

Abstract
Viruses are the most common biological entities in the oceans by an order of magnitude. However, very little is known about their diversity. Here we report a genomic analysis of two uncultured marine viral communities. Over 65% of the sequences were not significantly similar to previously reported sequences, suggesting that much of the diversity is previously uncharacterized. The most common significant hits among the known sequences were to viruses. The viral hits included sequences from all of the major families of dsDNA tailed phages, as well as some algal viruses. Several independent mathematical models based on the observed number of contigs predicted that the most abundant viral genome comprised 2–3% of the total population in both communities, which was estimated to contain between 374 and 7,114 viral types. Overall, diversity of the viral communities was extremely high. The results also showed that it would be possible to sequence the entire genome of an uncultured marine viral community.
Marine viruses, the majority of which are phages, have enormous influences on global biogeochemical cycles (1), microbial diversity (2, 3), and genetic exchange (4). Despite their importance, virtually nothing is known about marine viral biodiversity or the evolutionary relationships of marine and nonmarine viruses (5–7). Addressing these issues is difficult because viruses must be cultured on hosts, the majority of which cannot be cultivated by using standard techniques (8). In addition, viruses do not have ubiquitously conserved genetic elements such as rDNA that can be used as diversity and evolutionary distance markers (9). To circumvent these limitations, we developed a method to shotgun clone and sequence uncultured aquatic viral communities.
Materials and Methods
Isolation of Viral Community DNA.
Marine viruses were isolated from 200 liters of surface seawater from Scripps Pier (SP, La Jolla, CA; May 2001) and the channel side of Fiesta Island in Mission Bay (MB, San Diego; June 2001) by using a combination of differential filtration and density-dependent gradient centrifugation. The water at the MB site is exchanged with each tidal cycle. Both the SP and MB sites experience increased levels of pollution during the rainy season, because of runoff from the surrounding city. The MB site routinely has more eukaryotic algae than does the SP site. Once collected, the water samples were initially filtered through a 0.16-μm Centramate tangential flow filter (TFF; Pall) to remove bacteria, eukaryotes, and large particles. Approximately 90% of the viruses, as determined by epifluorescent microscopy (10), and most of the water, passed through the filter and were collected in a separate tank. Subsequently, the viruses in the filtrate were concentrated by using a 100-kDa TFF filter until the final sample volume was <100 ml (≈5,000× concentration). Recovery of viruses during this step was essentially 100%. After the TFF, the phage concentrate was loaded onto a cesium chloride (CsCl) step gradient, ultracentrifuged, and the 1.35–1.5 g/ml fraction was collected. This fraction contains the majority of the viral DNA as determined by pulse field gel electrophoresis (11); however, this method will not recover all viruses (e.g., large eukaryotic viruses and ssRNA phages). After CsCl purification, the viruses were lysed by using a formamide extraction, and the DNA was recovered by an isopropanol precipitation and a cetyltrimethylammonium bromide (CTAB) extraction (12).
Construction of the Shotgun Library.
The amount of viral DNA in an environmental sample is very low (≈10 μg/100 liters). Viral genomes often contain modified nucleotides that cannot be directly cloned into Escherichia coli. Additionally, because viral genomes contain genes (e.g., holins, lysozyme) that must be disrupted before cloning, we have not been able to create a representative cosmid library from these communities. We have circumvented these problems by randomly shearing the total marine viral community DNA (HydroShear, GenMachine, San Carlos, CA), end-repairing, ligating dsDNA linkers to the ends, and amplifying the fragments by using the high-fidelity Vent DNA polymerase. The resulting fragments were ligated into the pSMART vector and electroporated into MC12 cells (Lucigen, Middleton, WI). We call these libraries LASLs for linker-amplified shotgun libraries. This method has been checked to ensure randomness as described (ref. 13, and our web site at www.sci.sdsu.edu/PHAGE/LASL/index.htm). A test library was constructed of coliphage λ DNA, and 100 fragments were sequenced without observing any chimeras. Additionally, we have recently sequenced two phage genomes, Vibriophage 16T and 16C, from a mixed lysate by using the LASL approach. No chimeric molecules were observed in this mixed library. Together, these three libraries represent >1,000 sequences. Therefore, it is highly unlikely that we are observing a significant number of chimeric sequences in our library (F.R. and A.M.S., unpublished results).
