


Abstract
A DNA fragment d(GCGAAAGCT), known to adopt a stable mini-hairpin structure in solution, has been crystallized in the space group I4122 with the unit-cell dimensions a = b = 53.4 Å and c = 54.0 Å, and the crystal structure has been determined at 2.5 Å resolution. The four nucleotide residues CGAA of the first half of the oligomer form a parallel duplex with another half through the homo base pairs, C2:C2+ (singly-protonated between the Watson– Crick sites), G3:G3 (between the minor groove sites), A4:A4 (between the major groove sites) and A5:A5 (between the Watson–Crick sites). The two strands remaining in the half of the parallel duplex are split away in different directions, and they pair in an anti-parallel B-form duplex with the second half extending from a neighboring parallel duplex, so that an infinite column is formed in a head-to-tail fashion along the c-axis. It seems that a hexa-ammine cobalt cation supports such a branched and bent conformation of the oligomer. One end of the parallel duplex is stacked on the corresponding end of the adjacent parallel duplex; between them, the guanine base of the first residue is stacked on the fourth ribose of another duplex.
INTRODUCTION
Since Watson and Crick (1) proposed a structural model for the double helix of DNA from their interpretation of available fiber-diffraction data, DNA has been assumed to always adopt the B-form conformation in vivo, and this assumption has been verified by many X-ray analyses of DNA fragments. An alternative conformation is the A-form (2), which is observed predominantly in RNA structures, but DNA can also have this conformation at low-humidity (3). In either form, the two DNA strands are positioned to form a right-handed helix. Another discovery was left-handed Z-DNA, which was found to form in alternating purine–pyrimidine sequences, (CG)n (4,5). This conformation is also involved in some biological systems (6). All three major duplex structures, however, consist of two sugar-phosphate backbones aligned in anti-parallel fashion with one strand in the 5′→3′ and the other strand in the 3′→5′ orientation. These two strands are bound to each other through Watson–Crick base pairings. In the case of RNAs, too, the alignment of the two chains is anti-parallel.
Homo pairs formed between the same kinds of bases were found in the X-ray crystal structures of d(CG) (7) and d(CA) (8) at acidic pH, the two strands being aligned in parallel fashion with the same strand polarity. These findings stimulated extensive structural studies on parallel duplexes. Robinson et al. (9,10) showed from NMR studies that the d(CGATCG) sequence forms a B-form anti-parallel duplex at neutral pH, as well as a parallel duplex with homo base pairs at low pH (<5), and that the d(CGA) sequence forms such a motif. The sequence d(TCGA) also forms a parallel-stranded duplex, but the first T residue does not interact directly with another T (11). The parallel-aligned strands have been observed in the structures of DNA duplexes with reverse Watson–Crick A:T base pairs (12), triplexes (13), tetraplexes (14) and the i-motif (15,16).
In general, the parallel duplex structure is difficult to find in natural DNA, because every system in vivo is established by using the Watson–Crick-type anti-parallel duplex between complementary strands. In some cases, as reported by Tchurikov and co-workers (17–19), however, parallel duplex formation is possible for specific functions in local regions of DNA. Such a parallel duplex has been also detected in Escherichia coli mRNA (20). To understand the biological functions of parallel duplexes, it is necessary to clarify their structural properties. Despite various indirect evidence for parallel-stranded duplexes, detailed structures have not yet been fully investigated in the crystalline state.
From extensive studies on the nucleotide sequence d(GCGAAAGC), we found that the DNA fragment has extraordinary properties with (i) abnormal mobility in electrophoresis (21), (ii) high thermo-stability (22), (iii) an unusual CD spectrum different from expected patterns (22), and (iv) robustness against nuclease digestion (23). The biological functions of such a fragment were discussed for G4 phage (24). For this specific sequence, a mini-hairpin structure was postulated from NMR and CD spectroscopy (22). The present nonamer, containing a thymidine residue at the ninth position, shows very similar properties but a higher thermo-stability when compared with the octamer (22), and was also expected to have a similar hairpin structure in solution (22). In contrast, the crystal structures of the halogeno-derivatives of the nonamer containing 5-bromo- or 5-iodo-cytidines as the second residue show a zipper-like duplex (25,26). To investigate the structural versatility of such a specific sequence in relation to its physicochemical properties, we determined the crystal structure of native d(GCGAAAGCT) by X-ray diffraction, and found not only that this sequence adopts another type of structure, but also, surprisingly, that half of the sequence forms a parallel duplex, whereas the other half forms an anti-parallel duplex. Here we describe the structural details of the novel parallel duplex and compare its conformation with the standard anti-parallel duplex.
