

Abstract
We investigated 18 AIDS hearts (5 with and 13 without cardiomyopathy) by using immunocytochemistry and computerized image analysis regarding the roles of HIV-1 proteins and tumor necrosis factor ligands in HIV cardiomyopathy (HIVCM). HIVCM and cardiomyocyte apoptosis were significantly related to each other and to the expression by inflammatory cells of gp120 and tumor necrosis factor-α. In HIVCM heart, active caspase 9, a component of the mitochondrion-controlled apoptotic pathway, and the elements of the death receptor-mediated pathway, tumor necrosis factor-α and Fas ligand, were expressed strongly on macrophages and weakly on cardiomyocytes. HIVCM showed significantly greater macrophage infiltration and cardiomyocyte apoptosis rate compared with non-HIVCM. HIV-1 entered cultured neonatal rat ventricular myocytes by macropinocytosis but did not replicate. HIV-1- or gp120-induced apoptosis of rat myocytes through a mitochondrion-controlled pathway, which was inhibited by heparin, AOP-RANTES, or pertussis toxin, suggesting that cardiomyocyte apoptosis is induced by signaling through chemokine receptors. In conclusion, in patients with HIVCM, cardiomyocytes die through both mitochondrion- and death receptor-controlled apoptotic pathways.
HIV cardiomyopathy (HIVCM) is a complication of HIV-1 infection with high cardiovascular morbidity and mortality in young and middle-aged adults. Its annual incidence was estimated by echocardiographic monitoring to be 15.9 cases per 1,000 asymptomatic Italian HIV-1-positive patients (1). Despite its growing importance, the molecular mechanisms of HIVCM pathogenesis remain poorly understood.
Immunopathological examination of AIDS heart tissues showed that HIVCM is strongly inflammatory and related to the staining density of the prostacyclin enzyme cyclooxygenase-2 and the macrophage marker CD68 (2, 3). Infected hearts showed a strong expression of the viral envelope protein gp120 and the regulatory protein Nef in infiltrating macrophages and T cells but only a weak or absent gp120 and Nef expression in cardiomyocytes (3). This observation argues against a productive infection of cardiomyocytes by HIV-1 as a primary mechanism of HIVCM, as previously proposed (4). Instead, it suggests that indirect mechanisms initiated by inflammatory cells are plausible, such as secretion of cytokines, in particular tumor necrosis factor-α (TNF-α) (5), nitric oxide (6, 7), and viral proteins, in particular gp120 (7, 8) and Tat (9). In agreement with an indirect model, transgenic mice expressing the entire HIV-1 coding sequences in T cells and monocyte/macrophages developed a multitude of complications (10), including multifocal cardiomyopathy (P. Jolicoeur, personal communication), which were found to be related to Nef expression (11). A simian study was also suggestive of the role of Nef, because simian immunovirus (SIV)mac 239 infection produced more severe immune suppression and cardiac fibrosis compared with SIVmac 239 Δ nef (12). In addition to HIV-1, opportunistic cardiotropic viruses, such as adenoviruses and cytomegalovirus (13), may contribute to HIVCM.
Cardiomyocyte apoptosis underlies many forms of heart disease, including viral cardiomyopathy (14, 15). The inducers of cardiomyocyte apoptosis include myocardial cell stretch, pressure overload, oxygen radicals, nitric oxide, cytokines such as TNF-α, and viruses (16). In failing human hearts, cardiomyocyte apoptosis was found to be abundant, but the mechanism was not apparent (17). In experimental murine Coxsackievirus myocarditis, Fas ligand/Fas pathway was noted to play a critical role (14). We hypothesized that apoptosis might underlie HIVCM, because HIV-1 can induce apoptosis of different cells both in vitro and in vivo (18). In this study, we examined the role of gp120, Nef, TNF-α, Fas ligand, and caspases in virus-induced apoptosis in vitro and in vivo. We showed that in vitro gp120 induces cardiomyocyte apoptosis by a mitochondrion-controlled pathway, but in vivo death receptor ligands from macrophages are a major cause of apoptosis, and the apoptotic signaling may occur through chemokine receptors.
Methods
Heart Tissues.
The Texas Repository for AIDS Neuropathogenesis Research and the Manhattan HIV Brain Bank provided paraffin-embedded formalin-fixed heart tissues from the left ventricle and plasma specimens of 22 patients with AIDS, of which nine were diagnosed as HIVCM (“HIVCM-positive hearts”), and 13 had no history or biochemical evidence of heart failure (“HIVCM-negative hearts”). The University of California, Los Angeles (UCLA), Tissue Resource Bank provided heart tissues from a normal patient (death from a motor vehicle accident) and four patients with non-HIVCM (three idiopathic and one adriamycin-induced). The UCLA Office for Protection of Research Subjects approved all work with human tissues and blood cells.
Primary Neonatal Rat Ventricular Myocyte (NRVM) Cultures.