Analysis of Sequences: Composition Analyses.
Clones from the SP library were sequenced with both forward and reverse primers, yielding a total of 1,061 sequences. Eight hundred and seventy-three clones from the MB library were sequenced only with the forward primer. These sequences were compared against GenBank by using TBLASTX (14, 15). A hit was consider significant if it had an E value of <0.001. Significant hits to GenBank entries were classified into the groups described in the text, based on sequence annotation. In cases where multiple significant hits were observed for a single query sequence, the sequence was preferentially classified as a phage or virus if these occurred within the top five hits. Mobile elements consisted of transposons, plasmids, insertion sequences, retrotransposons, unstable genetic elements, and pathogenicity islands. Bacterial hits were examined manually according to a list provided by Sherwood Casjens (University of Utah, Salt Lake City) to look for potential hits to prophage sequences. Significant hits to phages were further classified into phage families according to The International Committee on Taxonomy of Viruses (ICTV) classification (16), with the one exception being that we considered P22 a Siphovirus.
Analysis of Sequences: Contig Analyses.
All sequences were initially analyzed by SEQUENCHER 3.0.6 (Gene Codes, Ann Arbor, MI) to identify contigs based on a minimum overlap of 20 bp with a 98% minimal mismatch percentage (MM%). Contigs were examined individually to ensure that any overlaps between fragments from the same clone were not considered contigs. Only contigs containing an overlapping sequence from two different clones were considered in the population analyses. To empirically determine the overlap size and MM% needed to differentiate between phage genotypes and groups during assembly, an in silico shotgun library was constructed and tested. The genomes used in this analysis were three Siphoviruses (coliphage λ, coliphage HK620, and coliphage N15), three Myoviruses (coliphage T4, coliphage P2, and coliphage Mu), four Podoviruses (coliphage T7, Yersinia phage YeO3–12, Roseophage SIO1, and Cyanophage P60), and one Corticovirus (Pseudoalteromonas phage PM2). This selection includes all of the marine phage in GenBank (P60, SIO1, and PM2), multiple phage that are very closely related (i.e., the λ-like Siphoviruses, or the T7-like Podoviruses), representatives from each of the major dsDNA phage groups, as well as many genomes that infect the same host E. coli (λ, HK620, N15, T4, P2, Mu, and T7). To construct the library for assembly, the 11 genome sequences were divided into 500-bp fragments and then reassembled by using different overlap sizes and MM%. Contigs that formed between fragments of different genomes were recorded.
At very low stringency (MM% = 80 with a 20-bp overlap), 25 contigs among phages from within the same group (Sipho-, Myo-, and Podovirus) were observed, as well as one contig between the Siphovirus coliphage λ, and the Myoviruses Mu and P2. Modestly raising the stringency to values typically used to assemble genomes (MM% = 85 with a 20-bp overlap) eliminated the formation of contigs between groups of phages, but not within the major phage groups (e.g., λ with HK620). A higher stringency of MM% = 97 with an overlap of 20 bp eliminated overlaps between any of the phage genomes in our test library. A very high stringency assembly of MM% = 98 with an overlap of 20 bp differentiates between the very closely related coliphage T3 and T7. In contrast to the effects of increasing stringency, increasing the overlap length to 100 bp still resulted in overlaps between fragments from different phage genomes at lower MM%. We concluded from these studies that at the high stringency assembly conditions (MM% = 97–100; overlap 20 bp) the assembly of two sequences into a contig suggested that they arose from the same phage or a very close relative. The observation of contigs at lower stringencies indicates that the two sequences belong to phages from the same major group, but are not necessarily from the same phage.
