

Abstract
Presenilin 1 (PS1), presenilin 2, and nicastrin form high molecular weight complexes that are necessary for the endoproteolysis of several type 1 transmembrane proteins, including amyloid precursor protein (APP) and the Notch receptor, by apparently similar mechanisms. The cleavage of the Notch receptor at the “S3-site” releases a C-terminal cytoplasmic fragment (Notch intracellular domain) that acts as the intracellular transduction molecule for Notch activation. Missense mutations in the presenilins cause familial Alzheimer's disease by augmenting the “γ-secretase” cleavage of APP and overproducing one of the proteolytic derivatives, the Aβ peptide. Null mutations in PS1 inhibit both γ-secretase cleavage of APP and S3-site cleavage of the Notch receptor. Mice lacking PS1 function have defective Notch signaling and die perinatally with severe skeletal and brain deformities. We report here that a genetic modifier on mouse distal chromosome 1, coinciding with the locus containing Nicastrin, influences presenilin-mediated Notch S3-site cleavage and the resultant Notch phenotype without affecting presenilin-mediated APP γ-site cleavage. Two missense substitutions of residues conserved among vertebrates have been identified in nicastrin. These results indicate that Notch S3-site cleavage and APP γ-site cleavage are distinct presenilin-dependent processes and support a functional interaction between nicastrin and presenilins in vertebrates. The dissociation of Notch S3-site and APP γ-site cleavage activities will facilitate development of γ-secretase inhibitors for treatment of Alzheimer's disease.
Presenilin 1 (PS1) plays a role in facilitating both Notch signaling and amyloid precursor protein (APP) processing (1; reviewed in ref. 2). Missense mutations in PS1 are the most common cause of familial early-onset cases of Alzheimer's disease (3) and augment γ-secretase-mediated production of the amyloidogenic Aβ42 peptide (4–7). Conversely, null and dominant negative PS1 mutations cause a marked reduction of γ-secretase cleavage of APP (8, 9).
Mice lacking PS1 (PS1-null mice) die perinatally with skeletal and brain deformities (10, 11) similar to Notch and paraxis mutant animals (12–16). PS1-null mice have poorly defined segmentation with malformed vertebrae, resulting in a kinked and shortened vertebral column (10, 11). The brains of PS1-null mice have fewer neural progenitor cells, substantially thinner ventricular zones, and bilateral cerebral cavitation of the subcortical regions of the temporal lobe and ventricular zone (10). The Notch phenotypes of PS1-null mice are due to defective proteolytic release of the Notch intracellular domain (NICD) from its membrane-bound receptor (1) by S3-site cleavage, a function considered analogous to γ-secretase cleavage of APP. The Notch phenotype, therefore, has been considered an indication of PS1 activity. Although the mechanism of PS1-dependent processing of APP and Notch is unclear, both presenilin 2 (PS2) (17) and nicastrin (18) are important components for these activities.
We reported a PS1-hypomorphic mouse line (19). On a 129/Sv background, the PS1-hypomorphic line displayed a Notch phenotype (high perinatal mortality with severe skeletal malformations and impaired APP processing similar to PS1-null mice) (10, 11). However, in contrast to PS1-null animals, a small proportion of the PS1-hypomorphic mice could survive for varying periods postnatally because of residual (<1% of wild type) PS1, and manifesting Notch skeletal deformities (19). Because this mutant mouse line can survive with limiting PS1, it provides a sensitive assay to investigate potential modifiers of PS1 function.
Here we report that breeding of the PS1-hypomorphic mice onto a C57BL/6J (designated B6 hereafter) × 129/SvJ (designated 129 hereafter) F2 background resulted in the emergence of Notch phenotypes that varied widely in severity. As expected, Notch phenotype severity correlated with differential Notch S3-site cleavage. However, no association between Notch phenotype severity and γ-secretase cleavage of APP was detected, consistent with presenilin-dependent cleavages of APP and Notch being distinct activities (20, 21). A modifier of the Notch phenotype, and by association Notch S3 cleavage, was mapped to mouse chromosome 1, at the locus containing PS2 and Nicastrin. Although no differences in the coding sequence of PS2 were detected between the 129 and B6 strains, two missense substitutions were identified in Nicastrin.
These results indicate that Notch S3-site and APP γ-site cleavage are distinct presenilin-dependent processes, thereby facilitating development of γ-secretase inhibitors for AD treatment. Furthermore, that variants in nicastrin can underlie differential presenilin-dependent Notch-S3 cleavage activities supports a functional interaction between nicastrin and the presenilins.
Methods
Mouse Breeding, Genotyping, and Phenotyping.
Crosses with 129/Sv and C57BL/6 mouse strains (The Jackson Laboratory) were performed with commercial stocks. Mice were genotyped by PCR of tail DNA either after death or at weaning, as described (19). All animals were phenotyped separately by two investigators at 3 weeks for degree of Notch skeletal deformities on a scale from 1 (mild) to 4 (most severe). The same investigators were responsible for all phenotyping to eliminate any possible variability in classification.
Measurement of NICD Production.