Primary NRVM cultures were prepared from neonatal 1- to 3-day-old Sprague–Dawley rats with full approval of the UCLA Animal Research Committee, essentially as described previously (19). Ventricles of ≈100 hearts were digested four times, 20 min each, in 30 ml of enzyme digestion solution containing 0.1% collagenase (Worthington)/12% pancreatin 4× (GIBCO/BRL)/0.06% Ads buffer (116.4 mM NaCl/5.4 mM KCl/5.6 mM dextrose/10.9 mM NaH2PO4/405.7 μM MgSO4/20 mM Hepes, pH 7.3). NRVMs were purified by using a Percoll (Pharmacia) step gradient in Ads buffer (comprising layers with final densities of 1.130, 1.082, and 1.059 g/ml) and washed in Ads buffer. Fibroblast cells were removed by aspiration. The myocytes (banding between the 1.082- and 1.059-g/ml layers) were finally washed in plating media (4:1 DMEM/M199/10% horse serum/5% neonatal calf serum/1% antibiotics) and plated at a density of 1 × 106 cells/60-mm dish or 104 cells in a four-well chamber slide coated with 1% gelatin. They were cultured in serum-free medium, a 1:1 mixture of DMEM/M199. The purity of cultured NRVMs was 95–100% when the number of cell nuclei [4′,6-diamidino-2-phenylindole (DAPI)-positive] was compared with the number of myocytes (MF20-positive).
Virus and Viral Proteins.
HIV-1 protein gp120SF2 was obtained from the AIDS Research and Reference Reagent Program, National Institute of Allergy and Infectious Diseases. HIV-1MN, either active or aldrithiol-inactivated, and control vesicles from uninfected cells were provided by J. Lifson (Science Applications International, Frederick, MD). The inactivated virus has an intact envelope but inactivated nucleocapsid (20). HIV-1NL4-3 was prepared by transfecting 293 cells with DNA of an infectious molecular clone (21). HIV-1NL4-3 incorporating Vpr-GFP (HIV-1NL4-3 Vpr-GFP) was produced by cotransfection of 293 cells with pNL4-3 DNA (10 μg) and expression vector coding for GFP-Vpr fusion protein (20 μg) (22). A pseudotyped luciferase-expressing NL HXB2 Env(+)LUC(+) virus was obtained by cotransfection of plasmid DNA-encoding envelope from a T-tropic HIV-1 clone HXB2 and pNL4-3 Env(−)LUC(+) into 293T cells (22).
Virus Assays.
In the luciferase assay, 106 NRVMs were infected with 3 × 107 RNA copies of NL HXB2 Env(+)LUC(+) and, at specified times postinfection, the cells were washed and lysed by using the Luciferase Cell Culture Lysis Reagent (Promega). The lysate was tested by using the Luciferase Assay Reagent (Promega) and scintillation counting (22).
For the RT-PCR assay, 106 cardiomyocytes were exposed for 4 h to 5 × 107 copies of HIV-1LAI that had been pretreated for 1 h at room temperature with 200 units/ml of RNase-free DNase I (Roche Molecular Biochemicals) in PBS containing 0.04 M MgCl2, washed four times, and cultivated. At indicated times, the cells were scraped, centrifuged, and the cell pellets were lysed in PCR buffer. After protein digestion with proteinase K (2 h at 56°C) and its inactivation (10 min at 95°C), the lysate was subjected to amplification with the primers for LTR R/U5 [(forward) 5′-GGC TAA CTA GGG AAC CCA CTG-3′, (reverse) 5′-CTG CTA GAG ATT TTC CAC ACT GAC-3′], and gag [(forward) 5′-CAG ATA TCC ACT GAC CTT TGG-3′, (reverse) 5′-GCT TAA TAC TGA CGC TCT CGC A-3′], as described (22).
Brain Natriuretic Peptide (BNP) Assay.
BNP, a peptide hormone whose plasma concentration is raised in congestive heart failure, was measured by a competitive enzyme immunoassay (American Laboratory Products, Windham, NH).
Immunocytochemistry and Immunofluorescence.
Formalin-fixed tissues were immunostained by DAKO LSAB 2 or EnVision Double Stain System kits (3). Cultured myocytes were stained by indirect immunofluorescence. The stained sections were examined at low power (×10 objective) to locate the area of the highest staining density, three high-power (×40 objective) images of this area were acquired, and the images were scanned by using IMAGE-PRO PLUS 4.1 (Media Cybernetics, Silver Spring, MD) (3).
The antibodies included those to sarcomere myosin (MF20, Developmental Studies Hybridoma Bank, University of Iowa), gp120 (clones ID6 and 5F7), Nef (EH1 ascites) (AIDS Research and Reference Reagent Program, National Institute of Allergy and Infectious Diseases), p10 fragment of caspase-9, Fas ligand (BioSource International, Camarillo, CA), TNF-α (Sigma), and CD68 and CD3 (DAKO). To study virus entry, cardiomyocytes were pretreated with cholera toxin B–Alexa 594 (Molecular Probes), methyl-β-cyclodextrin, heparin, 5-(N,N-dimethyl)-amiloride (all from Sigma), AOP-RANTES (gift of A. Proudfoot, Serono, Geneva), SDF-1 (PeproTech, Rocky Hill, NJ), U0126 (Promega), or pertussis toxin (List Biological Laboratories, Campbell, CA), as indicated.
Apoptosis Assays.