MATERIALS AND METHODS
Synthesis and crystallization
A DNA oligomer with the sequence d(GCGAAAGCT) was synthesized on a DNA synthesizer. The nonamer was purified by HPLC, and converted to the Na+ salt on a cation-exchange column. Finally, the cation was removed by gel-filtration chromatography. Crystallization conditions were surveyed at room temperature by the hanging-drop vapor diffusion method with a screening kit reported by Berger et al. (27) for nucleic acids, each droplet (4 µl initial volume) being equilibrated with 700 µl of reservoir solution. Although some crystals only appeared under conditions containing Co(NH3)6Cl3 with Mg acetate and NaCl, their size was too small for X-ray analysis. We found that adding 2,6-diaminopyridine and a small quantity of n-decanoyl-N-methylglucamide, purchased from DOJINDO Laboratories Co., Ltd, facilitated crystal growth. After further optimization of crystallization conditions, suitable crystals for X-ray experiments were obtained at room temperature in the following conditions. A droplet was prepared by merging 2 µl of a 3 mM DNA solution including 24 mM 2,6-diaminopyridine and 2 µl of a reservoir solution (pH 6.0) containing 20 mM MgCl2, 80 mM NaCl, 30 mM Co(NH3)6Cl3, 25% 2-methyl-2,4-pentanediol and 0.17% n-decanoyl-N-methylglucamide in 40 mM Na-cacodylate buffer. Crystals were grown up to 0.15 × 0.15 mm in size for 2 weeks. Fresh crystals were mounted in nylon cryoloops (Hampton Research) with the reservoir solution containing 30% (v/v) 2-methyl-2,4-pentanediol as a cryoprotectant and stocked in liquid nitrogen.
Data collection
For MAD phasing using the anomalous scattering of cobalt atoms, X-ray data were taken at BL18b of Photon Factory in Tsukuba. Three different wavelengths, 1.00, 1.6049 and 1.6054 Å, were employed, based on XAFS measurements. A crystal specimen was cooled at 100 K and X-ray diffraction patterns were collected on a CCD detector (Quantum 4z) positioned 250 mm (for 1.00 Å) or 150 mm (for 1.6 Å) from the crystal. Diffraction patterns, using 4 or 5° oscillation, with 90 s exposure per frame, and a total range 180°, were processed at 2.9 or 3.1 Å resolution by the program DPS/MOSFLM (28–31). The three data sets were scaled separately using the programs SCALA, TRUNCATE and SCALEIT of the CCP4 suite (32).
Another data set was collected for structure refinement at 100 K with synchrotron radiation (λ = 0.900 Å) at BL44XU of SPring-8 in Harima. Intensity data were recorded on a 3 × 3-arrayed CCD detector (PX210; Oxford Inst. Co.) positioned 243 mm from the crystal. Each frame was taken with 1° oscillation and 15 s exposure. In total, 180 frames were processed at 2.4 Å resolution with the same program mentioned above. To compensate for overloaded reflections, the data set was merged with the data taken with λ = 1.00 Å. 1676 unique reflections with 100% completeness were obtained with Rmerge = 6.8%. The statistics of the data collection and crystal data are summarized in Table 1.