To determine how many errors were introduced during cloning and sequencing, the DNA polymerase from coliphage T7 was PCR amplified and cloned, and 19 clones were sequenced and analyzed by using the same protocols as described for the uncultured marine viral samples. The resulting sequences were then globally aligned by using clustal x (17), and the number of gaps and miscalled bases that occurred in the area where all of the sequences overlapped were counted. For each occurrence of a miscalled base, an error was recorded. If one position in the alignment had a miscalled base in two of the sequences, then two errors were recorded. From a total of 12,502 positions investigated, 25 miscalled bases and 8 gaps were observed. Therefore, the average error in this area was 0.26%. The number of errors associated with the ends, where not all of the sequences overlapped, was determined based on comparisons with the published T7 sequence. At the 5′ end, there was one miscalled base in 273 positions (0.366%). At the 3′ end, there were 7 miscalled bases and 5 gaps in 739 positions (1.62%). Therefore, by using our cloning and bioinformatics protocols, the sequence data has an error rate of 0.26–1.62%. Given this limitation, two DNA sequences from the same phage would be expected to be at least 98–99% identical.
Results and Discussion
Identity of Uncultured Marine Viruses.
Shotgun libraries were made from two near-shore marine viral communities (SP and MB). Of the sequence fragments obtained from both samples (1,061 from SP and 873 from MB), the majority showed no significant hits (E < 0.001) to previously reported sequences in GenBank (Fig. 1A). This finding suggested that much of the diversity in the viral communities was previously uncharacterized. Significant hits to GenBank entries were classified as phages, viruses, mobile elements, repeat elements, Bacteria, Archaea, or Eukarya as described in Materials and Methods. In both marine communities, viruses were the most common known hit, and the majority (SP = 90% and MB = 75%) of the virus hits were most similar to phages (Fig. 1B).
Fig 1.

Genomic overview of a near-shore viral community based on sequence similarities. (A) Number of sequences from the uncultured shotgun libraries with a significant hit (E < 0.001) to GenBank. (B) Distribution of significant hits among major classes of biological entities. (C) Families of phages represented in the libraries. (D) Types of mobile elements found in the two libraries.
Significant hits to all of the major families of dsDNA tailed phages were observed in both libraries (Fig. 1C). In both communities, the phage genome with the most top hits was the marine phage Roseophage SIO1 (7). In addition, three of the four phages with the most top hits were Podoviruses (Roseophage SIO1, coliphage T7, and Cyanophage P60). Overall, Podoviruses dominated the known phage hits from the SP community, whereas Podoviruses and Siphoviruses contributed equally to the MB community. In addition, we observed sequences related to groups that, to our knowledge, have never before been reported in the marine environment, including coliphage λ and the Microviridae. The majority (>56%) of the significant phage hits were similar to genes of known function (Table 1). The known hits included DNA and RNA polymerases, helicases, DNA maturation proteins, packaging genes, terminases, and a variety of structural proteins.
Table 1.
Categories of phage proteins with significant hits in the uncultured libraries
| Protein category | SP | MB |
|---|---|---|
| Unknown | 41 | 20 |
| Structural | 20 | 11 |
| Terminase | 8 | 16 |
| Helicase | 6 | 5 |
| DNA maturation | 4 | 0 |
| RNA polymerase | 3 | 1 |
| DNA polymerase | 3 | 1 |
| Methyltransferase | 1 | 1 |
| Packaging | 1 | 7 |
| Recombinase | 1 | 1 |
| Host specificity | 1 | 0 |
| Polynucleotide kinase | 1 | 0 |
| Protease | 1 | 1 |
| DNA-dependent ATPase | 1 | 0 |
| Lytic enzyme | 1 | 0 |
| Morphogenesis | 1 | 0 |
| Endosialidase | 1 | 0 |
| Endonuclease | 0 | 1 |
| Endolysin | 0 | 1 |
| Exonuclease | 0 | 2 |
| Lysozyme | 0 | 1 |
| Phage resistance protein | 0 | 1 |
| PhoH-like | 0 | 1 |
| Total | 95 | 71 |
Although the majority of the diversity in both marine communities appears to be uncharacterized, the fraction that is similar to known sequences suggests fundamental differences between the two samples. The viral community from SP appears more “bacterial” in origin, whereas the MB sample is more “eukaryotic.” There are more eukaryotic hits in the MB sample, as well as a large number of repeat sequences (Fig. 1B). Although it is possible that these repeat regions are indicative of mobility (because mobile elements are frequently flanked by repeat regions), it is more likely that these repeats are representative of the repeats commonly found in eukaryotic genomes. Whether these repeats are from eukaryotic-derived viruses or contaminating DNA is unclear (see below). The former possibility is supported by the fact that more hits to viruses that infect eukaryotes were seen in the MB sample. Another supporting fact comes from the types of mobile hits that were observed (Fig. 1D). The majority (80%) of the mobile hits from the SP library were to bacterial plasmids, whereas most (57%) of the mobile hits from the MB library were to retrotransposons, which are common in eukaryotic genomes. The retrotransposon hits included both LTR and nonLTR retrotransposons. Together, these data suggest that there is a eukaryotic quality to the viral community from the MB library that is perhaps indicative of an algal virus bloom.