Apoptosis of cardiomyocytes and NRVMs was detected by the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) technique (Apoptosis Detection Kit with dUTP, Promega) by using the FITC-conjugated nucleotide mix, and simultaneous staining with DAPI or Hoechst 33342 (Molecular Probes). Apoptotic inhibitors included a general caspase inhibitor, Z-VAD-FMK, a preferential caspase-8 inhibitor, Z-IETD-FMK, and a preferential caspase-9 inhibitor, Z-LEHD-FMK (Enzyme Systems Products, Livermore, CA) (23). For detection of mitochondrial features of apoptosis, live cells were stained by using Hoechst 33342 (2 μM) and 5,5′′,6,6′′-tetrachloro-1,1′′,3,3′′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1, Molecular Probes) (2 μM) for 30 min at 37°C in complete medium. Cytochrome c release was determined by confocal microscopy in cells fixed with 4% paraformaldehyde and stained with monoclonal anticytochrome c antibody.
Western Blotting.
An equal volume of tissue from each heart was extracted by dounce homogenization in ice-cold cell lysis buffer. An equal amount of protein (40 μg) was run on 7.5% SDS–acrylamide gel, transferred to polyvinylidene membranes, incubated with primary and secondary antibodies, and developed by chemiluminescence, as described (24). Mouse anti-human caspase-8 (p18) (Biosource) was used as a primary antibody.
Statistical Analysis.
The significance of data was determined by t test analysis performed with the statistical software spss, Version 10.0 (SPSS, Chicago) and, when indicated, by nonparametric analysis using the Mann–Whitney U test (U test). The interaction of gp120 and TNF-α in apoptosis was analyzed by median regression (25).
Results
Because the relative importance of gp120, Nef and TNF-α, and apoptosis in HIVCM is unknown, we examined these parameters in the hearts of HIV-1-positive patients with and without HIVCM. Our previous study identified gp120 and Nef in macrophages and T cells but not cardiomyocytes within the infected heart (3), suggesting that these proteins might act in a paracrine rather than an autocrine fashion.
HIVCM Is Associated with gp120 Expression and Cardiomyocyte Apoptosis, and the Apoptosis Is Related to both gp120 and TNF-α.
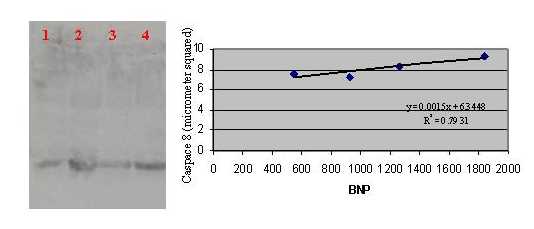
We examined apoptosis and HIV-1 proteins in the heart tissues of 18 HIV-1-positive patients, 5 HIVCM-positive patients (mean age 40 years), and 13 patients HIVCM-negative (mean age 44 years) (see Table 1, which is published as supporting information on the PNAS web site, www.pnas.org). The hearts of HIVCM-positive patients generally showed increased mass [mean weight: HIVCM-positive = 580 ± 220 g; HIVCM-negative = 346 ± 57 g (P = 0.002)], fibrosis [mean: HIVCM-positive = 162,008 ± 290,568 pixels (px); HIVCM-negative = 9,913 ± 5,147 px (P = 0.09)], and elevated BNP serum levels [mean: HIVCM-positive = 1,100 ± 703 units; HIVCM-negative = <342 units (P = 0.004)] (Fig. 1). HIVCM-positive hearts had a significantly higher expression of gp120 [mean: HIVCM-positive 153 ± 146 px, HIVCM-negative = 5.2 ± 8.1 px (U test, P = 0.08; t test, P = 0.008)], and TNF-α [mean: HIVCM-positive = 132 ± 215 px, HIVCM-negative 6.3 ± 12.9 px, (U test, P = 0.003; t test, P = 0.051)]. Nef expression was not significantly different between the two groups [mean: HIVCM-positive 65 ± 106 px, HIVCM-negative = 72 ± 33 px (U test, P = 0.315; t test P = 0.916)]. HIVCM was significantly associated with cardiomyocyte apoptosis [mean: HIVCM-positive 82 ± 51 px, HIVCM-negative = 7.4 ± 16.0 (U test, P = 0.008; t test, P = 0.004)]. When stepwise median regression was performed with apoptosis as the dependent variable and gp120 and TNF-α as independent variables, there was a significant relation with both (R = 0.5, P < 0.001).
Fig 1.

Relation of gp120, Nef, and TNF-α to HIV-1 cardiomyopathy and cardiomyocyte apoptosis. The hormone BNP was determined in the plasma of 12 patients by using a competitive enzyme immunoassay (A). The left ventricular sections from 18 AIDS hearts were stained by the trichrome technique to detect fibrosis (B), immunocytochemistry with anti-gp120, -Nef (C), and -TNF-α (E) , and the TUNEL and DAPI techniques (D) (note bright contracted nuclei corresponding to TUNEL-positive apoptotic nuclei). Maximum staining was identified at low power, and then three high-power fields were scanned in a predetermined order by using IMAGE PRO software. Zero value indicates that no positive staining was found anywhere. The mean ± SD for each group (HIVCM-positive n = 5 and HIVCM-negative, n = 13) was plotted on the y axis and the data were analyzed by the t test (P values: BNP = 0.004, fibrosis = 0.091, gp120 = 0.008, Nef = 0.315, apoptosis = 0.004, TNF-α = 0.051), and the Mann–Whitney nonparametric test (P values: BNP = 0.001, fibrosis = 0.04, gp120 = 0.08, Nef = 0.3315, apoptosis = 0.004, TNF-α = 0.003).