Primary fibroblast cultures were established from 6-day-old mice (genotypes determined postmortem). At this age the severe Notch phenotypes were unmistakable by visual inspection (classified as Notch-severe) (Fig. 2a), and the mice appearing wild-type but genotyped as PS1−/−, were designated Notch-mild. Mice with an intermediate phenotype were excluded. The cultures were transfected with a pCS2 plasmid expressing myc-tagged NotchΔE as described (22, 23), labeled with [35S]methionine and cysteine for 20 min, and chased for 0 or 60 min. The cells were then lysed and the lysates immunoprecipitated with an anti-myc antibody, separated by SDS/PAGE, the gels were dried and exposed to Kodak Biomax film and the full-length NotchΔE and NICD band intensities were measured by densitometry by using NIH IMAGE 1.62. Background values were subtracted from all band intensity measurements. All data were normalized by dividing NICD band intensity by combined NICD+NotchΔE intensity. To compare across replications, relative ratios of NICD/NotchΔE+NICD were divided by the corresponding ratio obtained for PS1+/+ samples assayed concurrently. All studies were performed in a double-blinded manner. All techniques have been described (23).
Fig 2.

Production of NICD by wild-type, PS1−/− Notch-mild and Notch-severe mice. (a) Six-day-old PS1−/− Notch-mild (Upper) and Notch-severe (Lower) mice showing reduced size and grossly deformed axial skeletons of the latter. (b) Cells from 6-day-old Notch-mild mice produced more NICD than cells from Notch-severe mice. (c) Comparison of relative NICD production (ratio of NICD/NICD+NotchΔE) from cells Notch-mild (n = 12) and Notch-severe (n = 18) mice shows a significant difference (P < 0.02) between the groups.
Aβ40 and Aβ42 ELISAs.
ELISAs to measure brain Aβ40 and Aβ42 levels were performed as described (19). Aβ assays could not be reliably performed on mice that died prematurely because of postmortem tissue degradation.
Western Blot Analysis.
Total mouse brain was prepared as described (19). After transfer to poly(vinylidene difluoride) membrane (Millipore), the Western blots were hybridized to polyclonal antibody 369 for APP-CTF and FL-APP and the anti-PS2 C-terminal polyclonal antibody G2L (24).
Histological Studies.
All histological materials and methods have been described (19).
Genome Scan and Microsatellite Typing.
DNA from Notch-severe and from Notch-mild B6 × 129 F2 PS1−/− mice was analyzed by typing of SSLP (simple sequence length polymorphism) markers distributed throughout the genome. Three to five markers distributed from the centromere to the telomere were typed for each chromosome for a total of 86 markers polymorphic (distinct product sizes) between the 129 and B6 strains. The concentration of DNA of each sample was determined and equal amounts of DNA from the 29 Notch-severe and 39 Notch-mild mice used in the genome scan were combined to prepare two pools that were assayed as described (25, 26). Linkage was suggested by reciprocal unequal allelic distribution between the two sample pools. For candidate linkages, the individual DNA samples were genotyped to confirm any significant deviation from random segregation.
Sequencing of Candidate Genes.
Brain RNA from B6 and 129 wild-type mice was made into cDNA by using the SuperScript II RNase H-Reverse Transcriptase (Life Technologies, Grand Island, NY) and amplified by PCR. The products containing the coding regions of PS2 and Nicastrin from the two strains were sequenced directly from PCR by using the ThermoSequenase Cycle Sequencing Kit (Pharmacia/Amersham) and primers PS2-V1 (5′-CTTCTGGAACTCCGGTTTTG-3′), PS2-V2 (5′-CAGGAGCATCTGTTTATTGGT-3′), PS2-V3 (5′-GGAGTCTTCTTCCATCTCTG-3′), PS2-V4 (5′-ACTGAGGATGCTGGTGGAAA-3′), PAMP-1 (5′-GAGGCAACATGGCTACGACT-3′), PAMP-2 (5′-CAGTCTCCTCAGGACAACTTC-3′), PAMP-3 (5′-GAGAACATCGACTCCTTCGTG-3′) and PAMP-4 (5′-AAGCAGGCCCAGAGACAGT-3′). Differences were confirmed on independent samples.
Statistical Analysis.
Comparisons of two sample populations were performed by the Student's t test. Significance of nonrandom segregation of genotypes was analyzed by χ2 statistics. Linkage analyses were performed by MAPMANAGER QT (ref. 27; Kenneth Manly, http://mapmgr.roswellpark.org/mmqt.html).
Results
Breeding.
The previously described 129 PS1-hypomorphic mice (19) were crossed with C57BL/6J (B6) mice to generate F1 hybrids heterozygous for the PS1 disruption (herein designated PS1+/−). PS1+/− B6 × 129 F1 mice were viable, fertile, and indistinguishable from their wild-type sibs. Intercrossing of F1 hybrids resulted in homozygous (PS1−/−) B6 × 129 F2 mice.
Viability and Phenotype of B6 × 129 F2 Ps1−/− Mice.