The significant hits to Bacteria, Eukarya, and Archaea may represent contamination in the libraries. However, we have accumulated numerous lines of evidence suggesting that this is not the case. In several clones from the SP library, which were sequenced from both ends, the hit from one end of the clone was to a phage, whereas the other end of the same clone had a significant hit to a bacterial or eukaryotic sequence. Second, blast searches with the predicted ORFs from the genomes of the marine phage Roseophage SIO1 (7) and Cyanophage P60 (18) resulted in 50% and 30% of the significant hits being bacterial in origin, respectively. Some of the bacterial hits may also represent uncharacterized prophages and their remnants in the bacterial genomes, or these sequences may come from transducing phages. Finally, the DNA isolation method included a cesium chloride purification of the viral particles to remove contaminating cells and free DNA (ref. 11 and www.sci.sdsu.edu/PHAGE/LASL/index.htm).
Population Modeling.
The samples used to make the shotgun libraries contained ≈2 × 1012 individual viral particles. The higher the diversity of these viruses, the lower the chances of sequencing overlapping fragments from the same viral genome would be. The sequence fragments from the two marine viral communities were assembled using MM% = 98 and an overlap of 20 bp. Based on the calculated error rate, these stringency conditions would prevent sequences from all but the most closely related phage genomes from assembling together (see Materials and Methods). By using these assembly criteria, the 1,061 sequences from the SP sample contained 17 contiguous sequences (contigs) made up of two fragments (2-contigs) and two 3-contigs, as well as 1,021 sequences that did not overlap with any other sequences (referred to as 1-contigs). Among the 873 sequences from the MB sample, 13 2-contigs and 2 3-contigs, as well as 841 sequences that did not overlap with any other sequences were observed. Approximately 3.5% of the total sequence fragments from both samples fell into contigs.
The small number of contigs observed suggested that diversity of the marine viral communities was high, and we therefore sought to model the populations based on the distribution of overlapping sequences. No existing mathematical models adequately described the observed number of contigs; therefore, one was derived from first principles. For this derivation, the following assumptions were made: (i) all of the genomes were 50 kb in length [the average length of marine viral genomes (11)]; (ii) all of the fragments were 663 bp long (the average fragment size in this study, see www.sci.sdsu.edu/PHAGE/LASL/index.htm); (iii) a minimum overlap of 20 bp was needed to form a contig (i.e., the parameters used in the assembly program sequencher); and (iv) fragments in the shotgun library were completely random and unbiased (ref. 19, and www.sci.sdsu.edu/PHAGE/LASL/index.htm).
For the derivation, we first considered a shotgun library from a single viral genotype. When n fragments are sequenced from a genome of length (L) bp, each fragment can be identified by its starting position on a line. Because L ≫ 1, edge effects are rather insignificant. In addition, edge effects also complicate the modeling significantly, and are therefore ignored in the sense that we assume there are L equally likely starting positions for each fragment. It follows that the distances between these randomly chosen points are exponentially distributed (19). Therefore, the probability that two starting points will be within a distance x of each other is 1 − exp(−αx), where 1/α is the average distance between starting points. Letting n represent the number of fragments sampled from one genome gives 1/α = L/n. The probability that two starting points on a genome of length L = 50,000 bp are not more than x = 663 − 20 = 643 bp apart (and thus form a contig) is
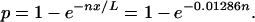 |
For each q-contig (where q equals the number of sequences in the contig), exactly q − 1 such overlaps are needed. This requires a nonoverlap gap followed by q − 1 overlaps followed by another nonoverlap gap. This occurs with the probability (1 − p)pq−1(1 − p).