To clarify which apoptotic pathways are involved in the hearts with HIVCM or non-HIVCM, we examined heart expression of the activated mitochondrial caspase, caspase-9, and the TNF superfamily ligand, Fas ligand.
Components of both Extrinsic and Intrinsic Apoptotic Pathways Are Present in HIVCM Hearts.
The p10 cleavage fragment of both caspase-9 and Fas ligand was significantly overexpressed in hearts with HIVCM (Fig. 2a) compared with hearts with non-HIVCM (Fig. 2b) or HIV-1 infection without HIVCM (Fig. 2c), and they were absent from a control heart. In HIVCM, their expression was robust on round inflammatory cells (Fig. 2d1) and weak on rare myocytes (Fig. 2d2). In addition, p18 fragment of caspase-8 was detected by Western blotting in left ventricular heart tissue of four patients with HIVCM in relation to the BNP plasma titer in each patient (see Fig. 4, which is published as supporting information on the PNAS web site).
Fig 2.

Cleaved caspase-9 and Fas ligand are overexpressed in HIVCM compared with non-HIVCM. Heart tissues from a control patient, three AIDS patients without cardiomyopathy, and four patients with non-HIV-1 cardiomyopathy were immunostained by using antiactive caspase-9 and anti-Fas ligand antibodies [×40 (a, b, e, and f); ×100 (c, d, g, and h)].
To further analyze the molecular mechanisms by which HIV-1 and gp120 induce cardiomyocyte apoptosis, we used NRVMs as a model, because they survive and function in vitro for at least 72 h and express apoptotic death on TNF-α stimulation (26). Rat cells are permissive for early virus products only after transfection with human CD4, chemokine receptors, and cyclin T1 (27). However, HIV-1 enters NRVMs by macropinocytosis (see below), which bypasses CD4 and chemokine receptors, and thus NRVMs may serve as a model of the nonproductive infection of human cardiomyocytes.
HIV-1 Binds to Ganglioside GM1 and Enters Cultured NRVMs by Macropinocytosis, Which Is Inhibited by Cholesterol Extraction, Heparin, and AOP-RANTES.
In brain and coronary endothelial cells, HIV-1 binds to ganglioside GM1 and enters by macropinocytosis susceptible to inhibition by the Na/K channel inhibitor dimethylamiloride, cholesterol-extracting agent cyclodextrin, the polyanion heparin, and U0126 (24). In cultured NRVMs, HIV-1-Vpr-GFP also bound to the ganglioside GM1, as seen by confocal microscopy 3 h postinfection (see Fig. 5, which is published as supporting information on the PNAS web site), and the same compounds, except U0126, blocked virus entry, suggesting that HIV-1 may use a similar entry mechanism in different cardiovascular cells. However, unlike in endothelial cells, the chemokine analogue AOP-RANTES had a significant inhibitory effect on entry into cultured NRVMs.
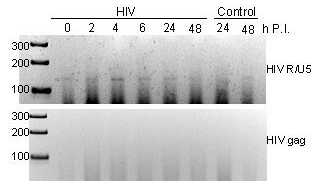
Virus entry into NRVMs did not lead to productive infection, as demonstrated by using luciferase and RT-PCR assays. The luciferase activity in NRVM lysates collected 3 and 4 days postinfection did not rise significantly above the background in any sample (negative data not shown). Reverse transcription was only detected with R/U5 primers, which amplify an early product, but not with gag primers amplifying a late product (see Fig. 6, which is published as supporting information on PNAS web site), indicating that, as in brain endothelial cells (22), HIV-1 infection in rat myocytes is abortive. Taken together, these results suggest that HIV-1 infection proceeds in a similar fashion in different heart cells.
HIV-1 and HIV-1 Envelope Protein gp120 Induce Apoptosis of Cultured Rat Myocytes Through a Mitochondrion-Controlled Pathway.
Both infectious HIV-1MN and noninfectious vaccine HIV-1MN with its nucleocapsid zinc finger inactivated by aldrithiol treatment but intact envelope (20) induced apoptosis of NRVMs (Fig. 3). This result suggested that the HIV-1 envelope was sufficient for apoptotic effect. This supposition was confirmed by experiments with recombinant gp120 (0.01–1 μg/ml), which induced NRVM apoptosis with rapid kinetics (49.6% after 5 min, 76.2% after 30 min, and 100% after 4 h). All three preparations of recombinant gp120 from T-tropic strains HIV-1MN, HIV-1SF2, and HIV-1IIIB/LAV had approximately equal apoptotic effects. NRVM apoptosis was also revealed by DAPI and Hoechst 33258 staining, which showed nuclear condensation and fragmentation (Fig. 3A).
Fig 3.