As described (19), only 7% (17/230) of surviving 129 mice were PS1−/−, constituting ≈30% of the homozygous mutant mice expected by Mendelian segregation. In contrast, 16% (148/943, 63% of the number expected) of the F2 B6 × 129 animals surviving postnatal were PS1−/−. Sixteen of the PS1−/− animals died before weaning (3 weeks of age) with severe skeletal deformities similar to that described for the 129 mice (19) and others (10, 11). The surviving F2 B6 × 129 Ps1−/− mice presented a wide spectrum of Notch axial skeletal deformities.
The surviving B6 × 129 F2 PS1−/− animals were phenotyped at 3 weeks for extent of Notch skeletal deformities on a scale from 1 (mild, indistinguishable from wild type) to 4 [severe, similar to 129 mice reported (19)]. The Notch phenotype 4 animals (n = 29; 22% of surviving PS1−/− animals, hereafter referred to as “Notch-severe”) were identified by severe vertebral column deformation (>50% reduction in length and at least four definable kinks resulting from malformed vertebrae), by gross physical examination (Fig. 1). Like the previously described 129 PS1−/− mice (19), the Notch-severe animals were significantly smaller than their wild-type sibs [3 weeks of age; PS1+/+, 14.8 ± 0.9 g vs. PS1−/−, Notch-severe, 12.2 ± 0.7 g (mean ± SEM of four males and four females of each group)]. Extensive vertebral body malformations were present throughout their spinal columns and 23 had paralysis of their hind limbs due to spinal cord compression and transection, as described (19). As with the 129 PS1−/− mice, the brains of the Notch-severe animals appeared histopathologically normal, although their cortical plates and lamination were slightly thinner (PS1+/+ and PS1−/−; 265.66 ± 4.79 and 242.81 ± 7.62 mM ± SD, respectively) than their wild-type sibs. The 16 PS1−/− animals with severe skeletal deformities (similar to the surviving Notch-severe animals) that died before 3 weeks were also classified as Notch-severe (for a total n = 45 animals) for the genetic studies.
Fig 1.
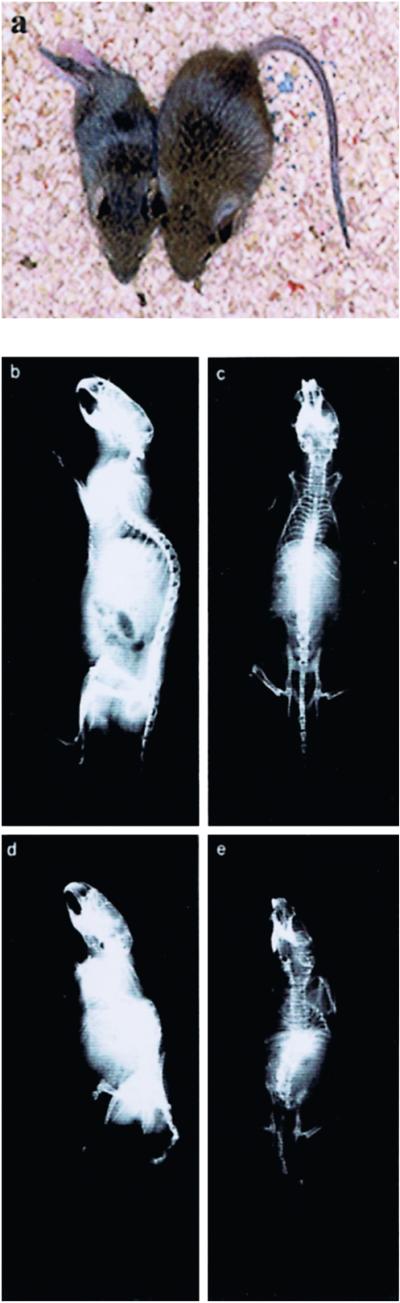
Gross phenotype of Notch-severe and Notch-mild PS1−/− mice. (a) Notch-severe (Left) and Notch-mild (Right) B6 × 129 F2 PS1−/− mice at 5 weeks of age. Identical to previously reported 129 PS1−/− animals, the Notch-severe mice were significantly smaller than their Notch-mild or wild-type sibs and presented a grossly deformed vertebral column. The represented Notch-severe animal also manifested back-end paralysis commonly observed in this group. (b and d) Ventral and lateral x-rays, respectively, of a 6-week-old Notch-severe mouse alongside its Notch-mild sib (c and e).
In contrast to the Notch-severe cohort, the B6 × 129 F2 PS1−/− animals scored as phenotype 1 (n = 39) (30% of surviving Ps1−/− animals, hereafter referred to as “Notch-mild”) were indistinguishable from wild type (Fig. 1). The size of these animals was similar to their wild-type sibs [3 weeks of age; PS1+/+, 14.8 ± 0.9 g vs. PS1−/−, Notch-mild, 13.9 ± 1.1 g (mean ± SEM of four males and four females of each group)], and their vertebral columns were histologically normal, having only rare, mildly misshapen vertebral bodies that did not alter gross vertebral column morphology (Fig. 1). The brains of representative Notch-mild mice were histologically normal, with cortical plate thickness similar to the wild-type sibs (PS1+/+ and PS1−/−, 265.66 ± 4.79 and 266.73 ± 59.27 μM ± SD, respectively). The remaining PS1−/− animals (scores 2 and 3) had intermediate phenotypes and were not included in further studies.