The probability that a randomly selected fragment is part of a q-contig is given by
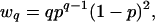 |
which is a negative binomial distribution. With n samples selected from this genome, the expected number of q-contig members will be
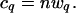 |
Now consider an environmental sample that is a mixture of M different viral genotypes, where each viral genotype i is represented by its population ni in the sample. The values of ni determine the corresponding probabilities of overlap to a neighboring segment pi according to Eq. 1 and probabilities of membership in a q-contig wqi according to Eq. 3. Each viral genotype will make its contribution to the observed q-contigs, with the expected number totaling
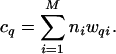 |
By matching the observed numbers of q-contig members in the sample, Cq, to the cq predicted by Eq. 4, it is possible to estimate the number ni of fragments from the ith species. The observed number of contigs in the uncultured marine libraries could not be adequately described by a small number of viral genomes (M < 20), or by assuming that all viral genotypes were evenly distributed. Thus, the observed values contain important information regarding both the evenness and the richness of the viral populations.
A variety of functions were tested to describe the observed distribution of contigs. These functions use two basic parametric forms, ni = f(i, a, b, n1, n2) for the number of segments sampled from species i, (i = 1, . . . , M). The parametric forms were either power-law, given by
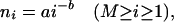 |
or the exponential function
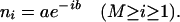 |
These are two of the basic functional forms of relative abundance distribution curves observed for biological populations (20). To examine the robustness of the predicted value of n for the most populous viral genotype, two additional models were explored for which the value of n1 was not determined by the functional form in Eqs. 5 or 6, but rather was allowed to take on whatever value gave the best fit. In these models Eqs. 5 and 6 were used only for i > 1.
As shown in Table 2, the exponential models consistently showed poorer fits to our data than the power-law models. The fits were obtained by minimizing the negative log likelihood function
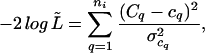 |
which is the sum of the variance-weighted, squared deviations from the observed Cq values. It corresponds to a quasi-likelihood function, L̃, which is a product of normal distributions with expected values given by the Cq and variances given by the sum of the binomial variances appropriate for each genotype
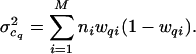 |
Table 2 lists the maximum likelihood values of the parameters obtained for the two data sets. The models are listed in order of preference, which was determined from the minimum weighted error found for the model and the number of parameters used. The power-law model (pow) was preferred over the augmented power-law model (n1pow), in accordance with Occam's razor. Note that only the power-law based models were able to determine a maximum likelihood estimate for M. The standard estimates of error were obtained from the variances estimated as the diagonal elements of the inverse of the second derivative matrix of −log(L̃) with respect to the parameters. For the SP sample, the parameter values (including standard estimates of error) were a = 21.72 ± 3.39, b = 0.64 ± 0.07, and M = 3,318 ± 2,116. For the MB sample, the parameter values were a = 23.22 ± 3.46, b = 0.73 ± 0.08, and M = 7,114 ± 8,691.
Table 2.
The maximum likelihood values of the parameters obtained for the two data sets
| Percent abundance of the most common virus | Weighted sum squared error | a | b | n1 | M | |
|---|---|---|---|---|---|---|
| SP | ||||||
| pow | 2.04 | 1.8 | 21.72 | 0.641 | n/a | 3,318 |
| n1pow | 1.73 | 1.5 | 30.60 | 0.739 | 14.2 | 6,850 |
| n1exp | 2.56 | 3.0 | 2.12 | 0.002 | 27.2 | n/a |
| exp | 0.36 | 10.6 | 3.82 | 0.004 | n/a | n/a |
| MB | ||||||
| pow | 2.66 | 2.1 | 23.22 | 0.729 | n/a | 7,114 |
| n1pow | 2.23 | 1.7 | 35.66 | 0.876 | 15.8 | n/a |
| n1exp | 3.25 | 3.3 | 1.75 | 0.002 | 28.3 | n/a |
| exp | 0.47 | 4.08 | 4.08 | 0.005 | n/a | n/a |
The models are listed in order of preference, which was determined from the minimum weighted error found for the model and the number of parameters used. n/a, Not applicable.
The percent in this category was obtained as the largest n × 100 divided by the number of fragments sequenced. Note that for the models with separately fit n1, the largest n is given by the maximum of the n1 value found and n2 is given by Eq. 5 or 6.