Apoptosis of cultured rat myocytes is induced by HIV-1 or gp120 through the mitochondrial pathway. (A) NRVMs were sham-infected (a), infected with HIV-1MN (b), or treated with gp120 (1 μg/ml) (c) and were stained with DAPI (Left), TUNEL (Center), antisarcomeric antibody MF-20 (Right, b), or anti-gp120 (Right, a and c). (B) NRVMs were treated as indicated with HIV-1 (active HIVMN or vaccine HIVMN) or gp120, intact, boiled or neutralized by antibody. gp120-treated cells were pretreated with the caspase-8 inhibitor Z-IETD-FMK (10 μM), caspase-9 inhibitor Z-LEHD-FMK (10 μM), the CCR5 antagonist AOP-RANTES (100 nM), pertussis toxin (Ptx) (1 μg/ml), or heparin (20 μg/ml), and stained by the TUNEL technique. The results were scanned and mean OD plotted. (C) NRVMs were either sham-infected or infected with HIV-1MN and were stained with Hoechst 33342 (a), JC-1 (b), and anticytochrome C (c). JC-1 staining (red and green) results were scanned. A high red/green ratio indicates a high mitochondrial membrane potential.
To determine the apoptotic pathway induced by gp120, we relied on inhibitors of specific caspases and staining with the dye JC-1 and antibody to cytochrome c. The caspase-9 inhibitor z-LEHD-FMK (Fig. 3B) completely blocked induction of apoptosis by gp120, whereas the preferential caspase-8 inhibitor s-IETD-FMK was ineffective. A reduced mitochondrial membrane potential, as shown by a low JC-1 red/green ratio, and release of cytochrome c in gp120-treated myocytes (Fig. 3C) supported a mitochondrion-dependent mechanism.
To exclude the possibility that apoptosis was due to the presence of endotoxin in recombinant gp120 preparations, we performed several control experiments. First, the endotoxin content in gp120 preparations was found below the sensitivity of the Limulus lysate test (<0.003 ng/ml). Second, heat inactivation of gp120 (100°C, 15 min) completely eliminated its apoptotic activity. Finally, apoptosis was blocked by treatment with gp120 monoclonal antibody (1:100, 30 min) (Fig. 3B).
To clarify the mechanisms of apoptosis induction, we investigated the effects of several agents previously demonstrated to inhibit gp120 interaction with target cells.
gp120-Induced Apoptosis Is Inhibited by AOP-RANTES.
Interaction of gp120 with CCR5 or CXCR4 on target human cells initiates signaling cascades (28, 29), which might initiate apoptosis in cardiovascular cells. Although the extent of homology between rat and human CCR5 is unknown, the observation that a low-level infection of rat cells by HIV-1 can be achieved (27) suggests that HIV-1 might use rat CCR5 or CCR3. We therefore tested whether AOP-RANTES, a CCR5 antagonist and a potent competitive inhibitor of gp120-CCR5 interaction (30, 31), and pertussis toxin, which ADP ribosylates Giα2 and Giα3 and thus inhibits G protein-coupled chemokine receptor signaling (32, 33), would inhibit gp120-induced apoptosis of NRVMs. AOP-RANTES strongly inhibited the induction of apoptosis by gp120 (Fig. 3B), supporting the hypothesis that gp120 signaling from CCR5 or CCR3 leads to NRVM apoptosis, as previously shown in T cells (34). CCR3 is overexpressed on cardiomyocytes in AIDS hearts (N.Q.L. and M.F., unpublished data), creating an opportunity for strong gp120 signaling. Consistent with the role of gp120- and chemokine-induced signaling, apoptosis was strongly inhibited by the polyanion heparin, which inhibits HIV-1 (35), Tat (36), and chemokine (37) binding to cells (Fig. 3B).
Discussion
The presence of either gp120, Nef, or both in each of 18 AIDS hearts in this study showed that HIV-1 may invade the heart even in patients without overt heart failure. Several etiological agents of HIVCM have been proposed but not proven, including HIV-1 itself (38), opportunistic cardiotropic viruses (13), abused drugs, and highly active antiretroviral therapy (5, 39–41). Although previous studies investigated the roles of gp120 (42), Tat (43), and Nef (44) in neurological complications and the effects of Tat (45), gp120 (45), and Nef (11) in transgenic animal models, none of these studies addressed the role of these HIV-1 proteins in AIDS heart.
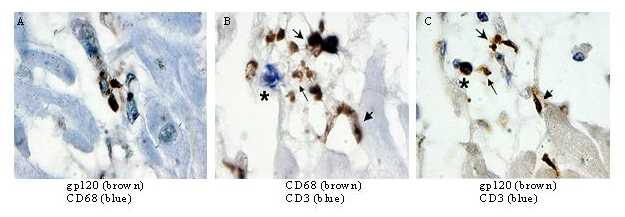
We show here that HIVCM is significantly related to the viral protein gp120 but not Nef, and cardiomyocyte apoptosis is a critical mechanism in HIVCM. In the AIDS heart, apoptosis was significantly related to gp120 and the extrinsic pathway ligand TNF-α. As previously demonstrated, in HIVCM, gp120 is expressed abundantly in macrophages and T cells but poorly or not at all in cardiomyocytes (see Fig. 7, which is published as supporting information on the PNAS web site) (3); furthermore, despite efficient entry, HIV-1 does not replicate in rat cardiomyocytes in vitro. Therefore, the proapoptotic stimuli, gp120 and TNF ligands, appear to originate from infected macrophages and lymphocytes, and cardiomyocytes may be affected secondarily.