Notch S3-Site Cleavage.
The differential phenotypic severity between the Notch-mild and Notch-severe PS1−/− animals could reflect differences in Notch S3-site cleavage [compromised in the absence of PS1 (1, 28)], or downstream elements of Notch signaling. To resolve this, capacity for NICD production in Notch-severe and Notch-mild animals was examined (Fig. 2a). Cells from Notch-mild mice produced significantly more NICD (42.7 ± 2.8% of wild type, n = 12) than did cells from Notch-severe mice (34.3 ± 1.9% of wild type, n = 18, P < 0.02) (Fig. 2 b and c). Because studies have shown that within this range small changes in NICD production can significantly compromise Notch signaling efficiency and therefore have profound effects on the Notch phenotype (22, 29), we concluded that the difference in Notch S3-cleavage between Notch-mild and Notch-severe conferred the phenotype distinction.
APP γ-Secretase Activity.
In contrast to the distinctions in Notch S3-site cleavage, no marked differences existed between Notch-severe and Notch-mild animals in γ-secretase cleavage of APP. This conclusion was robust, regardless of whether γ-secretase activity was assessed by accumulation of its substrate [i.e., APP-C83 (α-secretase stub) and APP-C99 (β-secretase stub)], or reduction in γ-secretase products (i.e., Aβ40 and Aβ42) (n = 4 mice per group). Both APP-C83 and APP-C99 fragments were barely detectable in PS1+/+ brain, but were equally elevated in Notch-severe and Notch-mild brains (Fig. 3a). Similarly, Aβ40 and Aβ42 were abundant in the brains of PS1+/+ mice (8.6 ± 0.35 and 3.0 ± 0.05 fmol/mg protein ± SEM, respectively) and PS1+/− mice (6.1 ± 0.6 and 2.4 ± 0.5 fmol/mg protein ± SEM, respectively) (Fig. 3b). In contrast, similar to PS1-null mice (8), the Notch-severe and Notch-mild brains had Aβ40 levels below detection, and relative to PS1+/+ brains (6.1 ± 0.6 fmol/mg protein), Aβ42 levels were dramatically reduced in the Notch-severe (0.66 ± 0.09 fmol/mg protein ± SEM) and Notch-mild samples (0.44 ± 0.06 fmol/mg protein ± SEM) (Notch-severe versus Notch-mild, P < 0.1)(Fig. 3b). Thus, although no change occurred in γ-secretase40, a slight reduction in γ-secretase42 activity in the Notch-mild compared with Notch-severe and PS1−/− brains was observed. Similar studies could not be performed on the animals that died before 3 weeks because of deterioration of brain tissue.
Fig 3.

APP processing by wild-type, PS1−/− Notch-mild and Notch-severe mice. (a) Western blot of wild-type, Notch-severe, and Notch-mild B6 × 129 F2 PS1−/− brain protein lysates incubated with an APP-specific antibody. The result shows equal amounts of full-length APP (FL-APP) among all three brains. Although no APP C-terminal fragment (CTF-APP) was observed in the wild-type brains, it was equally evident among all Notch-severe and Notch-mild brains. (b) Results of ELISAs for Aβ40 and Aβ42 from PS1+/+, PS1+/−, and Notch-severe and Notch-mild PS1−/− brains.
Genetic Studies.
Because 129 mice displayed a Notch-severe phenotype (19), the allele(s) of the modifier(s) conferring the Notch-mild phenotype were likely from by the B6 strain. To map the location(s) of this modifier(s), a genome scan of the surviving Notch-severe (n = 29) and Notch-mild (n = 39) mice by pooled sample PCR (25, 26) was performed. The only locus producing significant B6 vs. 129 allelic distortions was at distal chromosome 1 (marker D1Mit459 at 102 cM). In the Notch-severe group, individual genotypes for D1Mit459 showed a nonrandom excess of 129 alleles (B6/B6:B6/129:129/129 = 2:14:13; P < 0.02, χ2), whereas in the Notch-mild group an excess of B6 alleles (B6/B6:B6/129:129/129 = 16:18:5; P < 0.005, χ2). No significant deviation from expected was observed between the groups for the proximal chromosome 1 marker D1Mit65 at position 8.4-cM (χ2, P < 0.5 and 0.25).