Table 1. Crystal data, statistics of data collection, and statistics of structure refinement.
| X-ray source | Synchrotron radiation at Photon Factory | Synchrotron radiation at SPring-8 | Structure refinement | |||
|---|---|---|---|---|---|---|
| Space group | I1422 | I4122 | Resolution range (Å) | 9–2.5 | ||
| Unit cell (Å) | a = b = 53.3, c = 53.9 | a = b = 53.4, c = 54.0 | Used reflections | 1333 (Fo > 3σ) | ||
| Asymmetric unita | 1 | 1 | R-factor (%)b | 21.5 | ||
| Wavelength (Å) | 1.00 (remote) | 1.6049 (peak) | 1.6054 (edge) | 0.900 | Rfree (%)c | 25.7 |
| Resolution (Å) | 38–2.9 | 38–3.1 | 38–3.1 | 27–2.4 | Number of DNA atoms | 184 |
| Observed reflections | 12 067 | 9902 | 9904 | 20 224 | Number of waters | 16 |
| Unique reflections | 973 | 806 | 809 | 1647 | Number of magnesium atoms | 1.5 |
| Completeness (%) | 100 | 99.9 | 100 | 98.3 | Number of cobalt hexamine molecules | 0.5 |
| In the outer shell (%) | 100 (3.06–2.9) | 100 (3.27–3.1) | 100 (3.27–3.1) | 100 (2.53–2.4) | RMSD from ideal geometry | |
| Rmerge (%)d | 6.4 | 8.4 | 7.7 | 5.4 | Bond lengths (Å) | 0.005 |
| Ranom (%)e | 2.3 | 5.3 | 4.0 | – | Bond angles (°) | 0.9 |
| Theoretical f′/f″ | 0.16/1.78 | –5.94/3.92 | –6.09/3.93 | – | Improper angles (°) | 1.3 |
| Refined f′/f″ | –0.339/1.241 | –6.007/3.329 | –7.201/2.031 | – | ||
aA single strand d(GCGAAAGCT).
bR-factor = 100 × Σ||Fo| – |Fc|| / Σ|Fo|, where |Fo| and |Fc| are the observed and calculated structure factor amplitudes, respectively.
cCalculated using a random set containing 10% of observations that were not included during refinement (55).
dRmerge = 100 × Σhklj|Ihklj – 〈Ihkl〉| / Σhklj〈Ihkl〉.
eRanom = 100 × Σhklj|Ihklj(+) – Ihklj(–)| / Σhklj[Ihklj(+) + Ihklj(–)].
Structure determination and refinement
From the three data sets with different wavelengths, a cobalt atom was uniquely found on the crystallographic 2-fold axis. Phases were estimated by the MAD method using the program SOLVE (33), the figure-of-merit being 0.64. Electron density was modified by a solvent flattening technique (solvent content 60.9%) with the program CNS (34). In the modified map, the phosphate-ribose backbone with the individual bases was easily traced. The molecular structure was constructed on a graphic workstation with the program QUANTA (Molecular Simulation Inc.).
The atomic parameters were refined with the program CNS (34) through a combination of rigid-body, simulated-annealing, crystallographic-conjugate-gradient-minimization refinements and B-factor refinements, followed by interpretation of an omit map at every nucleotide residue. No restraints were applied between paired nucleotides or to the sugar puckering. Only the 5′-terminal G1 residue was difficult to assign due to rather poor density, but after several steps of refinement, it appeared in an Fo – Fc map. Once the two magnesium atoms were found, one at a general position and the other on the 2-fold axis, these were added to the refinement along with several water molecules. By taking hydrogen-bond interactions with surrounding atoms into consideration, water molecules octahedrally coordinated to the two magnesium atoms and ammonium cations coordinated around the cobalt atom could be reasonably assigned. In the final refinement, reflections in the outer shell beyond 2.5 Å resolution were suppressed due to extremely low I / σ values (<3). The statistics of the structure refinement are summarized in Table 1. Figure 1 shows local 2Fo – Fc maps for the homo base pairs. All local helical parameters, including torsion angles and pseudorotation phase angles of ribose rings, were calculated by using the program NUPARAM (35). Figure 1 was drawn with the program O (36), Figures 2 and 5 with the program MOLSCRIPT (37) and Figure 4 with the program RASMOL (38).
Figure 1.

Local 2Fo – Fc maps for phosphate backbones (A) and homo base pairs (B–E). Densities are contoured at 2σ level for (A), 1.5σ for (B and C), and 0.8σ for (D and E). Broken lines indicate possible hydrogen bonds.
Figure 2.

A stereo-pair diagram of the parallel duplex with homo base pairs. The two halves of the strands branch away from the parallel region in different directions. The superscript P indicates the counter strand aligned in parallel fashion.