Because of variation in this parameter, our estimate of the population size should be taken as an indication of the order of magnitude for the number of virus types.
Effect of Varying Average Genome Size on Model Predictions.
To study the effect of the genome size on the predictions of the model, the power-law model was analyzed under the assumptions that the genomes of all of the viruses were either 25 or 120 kb, instead of the 50-kb average size used for the analysis presented above. The a parameter in the power-law for both data sets roughly halved for the 25-kb simulation, which translates into half the number of the most abundant viral genotype. Conversely, by using 120 kb for the phage size roughly doubled the a parameter value, thus doubling the abundance of the most common viral genotype compared with a 50-kb genome size assumption. The best fit b parameter was very similar for all genome sizes. Because of the error associated with the M parameter, there was less predictability for this parameter as the genome size varied, but most calculations found M similar in size to the predicted value for the 50-kb genome size given in Table 2.
Marine Viral Community Diversity.
The rank abundance curves for the power-law model of both samples are shown in Fig. 2. This model predicted a total of 3,318 viral types in the SP community and 7,114 viral types in the MB community. All of the models shown in Table 2, except the exponential model (exp), which has a poor fit, predicted that the most abundant viral genome comprised 2–3% of the population in both communities. The robustness of this result lends considerable credibility to its accuracy. In both samples, only three viral genomes comprise more than 1% of the population. Because the entire 200-liter water samples contained ≈2 × 1012 viruses, the models predict that there were ≈5 × 1010 individuals of the most abundant viral genotype. Assuming the most abundant genotype was a phage and an average burst size of 24 (2), ≈2 × 109 bacteria would have been lysed to produce this phage population. Therefore, the most abundant phage must have been capable of infecting ≈1% of the total bacterial population (≈2 × 1011 cells) at one point in time.
Fig 2.

The rank abundance curves for the viral communities as predicted by the power-law model. The curve is shown in red and the standard estimates of error are shown in black.
An estimate of the viral genotype richness M of the population was also obtained by using the nonparametric estimator Chao1 (21) by considering each q-contig as an operational taxonomic unit (OTU), where q equals the number of times that a certain OTU was observed. The Chao1 index is particularly useful for data sets skewed toward the low-abundance classes (22). The Chao1 estimator predicts that there are 31,700 fragments in the SP sample and 28,059 fragments in the MB sample. Because it takes ≈75 fragments of 663 bp to constitute an average sized marine phage genome of 50 kb, Chao1 predicts 423 different viral genomes in the SIO51 sample and 374 different viral genomes in the MB61 sample. Because Chao1 underestimates true richness when sample sizes are small (22), these values should be considered lower boundaries for viral richness. Combining the lower bound from Chao1 and the estimate from our power-law model, we estimate that there are between 423 and 3,318 viral genotypes in the SP sample, and between 374 and 7,114 viral genotypes in the MB sample. The predicted diversity for these communities was extremely high, with a Shannon Index of 7.56 for the SP community, and Shannon Index of 7.99 for the MB community (23).
Given the fact that the two samples came from different environments, and the viral community composition appears to be quite different, it is striking that the population diversity for both samples is so similar. In these 200-liter seawater samples, which each contained ≈1012 virus particles, both parametric and nonparametric analyses predicted <104 different viral types. A contig between sequence fragments from the two samples was also found (data not shown), which means that we will be able to estimate intersample diversity with larger sequencing efforts. This makes the task of estimating oceanic viral diversity a tractable problem.
The most abundant virus in both marine communities comprised 2–3% of the total population. If this genome is ≈50 kb, only 25,000 clones would have to be sequenced to get 10× coverage of this virus. Similarly, ≈500,000 clones would have to be sequenced to get 10× coverage of the 100 most abundant viruses. The Joint Genome Institute is currently sequencing ≈206 96-well plates per day (May 1, 2002, at www.jgi.doe.gov/). This is a total of ≈19,776 clones per day. Therefore, the sequence of the most abundant virus could be obtained with 1 day's worth of sequencing effort, and the 100 most abundant viruses could be sequenced in 1 month. The current library contains ≈1 million clones, with enough viral DNA remaining to create 15 more libraries of this size. Assuming our diversity estimates are correct, there is enough DNA in our sample to sequence the genome of every virus in the marine communities, even though this would require extraordinarily large sequencing efforts. These results show that it is not only possible to sequence the entire genome of an uncultured marine virus by using this approach, but also to sequence the entire genome of an uncultured marine viral community.