Localization of the modifier was refined with markers spanning mid to distal chromosome 1 (D1Mit215 (47-cM), D1Mit306 (58.7-cM), D1Mit501 (79-cM), D1Mit15 (87.9-cM), D1Mit206 (95.8-cM), D1Mit150 (100-cM), D1Mit459 (102-cM), and D1Mit293 (109.6-cM) (www.informatics.jax.org) (Fig. 4a). This analysis placed the confidence interval (LODMax-1) between D1Mit15 and the telomere (87.9–112 cM or 168–198 Mb from the centromere) with a maximum LOD score (z = 6.1) at marker D1Mit150 at 178 Mb (Fig. 4a). The closely flanking markers D1Mit206 and D1Mit459 gave slightly lower LOD scores of 5.8 and 5.9, respectively; however, these values were based on a smaller database because several of the DNA samples could not be genotyped for these markers at the later date because of degradation. Nevertheless, assuming the D1Mit206 and D1Mit459 genotypes for the missing samples corresponded to D1Mit150 (no recombinants were detected between these markers among the samples that could be genotyped) D1Mit206 and D1Mit459 would also reside at the peak LOD score.
Fig 4.

Linkage of the modifier to chromosome 1. (a) A genome scan of the Notch-severe and Notch-mild mice mapped a modifier of the Notch phenotype to distal chromosome 1. Genetic analysis of the locus showed the maximum LOD score was 6.1, identified by D1Mit150 at position 100-cM. (b) Physical definition of the modifier confidence interval showing placement of Nicastrin and PS2.
Characterization of Modifier Candidates.
On the basis of functions of known genes within the confidence interval (from the Celera Mouse Genome Database (www.celera.com), only PS2 and Nicastrin were considered strong candidates for the modifier because of their direct participation in Notch S3-site cleavage. Similar to PS1, missense mutations in PS2 modulate Aβ secretion to cause AD. In addition, PS2, like PS1, complements for loss of Sel-12 in Notch signaling (30, 31), and mice lacking both PS1 and PS2 have more severe Notch signaling deficits (32) and absent γ-secretase cleavage of APP (33), compared with deficiency of PS1 alone. Synteny with the human locus placed murine PS2 on distal chromosome 1 and alignment with the Celera Mouse Genome Database localized it to 184 Mb from the centromere, between markers D1Mit150 and D1Mit459, within the confidence interval (Fig. 4b). Analysis of the PS2 coding sequence between the 129 and B6 strains detected no variations. Moreover, Western blot analysis of brains of 4- to 6-week-old mice showed no detectable difference in PS2 protein levels or processing among Notch-severe and Notch-mild mice (Fig. 5). However, all PS1−/− animals showed elevated levels of PS2 protein compared with their wild-type sibs (Fig. 5), consistent with its coordinate regulation with PS1 (34). A similar investigation at representative Notch developmental stages was not possible because of insufficient tissue and inability to distinguish phenotypes. Nevertheless, on the basis of these results we concluded that PS2 was unlikely to be the modifier.
Fig 5.

Quantitation of PS2 protein. The same Western blot of wild-type, Notch-severe, and Notch-mild B6 × 129 F2 PS1−/− brain lysates shown in Fig. 3a was incubated with a PS2 C-terminal antibody. A signal corresponding to the PS2-CTF is seen in the wild-type lanes, whereas a more intense signal is evident in the lanes corresponding to the mutant animals. No consistent difference is observed between the Notch-severe and Notch-mild animals. Hybridization with the Rab8 antibody is shown for control of protein loading (identical to that in Fig. 3a).
Nicastrin is a component of the presenilin complexes and plays an important role in Notch S3 and APP cleavage (18, 23, 35–42). An EST corresponding to Nicastrin was previously mapped to 95 cM of mouse chromosome 1 (43). We confirmed its localization to the peak LOD interval at position 172 Mb, adjacent to D1Mit206 (Fig. 4b) through the Celera Database. Sequencing of the nicastrin-coding region from the 129 and B6 strains identified two missense substitutions: residue 21 (129 = Phe-21; B6 = Ser-21) corresponding to a N-terminal hydrophobic domain (18), and residue 678 within the putative membrane-spanning domain (129 = Iso678; B6 = Thr-678) (44).
Discussion
We describe the mapping of a modifier of the PS1-deficiency Notch phenotype to distal chromosome 1 in mice. The B6 allele of this modifier results in markedly milder axial skeletal deformities than its 129 counterpart. Although the mode of inheritance of this modifier is unknown, several lines of reasoning argue against it being a simple Mendelian trait. First, the ratios of Notch-severe and Notch-mild mice are not consistent with a simple dominant or recessive model. Second, allelic segregation of the modifier locus is not completely associated with phenotype in both the Notch-severe and Notch-mild groups. Third, the continuous spectrum of phenotypic variability is unlikely the result of a single genetic contribution. The data, therefore, support a more complex genetic model for the modifier, involving multiple loci and/or epigenetic factors. Identification of other modifier loci and investigation of the Notch phenotype on a congenic B6 background is required to gain further insight into the genetic model.