Figure 5.

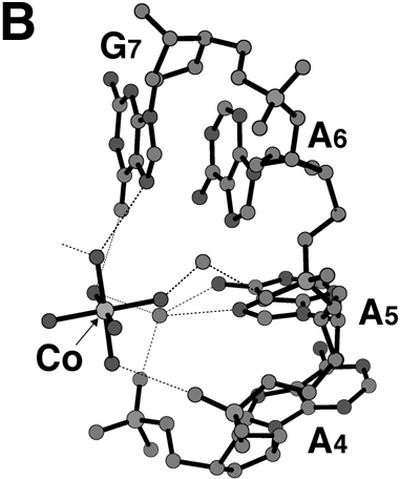
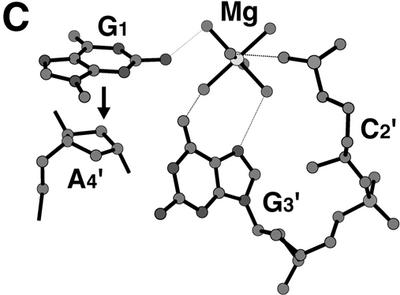
Bound cations stabilizing the d(GCGAAAGCT) structure. (A) A junction between the parallel and the anti-parallel duplexes. A hydrated magnesium cation is bound to the two T9 residues to stabilize their stacking interaction. Broken lines indicate possible hydrogen bonds. (B) A hexa- ammine cobalt cation bound to the bent corner joining the parallel and the anti-parallel duplexes. Broken lines indicate possible hydrogen bonds. This diagram shows half of the interactions. The other half is related by the 2-fold symmetry, so that the cobalt atom is centered in the spherical hole of the two bent strands. (C) Stacking interaction of the G1 residue with the ribose moieties of the neighboring A4 residue. The hydrated magnesium cation bridges the G1, G3′ and C2′ residues of different strands to stabilize the stacking of the two parallel duplexes.
Figure 4.

Hydrogen bond formation (broken lines) between the homo bases. Values indicate atomic distances (Å) and angles (°). The crystallographic 2-fold axis is indicated with a black spot at the center of the base pair.
RESULTS
Crystal packing of the two duplex regions
The Mathew’s criterion for protein crystals (39) is also applicable to nucleic acid crystals as a general rule (40); from this, the number of nonamers in the asymmetric unit is estimated to be 1. Initially the structure was expected to adopt one of two possible structures, hairpin-type (stem–loop) or zipper-like duplex, because these have been already assigned from NMR studies (22) and X-ray analysis of a bromo-derivative (25). However, an electron density map of the present nonamer clearly shows that the conformation of the phosphate backbone is quite different (see Fig. 1). The first 5′-terminal guanosine residue G1 protrudes into the solvent region and its density is slightly broadened even on the final 2Fo – Fc map, suggesting its flexibility or disorder in the crystal. Figure 2 shows that the next four residues, C2, G3, A4 and A5, form a right-handed helix with those of another strand, related by a crystallographic 2-fold symmetry along the face-diagonal axis of the tetragonal lattice. The two chains are associated and oriented in parallel alignment. The remaining halves of the two strands, containing the A6, G7, C8 and T9 residues, split away in opposite directions along the c-axis. Around this axis, one half is further coupled with another half of the adjacent parallel duplex, related by a crystallographic 41 symmetry, as shown schematically in Figure3A. These two halves are associated in an anti-parallel alignment to form another duplex through normal, Watson–Crick base pairs. This coupling is repeated in a head-to-tail manner to form a long column along the c-axis, so that the parallel-stranded regions extrude at right angles from the surface of the column, like knobs. The anti-parallel columns are linked together by facing the flat ends of the parallel-stranded regions, and forming base–base stacking interactions between C2 residues, as shown in Figure 3B. This linkage is further stabilized by hydrophobic interactions of the 5′-terminal G1 residues. The guanine moiety of the major conformer, which is stacked on the ribose ring of the fourth residue A4, covering the hydrophobic face of the ring in a hydrophilic environment. Such an interaction has already been observed in nucleic acid crystals (41). Between the four columns of anti-parallel duplexes, there is a long channel for solvent molecules, extending along the c-axis, as shown in Figure 3B. The water molecules in this channel were difficult to locate.