Acknowledgments
We thank Steven Rayhawk, Colleen Kelly, Ben Felts, James Nulton, Sherwood Casjens, and Richard Long for helpful conversations and suggestions related to this work. B.A. thanks San Diego State University for its hospitality. This work was sponsored by National Science Foundation Small Grant for Exploratory Research OCE01-16900.
Abbreviations
SP, Scripps Pier
MB, Mission Bay
LASL, linker-amplified shotgun library
MM%, minimal mismatch percentage
References
- 1.Fuhrman J. A. (1999) Nature 399, 541-548. [DOI] [PubMed] [Google Scholar]
- 2.Wommack K. E. & Colwell, R. R. (2000) Microbiol. Mol. Biol. Rev. 64, 69-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bratbak G., Heldal, M., Thingstad, T. F. & Tuomi, P. (1996) FEMS Microbiol. Ecol. 19, 263-269. [Google Scholar]
- 4.Paul J. H. (1999) J. Mol. Microbiol. Biotechnol. 1, 45-50. [PubMed] [Google Scholar]
- 5.Fuller N. J., Wilson, W. H., Joint, I. R. & Mann, N. H. (1998) Appl. Environ. Microbiol. 64, 2051-2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hambly E., Tetart, F., Desplats, C., Wilson, W. H., Krisch, H. M. & Mann, N. H. (2001) Proc. Natl. Acad. Sci. USA 98, 11411-11416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rohwer F., Segall, A., Steward, G., Seguritan, V., Breitbart, M., Wolven, F. & Azam, F. (2000) Limnol. Oceanogr. 42, 408-418. [Google Scholar]
- 8.Fuhrman J. A. & Campbell, L. (1998) Nature 393, 410-411. [Google Scholar]
- 9.Rohwer F. & Edwards, R. (2002) J. Bacteriol. 184, 4529-4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noble R. T. & Fuhrman, J. A. (1998) Aquatic Microbial Ecol. 14, 113-118. [Google Scholar]
- 11.Steward G. F., Montiel, J. L. & Azam, F. (2000) Limnol. Oceanogr. 45, 1697-1706. [Google Scholar]
- 12.Sambrook J., Fritsch, E. F. & Maniatis, T., (1989) Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Lab. Press, Plainview, NY).
- 13.Rohwer F., Seguritan, V., Choi, D. H., Segall, A. M. & Azam, F. (2001) BioTechniques 31, 108-118. [DOI] [PubMed] [Google Scholar]
- 14.Altschul S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997) Nucleic Acids Res. 25, 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Altschul S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990) J. Mol. Biol. 215, 403-410. [DOI] [PubMed] [Google Scholar]
- 16.Murphy F. A., Fauquet, C. M., Bishop, D. H. L., Ghabrial, S. A., Jarvis, A. W., Martelli, G. P., Mayo, M. A. & Summers, M. D., (1995) Virus Taxonomy: Sixth Report of the International Committee on the Taxonomy of Viruses (Springer, New York), Vol. 10, pp. 586. [Google Scholar]
- 17.Thompson J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F. & Higgins, D. G. (1997) Nucleic Acids Res. 24, 4876-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen F. & Lu, J. (2002) Appl. Environ. Microbiol. 68, 2589-2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feller W., (1966) An Introduction to Probability Theory and Its Applications (Wiley, New York).
- 20.Ulrich W. (2001) Pol. J. Ecol. 49, 145-157. [Google Scholar]
- 21.Chao A. (1984) Scand. J. Stat. 11, 783-791. [Google Scholar]
- 22.Hughes J. B., Hellmann, J. J., Richetts, T. H. & Bohannan, B. J. M. (2001) Appl. Environ. Microbiol. 67, 4399-4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shannon C. E. & Weaver, W., (1963) The Mathematical Theory of Communication (Univ. of Illinois Press, Urbana).