The mechanism of the Notch phenotype modifier is through differential Notch S3-site cleavage. Based on previous studies, the observed difference in NICD production is sufficient to account for the distinct Notch phenotypes, because within the range observed between the Notch-mild and Notch-severe mice, small changes in NICD production can significantly compromise Notch signaling and have a profound effect on the Notch phenotype (22, 29). For example, by using the HES-luciferase reporter assay for NICD production, wild-type Notch supports normal signaling and the equivalent wild-type NotchΔE construct generates ≈60% relative HES-Luciferase activation, whereas the hypofunctional V1744L Notch mutant causes an embryonic lethal Notch phenotype and the equivalent V1744L NotchΔE mutant generates ≈40% relative HES-Luciferase activation (22, 29). These results indicate that subtle differences in NICD at this level have profound effects on Notch signaling.
The PS2 and nicastrin genes, which map to the critical locus, are considered strong candidates for the modifier. Although no sequence differences in PS2 between the B6 and 129 strains were identified, this finding does not conclusively exclude it from consideration as the modifier. It could be argued that strain-specific differences in PS2 regulatory sequences can cause variations in its spatial-temporal expression restricted either to specific developmental stages or subpopulation of progenitor cells. However, this limitation of the modifier effect is not congruent with the observation that significant differences in Notch-S3 cleavage were detected in postnatal fibroblast. Thus, we concluded that PS2 is unlikely to be the modifier.
Nicastrin, a type 1 membrane protein, interacts with both PS1 and PS2 to facilitate APP and Notch S3 cleavage (18, 23, 35–42). Two missense substitutions were identified in residues of nicastrin that correspond to a N-terminal hydrophobic domain that may act as a signal peptide or membrane-associated domain (44), and within its putative transmembrane domain (18). The Ile-678 residue (within the transmembrane domain) is conserved in humans and rodents, but not in invertebrates, and Ser-21 (within the N-terminal hydrophobic domain) is conserved in humans, rodents and Drosophila melanogaster. Substitutions in one or both of these residues might slightly alter nicastrin function. For instance, the Phe-21 variant in the Notch-severe mice might subtly alter interactions of nicastrin with the other components of the PS1 complex. With limiting PS1, this subtle alteration could further compromise the formation of functional PS1/nicastrin complexes, leading to the differences in Notch cleavage between the Notch-severe and Notch-mild phenotypes. However, because this modifier confers only small effects on NICD production, its confirmation and mechanistic elucidation needs to be studied in mice or cells lacking endogenous nicastrin.
Of particular importance, our results indicate a disparate effect on S3-cleavage of Notch (the Notch-mild B6 allele increased S3-cleavage) versus γ-secretase cleavage of APP (the Notch-mild allele caused no change in γ-secretase40, and a nonsignificant reduction in γ-secretase42 activity) by the modifier. These results provide genetic support for the concept that S3-cleavage of Notch (≈2–5 residues inside the cytoplasmic face of the membrane) and APP γ-secretase cleavage (in the middle of the transmembrane domain) are distinct presenilin-dependent activities (2, 21, 45, 46). Previous data have shown that missense substitutions elsewhere in nicastrin (i.e., in the 312–369 DYIGS domain) modulate Aβ production more profoundly than S3-cleavage of Notch (18, 23). Moreover, haplotypes around nicastrin associate with increased risk for AD (47). Two conclusions can be drawn from these observations. First, mutations in different domains of nicastrin have differential effects on S3- and γ-cleavage, implying their distinct functional properties, and presumably interactions with different partners. Second, therapeutic agents might be developed that selectively affect γ-secretase over Notch S3-cleavage.
Acknowledgments
We thank Dr. Marc Mercken (Johnson and Johnson Pharmaceutical Research and Development/Janssen Pharmaceutica) for use of his anti-Aβ mAbs. We acknowledge the support of the Canadian Institutes of Health Research (to R.R. and P.S.G.-H.), the Howard Hughes Medical Research Institute, the Canadian Genetic Diseases Network (to P.S.G.-H.), the Alzheimer's Society of Ontario, and National Institutes on Aging Grant AG 17617 (to R.A.N. and P.M.M.).