Figure 3.
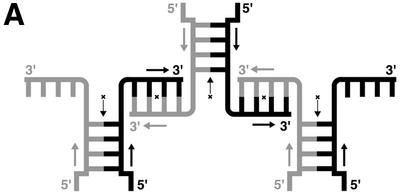
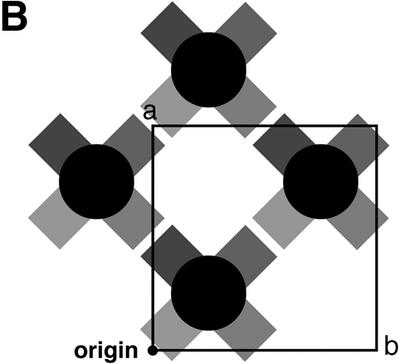
A coupling motif formed by the two arms extending out from the parallel duplexes, to form a long column of anti-parallel duplexes (A) and a large channel extending along the c-axis, presumably occupied by water (B). The crystallographic 2-fold axes are indicated with an arrow and a cross. The parallel duplex regions protrude from the column like knobs at right angles. The flat end of each parallel duplex is stacked with another flat end of the adjacent parallel duplex.
Base-pair formation
Figures 1B–E and 4 show the homo base pairs found in the parallel duplex region; the hydrogen bond distances and angles are also indicated. These base pairings stabilize the parallel duplex region. The first C:C+P pair involves three hydrogen bonds between N4 and O2P, between N3 and N3P, and between O2 and N4P (P indicates the parallel strand). In this case, the C:C+ pair must be protonated at N3 of either C residue, with disordering between N3 and N3P. The second G:GP pair involves N2–H…N3P and N2P–HP…N3 hydrogen bonds between the minor groove sites. These two base pairs are of the same types as those reported in several X-ray structures (7,42,43). The third A:AP pair involves the two N6–H…N7 hydrogen bonds between major sites. This paring is known to occur between the protonated adenines (44,45) and between the neutral forms (46). The latter case is detected in d(TCGA) at pH 4.0 by NMR measurement (11). Since the present crystal was obtained at pH 6.0, the adenine moiety of the A3 residue is not protonated. The three base-pair formations, described above, are identical to those derived from NMR analyses for d(CGA) (10) and d(TCGA) (11).
In the fourth A:AP pair, the pairing occurs between the Watson–Crick sites, through two N6–H…N1 hydrogen bonds. This pairing is also the same as that found in crystals of small molecules (47) and oligo-nucleotides (48). In all base pairings of the parallel duplex region, the two bases or two nucleotide residues are images of each other under the crystallographic 2-fold symmetry. On the other hand, the anti-parallel duplex region is formed by four base pairs, A6:T9A, G7:C8A, C8:G7A and T9:A6A, with the normal Watson–Crick types (A indicates the anti-parallel strand); the distances and angles for hydrogen bond formations are in standard ranges (49).
Conformation of the parallel duplex
A defining feature of the parallel-stranded duplex is the backbone conformation, forming a right-handed helix. Table 2 shows values of representative helical parameters. The average values of the twist angle, Dz, and the C1…C1 distance in the parallel region seem close to those of the anti-parallel region, which has a typical B-form conformation (49). However, the individual values fluctuate highly in the parallel region depending on the pairing types. Both the C2:C2+ and A5:A5 homo pairs are formed at the Watson–Crick pairing sites, resulting in a shorter C1…C1 distance at the C2:C2+ pair and a longer one at the A5:A5 pair. The second G3:G3 pair occurs in the minor groove sites so that the C1…C1 distance is the shortest. The C1…C1 distance of the A4:A4 pair formed in the major groove sites is similar to the normal distance, 10.8 Å. From the first pair to the second pair, the twist angle is only 13°, reflecting a small deviation from the Watson–Crick pairing site to the minor groove site. But the largest movement is found in going from the second step to the third, since the pairing changes from the minor groove site to the major groove site. The fourth pair requires a small twist, due to the change from the major site to the Watson–Crick site. Depending on the pairing sites, the stacking distances are also changed. The largest differences are found in the opening angles. In the parallel region, the paired bases must occupy a trans position with respect to each other. Furthermore, the two bases in the present crystal are exactly related by the crystallographic 2-fold symmetry so that the opening angles should be just 180°. Large propeller twists occur at the G:G pair in the minor site and at the A:A pair in the major site, but they are not abnormal values. Such a twisting is required to release the close contacts of other regions and the geometrical constraints for hydrogen bonding (41).