In previous studies in immune and neural cells, apoptosis was induced by HIV-1 proteins, gp120 (46, 47), Tat (48, 49), Nef (50, 51), and vpr (52). gp120 produced apoptosis in T cells by Fas signaling (53) but also by mitochondrial membrane depolarization independently of Fas signaling (46). The latter apoptosis was inhibited by the CXCR4 ligand SDF-1 (54). In another study, the mechanism of cell death diverged between R5 HIV env, being caspase-8-dependent, and X4 HIV-1 env (55), being caspase-8- and -9-independent. HIV-1 Tat (56) and Nef (50, 57) proteins protected infected cells but caused apoptosis of bystander cells. Tat induced apoptosis of endothelial cells through caspase activation but not by TNF-α or Fas pathways (58). Vpr induced apoptosis through caspase activation (59). Thus, whereas several HIV-1 proteins have been shown to induce in vitro apoptosis through different pathways, the impact of these proteins on apoptosis in vivo has not been resolved.
In cultured rat myocytes, recombinant gp120 alone was sufficient to induce apoptosis by a mitochondrion-controlled pathway. The virus avidly bound to the myocytes through the ganglioside GM1 and entered these cells by macropinocytosis (22). AOP-RANTES inhibited cardiomyocyte apoptosis induced by gp120MN, which is a CXCR4-using envelope protein. This result suggests that a signal from AOP-RANTES through CCR3 or CCR5 interferes with the apoptosis-inducing signal from gp120MN through CXCR4. Heparin inhibited both virus entry and apoptosis, suggesting that virus entry could be a significant proaptotic stimulus. However, sulfated polysaccharides, including the heparan sulfate proteoglycans syndecans, bind both to the virus and the chemokines with basic residues, including RANTES and SDF-1 (35, 37), suggesting that heparin could inhibit induction of apoptosis by binding chemokines rather than the virus. Regardless of the underlying mechanisms, robust effects of several inhibitors on HIV-1-induced cardiomyocyte apoptosis suggest interesting opportunities for management of HIVCM.
In conclusion, gp120 alone initiates a mitochondrion-controlled pathway of apoptosis in cultured myocytes, whereas the apoptotic signaling in human cardiomyocytes in vivo involves the components of both mitochondrial and extrinsic pathways.
Supplementary Material
Acknowledgments
We thank J. Reed for advice concerning caspase inhibitors, M. Schibler for assistance with confocal microscopic imaging, S. Antrobus and R. Ross for neonatal rat cardiomyocytes, V. Kermani (Immunobiogene, Cardiovasular Research Laboratory, University of California at Los Angeles, West Hills, CA) for BNP assays, and M. Jegapragasan for skillful technical assistance. This work was supported by Grants HL63639 and HL63065 from the National Institutes of Health (to M.F.), Grants AI42557 and AI50461 from the National Institutes of Health (to W.P.), and the Laubisch Endowment (to K.P.R. and W.R.M.). The Manhattan HIV Brain Bank, New York (R24MH59724), and the Texas Repository for AIDS Neuropathogenesis Research, University of Texas, Galveston (R24MH59656), of the National NeuroAIDS Tissue Consortium provided the heart tissues of HIV-1-positive patients. The AIDS Research and Reference Reagent Program, National Institute of Allergy and Infectious Diseases, National Institutes of Health, provided the listed HIV-1 strains and antibodies.
Abbreviations
HIVCM, HIV cardiomyopathy
TNF-α, tumor necrosis factor-α
NRVM, neonatal rat ventricular myocyte
BNP, brain natriuretic peptide
TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
DAPI, 4′,6-diamidino-2-phenylindole
U test, Mann–Whitney U test
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Barbaro G., Fisher, S. D. & Lipshultz, S. E. (2001) Lancet Infect. Dis. 1, 115-124. [DOI] [PubMed] [Google Scholar]
- 2.Fiala M., Liu, N. Q., Reddy, S. & Graves, M. (2001) Alzheimer's Rep. 4, 1-7. [Google Scholar]
- 3.Liu Q. N., Reddy, S., Sayre, J. W., Pop, V., Graves, M. C. & Fiala, M. (2001) AIDS Res. Hum. Retroviruses 17, 1423-1433. [DOI] [PubMed] [Google Scholar]
- 4.Grody W., Cheng, L. & Lewis, W. (1990) Am. J. Cardiol. 66, 203-206. [DOI] [PubMed] [Google Scholar]
- 5.Lewis W. (2000) Prog. Cardiovasc. Dis. 43, 151-170. [DOI] [PubMed] [Google Scholar]