Abbreviations
PS1, presenilin 1
PS2 presenilin 2, APP, amyloid precursor protein
NICD, Notch intracellular domain
AD, Alzheimer's disease
Aβ, amyloid-beta
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.De Strooper B., Annaert, W., Cupers, P., Saftig, P., Craessaerts, K., Mumm, J. S., Schroeter, E. H., Schrijvers, V., Wolfe, M. S., Ray, W. J., et al. (1999) Nature 398, 518-522. [DOI] [PubMed] [Google Scholar]
- 2.Sisodia S. S. & St George-Hyslop, P. H. (2002) Nat. Rev. Neurosci. 3, 281-290. [DOI] [PubMed] [Google Scholar]
- 3.Sherrington R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., Chi, H., Lin, C., Li, G., Holman, K., et al. (1995) Nature 375, 754-760. [DOI] [PubMed] [Google Scholar]
- 4.Borchelt D. R., Thinakaran, G., Eckman, C. B., Lee, M. K., Davenport, F., Ratovitsky, T., Prada, C. M., Kim, G., Seekins, S., Yager, D., et al. (1996) Neuron 17, 1005-1013. [DOI] [PubMed] [Google Scholar]
- 5.Duff K., Eckman, C., Zehr, C., Yu, X., Prada, C. M., Perez-tur, J., Hutton, M., Buee, L., Harigaya, Y., Yager, D., et al. (1996) Nature 383, 710-713. [DOI] [PubMed] [Google Scholar]
- 6.Scheuner D., Eckman, C., Jensen, M., Song, X., Citron, M., Suzuki, N., Bird, T. D., Hardy, J., Hutton, M., Kukull, W., et al. (1996) Nat. Med. 2, 864-870. [DOI] [PubMed] [Google Scholar]
- 7.Citron M., Westaway, D., Xia, W., Carlson, G., Diehl, T., Levesque, G., Johnson-Wood, K., Lee, M., Seubert, P., Davis, A., et al. (1997) Nat. Med. 3, 67-72. [DOI] [PubMed] [Google Scholar]
- 8.De Strooper B., Saftig, P., Craessaerts, K., Vanderstichele, H., Guhde, G., Annaert, W., Von Figura, K. & Van Leuven, F. (1998) Nature 391, 387-390. [DOI] [PubMed] [Google Scholar]
- 9.Wolfe M. S., Xia, W., Ostaszewski, B. L., Diehl, T. S., Kimberly, W. T. & Selkoe, D. J. (1999) Nature 398, 513-517. [DOI] [PubMed] [Google Scholar]
- 10.Shen J., Bronson, R. T., Chen, D. F., Xia, W., Selkoe, D. J. & Tonegawa, S. (1997) Cell 89, 629-639. [DOI] [PubMed] [Google Scholar]
- 11.Wong P. C., Zheng, H., Chen, H., Becher, M. W., Sirinathsinghji, D. J., Trumbauer, M. E., Chen, H. Y., Price, D. L., Van der Ploeg, L. H. & Sisodia, S. S. (1997) Nature 387, 288-292. [DOI] [PubMed] [Google Scholar]
- 12.Kusumi K., Sun, E. S., Kerrebrock, A. W., Bronson, R. T., Chi, D. C., Bulotsky, M. S., Spencer, J. B., Birren, B. W., Frankel, W. N. & Lander, E. S. (1998) Nat. Genet. 19, 274-278. [DOI] [PubMed] [Google Scholar]
- 13.Hrabe de Angelis M., McIntyre, J., 2nd & Gossler, A. (1997) Nature 386, 717-721. [DOI] [PubMed] [Google Scholar]
- 14.Swiatek P. J., Lindsell, C. E., del Amo, F. F., Weinmaster, G. & Gridley, T. (1994) Genes Dev. 8, 707-719. [DOI] [PubMed] [Google Scholar]
- 15.Conlon R. A., Reaume, A. G. & Rossant, J. (1995) Development (Cambridge, U.K.) 121, 1533-1545. [DOI] [PubMed] [Google Scholar]
- 16.Burgess R., Rawls, A., Brown, D., Bradley, A. & Olson, E. N. (1996) Nature 384, 570-573. [DOI] [PubMed] [Google Scholar]
- 17.Rogaev E. I., Sherrington, R., Rogaeva, E. A., Levesque, G., Ikeda, M., Liang, Y., Chi, H., Lin, C., Holman, K., Tsuda, T., et al. (1995) Nature 376, 775-778. [DOI] [PubMed] [Google Scholar]
- 18.Yu G., Nishimura, M., Arawaka, S., Levitan, D., Zhang, L., Tandon, A., Song, Y. Q., Rogaeva, E., Chen, F., Kawarai, T., et al. (2000) Nature 407, 48-54. [DOI] [PubMed] [Google Scholar]
- 19.Rozmahel R., Huang, J., Chen, F., Liang, Y., Nguyen, V., Ikeda, M., Levesque, G., Yu, G., Nishimura, M., Mathews, P., et al. (2002) Neurobiol. Aging 23, 187-194. [DOI] [PubMed] [Google Scholar]
- 20.Kulic L., Walter, J., Multhaup, G., Teplow, D. B., Baumeister, R., Romig, H., Capell, A., Steiner, H. & Haass, C. (2000) Proc. Natl. Acad. Sci. USA 97, 5913-5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petit A., St George-Hyslop, P., Fraser, P. & Checler, F. (2002) Biochem. Biophys. Res. Commun. 290, 1408-1410. [DOI] [PubMed] [Google Scholar]
- 22.Schroeter E. H., Kisslinger, J. A. & Kopan, R. (1998) Nature 393, 382-386. [DOI] [PubMed] [Google Scholar]