Table 2. Local helical parameters and intra base-pair parameters.
| Base pair | Inclina | Tip | Twist | Dzb | Propa | Bucka | Opena | C1…C1b | |
|---|---|---|---|---|---|---|---|---|---|
| Parallel | |||||||||
| 2 | C:C | –6 | 4 | 180 | 9.5 | ||||
| 0 | 0 | 13 | 3.6 | ||||||
| 3 | G:G | 17 | –23 | 180 | 7.4 | ||||
| 0 | 0 | 80 | 3.2 | ||||||
| 4 | A:A | 27 | 27 | 180 | 10.8 | ||||
| 0 | 0 | 21 | 3.8 | ||||||
| 5 | A:A | –2 | –1 | 180 | 13.3 | ||||
| Anti-parallel | |||||||||
| 6 | A:T | –18 | –2 | 15 | 10.4 | ||||
| 0 | –4 | 36 | 3.4 | ||||||
| 7 | G:C | –15 | –3 | 21 | 10.8 | ||||
| –9 | 0 | 42 | 3.2 | ||||||
| 8 | C:G | –15 | 3 | 21 | 10.8 | ||||
| 0 | 4 | 36 | 3.4 | ||||||
| 9 | T:A | –18 | 2 | 15 | 10.4 | ||||
| Average value | |||||||||
| Parallel | 0 | 0 | 38 | 3.5 | 19 | 2 | 180 | 10.3 | |
| Anti-parallel | –3 | 0 | 38 | 3.3 | –16 | 0 | 18 | 10.6 |
aInclin, inclination angle (°); Prop, propeller twist angle (°); Buck, buckle angle (°); Open, opening angle (°) (35).
bDistance in Å.
The torsion angles in the ribose phosphate backbone are indicated in Table 3. They fluctuate within an allowed range, with a similar behavior in the parallel and the anti-parallel regions. The conformational feature of the parallel region is similar to those of the NMR structures (10,11), including some differences at both ends of the duplexes; the root mean square deviations (RMSDs) are within 1.4 Å when the corresponding parts are superimposed on each other. In the anti-parallel region, every residue, except for the terminal T9 residue, adopts a C2′-endo conformation, with a normal fluctuation (41). The T9 residue adopts a C4′-exo conformer, due to a close contact with a hydrated magnesium cation, as shown in Figure 5A. Even in the parallel region, however, the second and third residues (forming the first and second pairs) adopt a C2′-endo conformation. In contrast, the fourth and fifth residues adopt a C3′-endo pucker. This drastic change occurs at the junction between the two regions, parallel and anti-parallel, at a point where they are largely bent, as shown in Figure 5A and B. The two A6 residues are split away from the parallel region to form Watson–Crick pairs with the neighboring T9 residues, which are almost perpendicular to the A5:A5 pair.
Table 3. Torsion angles (°) and sugar conformation.
| Sequence | P–O5′ | O5′–C5′ | C5′–C4′ | C4′–C3′ | C3′–O3′ | O3′–P | C1′–N | Phasea | Pucker |
|---|---|---|---|---|---|---|---|---|---|
| α | β | γ | δ | ε | ζ | χ | θ | ||
| G | – | – | 160 | 149 | –134 | 156 | –124 | –133 | C4′-endo |
| Parallel | |||||||||
| C | –57 | 165 | 57 | 137 | –174 | –69 | –120 | 150 | C2′-endo |
| G | –74 | –176 | 49 | 139 | –155 | 154 | –82 | 154 | C2′-endo |
| A | –59 | 135 | 55 | 95 | –136 | –65 | –175 | 31 | C3′-endo |
| A | –67 | 172 | 43 | 87 | –145 | –69 | –124 | 15 | C3′-endo |
| Anti-parallel | |||||||||
| A | –74 | –156 | 51 | 147 | –162 | –98 | –114 | 166 | C2′-endo |
| G | –62 | 170 | 39 | 150 | –124 | 174 | –86 | 153 | C2′-endo |
| C | –49 | 132 | 45 | 130 | –173 | –110 | –110 | 141 | C1′-exo |
| T | –68 | 173 | 49 | 88 | – | – | –124 | 44 | C4′-exo |
aPseudo-rotation phase angle of ribose ring.