- 6.Barbaro G., Di Lorenzo, G., Soldini, M., Giancaspro, G., Grisorio, B., Pellicelli, A. & Barbarini, G. (1999) Circulation 100, 933-939. [DOI] [PubMed] [Google Scholar]
- 7.Kan H., Xie, Z. & Finkel, M. S. (2000) Am. J. Physiol. 279, H3138-H3143. [DOI] [PubMed] [Google Scholar]
- 8.Chen F., Shannon, K., Ding, S., Silva, M. E., Wetzel, G. T., Klitzner, T. S. & Krogstad, P. (2002) AIDS Res. Hum. Retroviruses 18, 777-784. [DOI] [PubMed] [Google Scholar]
- 9.Raidel S. M., Haase, C., Jansen, N. R., Russ, R. B., Sutliff, R. L., Velsor, L. W., Day, B. J., Hoit, B. D., Samarel, A. M. & Lewis, W. (2002) Am. J. Physiol. 282, H1672-H1678. [DOI] [PubMed] [Google Scholar]
- 10.Hanna Z., Kay, D. G., Cool, M., Jothy, S., Rebai, N. & Jolicoeur, P. (1998) J. Virol. 72, 121-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanna Z., Kay, D. G., Rebai, N., Guimond, A., Jothy, S. & Jolicoeur, P. (1998) Cell 95, 163-175. [DOI] [PubMed] [Google Scholar]
- 12.Shannon R. P., Simon, M. A., Mathier, M. A., Geng, Y. J., Mankad, S. & Lackner, A. A. (2000) Circulation 101, 185-193. [DOI] [PubMed] [Google Scholar]
- 13.Bowles N. E., Kearney, D. L., Ni, J., Perez-Atayde, A. R., Kline, M. W., Bricker, J. T., Ayres, N. A., Lipshultz, S. E., Shearer, W. T. & Towbin, J. A. (1999) J. Am. Coll. Cardiol. 34, 857-865. [DOI] [PubMed] [Google Scholar]
- 14.Seko Y., Kayagaki, N., Seino, K., Yagita, H., Okumura, K. & Nagai, R. (2002) J. Am. Coll. Cardiol. 39, 1399-1403. [DOI] [PubMed] [Google Scholar]
- 15.Alter P., Jobmann, M., Meyer, E., Pankuweit, S. & Maisch, B. (2001) Cardiovasc. Pathol. 10, 229-234. [DOI] [PubMed] [Google Scholar]
- 16.MacLellan W. R. & Schneider, M. D. (1997) Circ. Res. 81, 137-144. [DOI] [PubMed] [Google Scholar]
- 17.Olivetti G., Abbi, R., Quaini, F., Kajstura, J., Cheng, W., Nitahara, J. A., Quaini, E., Di Loreto, C., Beltrami, C. A., Krajewski, S., et al. (1997) N. Engl. J. Med. 336, 1131-1141. [DOI] [PubMed] [Google Scholar]
- 18.Yang Y. & Ashwell, J. D. (2001) Apoptosis 6, 139-146. [DOI] [PubMed] [Google Scholar]
- 19.Paradis P., MacLellan, W. R., Belaguli, N. S., Schwartz, R. J. & Schneider, M. D. (1996) J. Biol. Chem. 271, 10827-10833. [DOI] [PubMed] [Google Scholar]
- 20.Rossio J. L., Esser, M. T., Suryanarayana, K., Schneider, D. K., Bess, J. W., Jr., Vasquez, G. M., Wiltrout, T. A., Chertova, E., Grimes, M. K., Sattentau, Q., et al. (1998) J. Virol. 72, 7992-8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Popik W. & Pitha, P. M. (2000) J. Virol. 74, 2558-2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu N. Q., Lossinsky, A. S., Popik, W., Li, X., Gujuluva, C., Robertson, J., Pushkarsky, T., Bukrinsky, M., Kriederman, B., Witte, M., et al. (2002) J. Virol. 76, 6689-6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li P., Nijhawan, D., Budihardjo, I., Srinivasula, S. M., Ahmad, M., Alnemri, E. S. & Wang, X. (1997) Cell 91, 479-489. [DOI] [PubMed] [Google Scholar]
- 24.Liu N. Q., Lossinsky, A. S., Popik, W., Li, X., Gujuluva, C., Kriederman, B., Roberts, J., Pushkarsky, T., Bukrinsky, M., Witte, M., et al. (2002) J. Virol. 76, 6689-6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harris A. (1988) Am. Stat. 4, 14-15. [Google Scholar]
- 26.Krown K. A., Page, M. T., Nguyen, C., Zechner, D., Gutierrez, V., Comstock, K. L., Glembotski, C. C., Quintana, P. J. & Sabbadini, R. A. (1996) J. Clin. Invest. 98, 2854-2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Keppler O. T., Yonemoto, W., Welte, F. J., Patton, K. S., Iacovides, D., Atchison, R. E., Ngo, T., Hirschberg, D. L., Speck, R. F. & Goldsmith, M. A. (2001) J. Virol. 75, 8063-8073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis C. B., Dikic, I., Unutmaz, D., Hill, C. M., Arthos, J., Siani, M. A., Thompson, D. A., Schlessinger, J. & Littman, D. R. (1997) J. Exp. Med. 186, 1793-1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Popik W., Hesselgesser, J. E. & Pitha, P. M. (1998) J. Virol. 72, 6406-6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Simmons G., Clapham, P. R., Picard, L., Offord, R. E., Rosenkilde, M. M., Schwartz, T. W., Buser, R., Wells, T. N. C. & Proudfoot, A. E. (1997) Science 276, 276-279. [DOI] [PubMed] [Google Scholar]
