) J. Phys. Chem. 76, 5128-5133. [Google Scholar]
- 16.Nicollian E. & Brews, J., (1982) MOS (Metal Oxide Semiconductor) Physics and Technology (Wiley, New York).
- 17.Hall E. A. H., (1991) Biosensors (Prentice Hall, Englewood Cliffs, NJ).
- 18.Hafeman D. G., Parce, J. W. & McConnell, H. M. (1988) Science 240, 1182-1185. [DOI] [PubMed] [Google Scholar]
- 19.Offenhäusser A. & Knoll, W. (2001) Trends Biotechnol. 19, 62-66. [DOI] [PubMed] [Google Scholar]
- 20.Zeck G. & Fromherz, P. (2001) Proc. Natl. Acad. Sci. USA 98, 10457-10462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Souteyrand E., Cloarec, J. P., Martin, J. R., Wilson, C., Lawrence, I., Mikkelsen, S. & Lawrence, M. F. (1997) J. Phys. Chem. B 101, 2980-2985. [Google Scholar]
- 22.Berney H., West, J., Haefele, E., Alderman, J., Lane, W. & Collins, J. K. (2000) Sens. Actuators B Chem. 68, 100-108. [Google Scholar]
- 23.Cooper E. B., Fritz, J., Wiegand, G., Wagner, P. & Manalis, S. R. (2001) Appl. Phys. Lett. 79, 3875-3877. [Google Scholar]
- 24.Berggren C., Bjarnason, B. & Johansson, G. (2001) Electroanalysis (N.Y.) 13, 173-180. [Google Scholar]
- 25.Decher G. (1997) Science 277, 1232-1237. [Google Scholar]
- 26.Bergveld P. (1996) Sens. Actuators A Phys. 56, 65-73. [Google Scholar]
- 27.Wetmur J. G. & Davidson, N. (1968) J. Mol. Biol. 31, 349-370. [DOI] [PubMed] [Google Scholar]
- 28.Williams A. P., Longfellow, C. E., Freier, S. M., Kierzek, R. & Turner, D. H. (1989) Biochemistry 28, 4283-4291. [DOI] [PubMed] [Google Scholar]
- 29.Belosludtsev Y., Belosludtsev, I., Iverson, B., Lemeshko, S., Wiese, R., Hogan, M. & Powdrill, T. (2001) Biochem. Biophys. Res. Commun. 282, 1263-1267. [DOI] [PubMed] [Google Scholar]
- 30.Cui Y., Wei, Q., Park, H. & Lieber, C. M. (2001) Science 293, 1289-1292. [DOI] [PubMed] [Google Scholar]
- 31.Dong Y., Pappu, S. V. & Xu, Z. (1998) Anal. Chem. 70, 4730-4735. [Google Scholar]
- 32.Andersen M. L. M., (1999) Nucleic Acid Hybridization (Springer, New York).