- 23.Chen F., Yu, G., Arawaka, S., Nishimura, M., Kawarai, T., Yu, H., Tandon, A., Supala, A., Song, Y. Q., Rogaeva, E., et al. (2001) Nat. Cell Biol. 3, 751-754. [DOI] [PubMed] [Google Scholar]
- 24.Tomita T., Takikawa, R., Koyama, A., Morohashi, Y., Takasugi, N., Saido, T. C., Maruyama, K. & Iwatsubo, T. (1999) J. Neurosci. 19, 10627-10634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Asada Y., Varnum, D. S., Frankel, W. N. & Nadeau, J. H. (1994) Nat. Genet. 6, 363-368. [DOI] [PubMed] [Google Scholar]
- 26.Rozmahel R., Wilschanski, M., Matin, A., Plyte, S., Oliver, M., Auerbach, W., Moore, A., Forstner, J., Durie, P., Nadeau, J., et al. (1996) Nat. Genet. 12, 280-287. [DOI] [PubMed] [Google Scholar]
- 27.Manly K. F. & Olson, J. M. (1999) Mamm. Genome 10, 327-334. [DOI] [PubMed] [Google Scholar]
- 28.Ray W. J., Yao, M., Mumm, J., Schroeter, E. H., Saftig, P., Wolfe, M., Selkoe, D. J., Kopan, R. & Goate, A. M. (1999) J. Biol. Chem. 274, 36801-36807. [DOI] [PubMed] [Google Scholar]
- 29.Huppert S. S., Le, A., Schroeter, E. H., Mumm, J. S., Saxena, M. T., Milner, L. A. & Kopan, R. (2000) Nature 405, 966-970. [DOI] [PubMed] [Google Scholar]
- 30.Levitan D., Doyle, T. G., Brousseau, D., Lee, M. K., Thinakaran, G., Slunt, H. H., Sisodia, S. S. & Greenwald, I. (1996) Proc. Natl. Acad. Sci. USA 93, 14940-14944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baumeister R., Leimer, U., Zweckbronner, I., Jakubek, C., Grunberg, J. & Haass, C. (1997) Genes Funct. 1, 149-159. [DOI] [PubMed] [Google Scholar]
- 32.Donoviel D. B., Hadjantonakis, A. K., Ikeda, M., Zheng, H., Hyslop, P. S. & Bernstein, A. (1999) Genes Dev. 13, 2801-2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herreman A., Serneels, L., Annaert, W., Collen, D., Schoonjans, L. & De Strooper, B. (2000) Nat. Cell Biol. 2, 461-462. [DOI] [PubMed] [Google Scholar]
- 34.Thinakaran G., Harris, C. L., Ratovitski, T., Davenport, F., Slunt, H. H., Price, D. L., Borchelt, D. R. & Sisodia, S. S. (1997) J. Biol. Chem. 272, 28415-28422. [DOI] [PubMed] [Google Scholar]
- 35.Chung H. M. & Struhl, G. (2001) Nat. Cell Biol. 3, 1129-1132. [DOI] [PubMed] [Google Scholar]
- 36.Esler W. P., Kimberly, W. T., Ostaszewski, B. L., Ye, W., Diehl, T. S., Selkoe, D. J. & Wolfe, M. S. (2002) Proc. Natl. Acad. Sci. USA 99, 2720-2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goutte C., Tsunozaki, M., Hale, V. A. & Priess, J. R. (2002) Proc. Natl. Acad. Sci. USA 99, 775-779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu Y., Ye, Y. & Fortini, M. E. (2002) Dev. Cell 2, 69-78. [DOI] [PubMed] [Google Scholar]
- 39.Kopan R. & Goate, A. (2002) Neuron 33, 321-324. [DOI] [PubMed] [Google Scholar]
- 40.Leem J. Y., Vijayan, S., Han, P., Cai, D., Machura, M., Lopes, K. O., Veselits, M. L., Xu, H. & Thinakaran, G. (2002) J. Biol. Chem. 277, 19236-19240. [DOI] [PubMed] [Google Scholar]
- 41.Levitan D., Yu, G., St George-Hyslop, P. & Goutte, C. (2001) Dev. Biol. 240, 654-661. [DOI] [PubMed] [Google Scholar]
- 42.Lopez-Schier H. & St Johnston, D. (2002) Dev. Cell 2, 79-89. [DOI] [PubMed] [Google Scholar]
- 43.Underhill D. A., Mullick, A., Groulx, N., Beatty, B. G. & Gros, P. (1999) Genomics 55, 185-193. [DOI] [PubMed] [Google Scholar]
- 44.Yang, D.-S., Tandon, A., Chen, F., Yu, G., Yu, H., Arawaka, S., Hasegawa, H., Duthie, M., Schmidt, S., Nixon, R. A., et al. (2002) J. Biol. Chem. M110871200. [DOI] [PubMed]
- 45.Ikeuchi T. & Sisodia, S. S. (2002) Neuromol. Med. 1, 43-54. [DOI] [PubMed] [Google Scholar]
- 46.Yu C., Kim, S. H., Ikeuchi, T., Xu, H., Gasparini, L., Wang, R. & Sisodia, S. S. (2001) J. Biol. Chem. 276, 43756-43760. [DOI] [PubMed] [Google Scholar]
- 47.Dermaut B., Theuns, J., Sleegers, K., Hasegawa, H., Van den Broeck, M., Vennekens, K., Corsmit, E., St George-Hyslop, P., Cruts, M., van Duijn, C. M. & Van Broeckhoven, C. (2002) Am. J. Hum. Genet. 70, 1568-15674. [DOI] [PMC free article] [PubMed] [Google Scholar]