- 31.Mack M., Luckow, B., Nelson, P. J., Cihak, J., Simmons, G., Clapham, P. R., Signoret, N., Marsh, M., Stangassinger, M., Borlat, F., et al. (1998) J. Exp. Med. 187, 1215-1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beals C. R., Wilson, C. B. & Perlmutter, R. M. (1987) Proc. Natl. Acad. Sci. USA 84, 7886-7890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amatruda T. T., Gerard, N. P., Gerard, C. & Simon, M. I. (1993) J. Biol. Chem. 268, 10139-10144. [PubMed] [Google Scholar]
- 34.Cicala C., Arthos, J., Rubbert, A., Selig, S., Wildt, K., Cohen, O. J. & Fauci, A. S. (2000) Proc. Natl. Acad. Sci. USA 97, 1178-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saphire A. C., Bobardt, M. D., Zhang, Z., David, G. & Gallay, P. A. (2001) J. Virol. 75, 9187-9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rusnati M., Tulipano, G., Spillmann, D., Tanghetti, E., Oreste, P., Zoppetti, G., Giacca, M. & Presta, M. (1999) J. Biol. Chem. 274, 28198-28205. [DOI] [PubMed] [Google Scholar]
- 37.Sweeney E. A., Lortat-Jacob, H., Priestley, G. V., Nakamoto, B. & Papayannopoulou, T. (2002) Blood 99, 44-51. [DOI] [PubMed] [Google Scholar]
- 38.Lipshultz S., Fox, C. & Perez-Atayde, A. (1990) Am. J. Cardiol. 66, 246-250. [DOI] [PubMed] [Google Scholar]
- 39.Lipshultz S. E. (1998) N. Engl. J. Med. 339, 1153-1155. [DOI] [PubMed] [Google Scholar]
- 40.Lewis W., Grupp, I. L., Grupp, G., Hoit, B., Morris, R., Samarel, A. M., Bruggeman, L. & Klotman, P. (2000) Lab. Invest. 80, 187-197. [DOI] [PubMed] [Google Scholar]
- 41.Lewis W. (2001) Ann. N.Y. Acad. Sci. 946, 46-56. [PubMed] [Google Scholar]
- 42.Jones M. V., Bell, J. E. & Nath, A. (2000) AIDS 14, 2709-2713. [DOI] [PubMed] [Google Scholar]
- 43.Bonwetsch R., Croul, S., Richardson, M. W., Lorenzana, C., Valle, L. D., Sverstiuk, A. E., Amini, S., Morgello, S., Khalili, K. & Rappaport, J. (1999) J. Neurovirol. 5, 685-694. [DOI] [PubMed] [Google Scholar]
- 44.Mankowski J. L., Flaherty, M. T., Spelman, J. P., Hauer, D. A., Didier, P. J., Amedee, A. M., Murphey-Corb, M., Kirstein, L. M., Munoz, A., Clements, J. E., et al. (1997) J. Virol. 71, 6055-6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reid W., Sadowska, M., Denaro, F., Rao, S., Foulke, J., Jr., Hayes, N., Jones, O., Doodnauth, D., Davis, H., Sill, A., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 9271-9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roggero R., Robert-Hebmann, V., Harrington, S., Roland, J., Vergne, L., Jaleco, S., Devaux, C. & Biard-Piechaczyk, M. (2001) J. Virol. 75, 7637-7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scorziello A., Florio, T., Bajetto, A. & Schettini, G. (1998) Cell. Signalling 10, 75-84. [DOI] [PubMed] [Google Scholar]
- 48.Li C. J., Friedman, D. J., Wang, C., Metelev, V. & Pardee, A. B. (1995) Science 268, 429-431. [DOI] [PubMed] [Google Scholar]
- 49.Nath A., Haughey, N. J., Jones, M., Anderson, C., Bell, J. E. & Geiger, J. D. (2000) Ann. Neurol. 47, 186-194. [PubMed] [Google Scholar]
- 50.Ameisen J. (2001) Nat. Med. 7, 1181-1182. [DOI] [PubMed] [Google Scholar]
- 51.Geleziunas R., Xu, W., Takeda, K., Ichijo, H. & Greene, W. C. (2001) Nature 410, 834-838. [DOI] [PubMed] [Google Scholar]
- 52.Patel C. A., Mukhtar, M. & Pomerantz, R. J. (2000) J. Virol. 74, 9717-9726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bottarel F., Feito, M. J., Bragardo, M., Bonissoni, S., Buonfiglio, D., DeFranco, S., Malavasi, F., Bensi, T., Ramenghi, U. & Dianzani, U. (1999) AIDS Res. Hum. Retroviruses 15, 1255-1263. [DOI] [PubMed] [Google Scholar]
- 54.Berndt C., Mopps, B., Angermuller, S., Gierschik, P. & Krammer, P. H. (1998) Proc. Natl. Acad. Sci. USA 95, 12556-125561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vlahakis S. R., Algeciras-Schimnich, A., Bou, G., Heppelmann, C. J., Villasis-Keever, A., Collman, R. C. & Paya, C. V. (2001) J. Clin. Invest. 107, 207-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCloskey T. W., Ott, M., Tribble, E., Khan, S. A., Teichberg, S., Paul, M. O., Pahwa, S., Verdin, E. & Chirmule, N. (1997) J. Immunol. 158, 1014-1019. [PubMed] [Google Scholar]
- 57.Wolf D., Witte, V. & Lafferty, B. (2001) Nat. Med. 7, 1217-1224. [DOI] [PubMed] [Google Scholar]
- 58.Park I. W., Ullrich, C. K., Schoenberger, E., Ganju, R. K. & Groopman, J. E. (2001) J. Immunol. 167, 2766-2771. [DOI] [PubMed] [Google Scholar]
- 59.Stewart S. A., Poon, B., Song, J. Y. & Chen, I. S. (2000) J. Virol. 74, 3105-3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}