The ribose ring of the first residue G1 adopts a C4′-endo conformation and the C5′–C4′ bond adopts an unusual torsion angle in order to extend the guanine moiety for stacking on the ribose ring of the neighboring A4 residue.
Ionic effect for stabilizing the structure
The two arms containing the A6, G7, C8 and T9 residues, which form an anti-parallel duplex as described above, are further related by the crystallographic 2-fold symmetry. On this symmetry axis, a hexa-ammine cobalt cation is bound to the center of the anti-parallel duplex, as seen in Figure 5B. The N7 and O6 atoms of the G7 residue are directly hydrogen bonded to two different ammonium cations separately. A water molecule, which is bound to the third ammonium cation, is hydrogen bonded to the phosphate oxygen of the A4 residue and to the N7 and N6 atoms of the A5 residue. Another water bridges the phosphate oxygen of the A6 residue and the third ammonium cation. With these interactions, the hexa-ammine cobalt cation thus supports bending of the nonamer at the junction between the parallel region and the anti-parallel region. The C3′-endo puckering of the A4 and A5 residues may be required for this bending.
The two hexa-hydrated magnesium cations were located. One is bound to the G3′ and C2′ residues through hydrogen bonds with coordinated water molecules, as shown in Figure 5C. The primed residues are those of the parallel duplex, stacked on the original duplex. The guanine moiety of the flipped G1 residue, which is covered by the ribose ring of A4′, is bound to the coordinated waters. Another magnesium cation is located on the 2-fold axis at the branching point of the two arms of the parallel duplex, where the two other arms, extending from different parallel duplexes, come together to form the two anti-parallel duplex regions, as shown in Figure 5A. The cation is bound to the A6:T9′P and T9′:A6P pairs, the two hydrated water molecules being hydrogen bonded to the two O2 atoms of these T9 residues. Thus, the stacking between the two base pairs is further stabilized by these interactions.
DISCUSSION
From NMR and CD measurements, the present d(GCG AAAGCT) nonamer shows extraordinary properties, similar to those of the d(GCGAAAGC) octamer (22). Therefore, it was expected to adopt a similar mini-hairpin structure in solution (22). To investigate the structure of the nonamer, we first crystallized its iodo-derivative d(GCXGAAAGCT) (CX = 2′-deoxy-5-iodocytidine) at pH 6.0 (50). Initially, this crystal structure was solved at 1.9 Å resolution, but it was difficult to distinguish between the two possible structures, hairpin dimer or duplex dimer. Recently, we have re-determined its structure at a higher resolution, 1.4 Å (26); this structure clearly indicates that the derivatives form a duplex dimer with a zipper-like structure, similar to the bromo-derivative solved earlier (25). The crystal structure of d(GCGAAAGC), which has been recently determined at a high 1.6 Å resolution (26), also shows that the octamer forms a zipper-like duplex when crystallized at pH 7.0. In either structure, the mini-hairpin and the zipper, however, the stem and the duplex parts are formed in anti-parallel alignment, as is usual in B-DNA. Although the present crystals were prepared under acidic conditions (at pH 6.0 in crystallization droplets) slightly above the proton dissociation constant of cytosine [pKa ≈ 4.5 (51)], hemi-protonation is induced on one cytidine residue, because the C:C pair has a negative hole at the center, which can more easily accept a proton to stabilize the pairing. Such phenomena have been observed in several cases (52). It is necessary to note that one (C2) of the two cytidine residues is hemi-protonated and the other cytidine residue C8 is not protonated at all under such mild acidic conditions. Therefore, it is reasonable to suppose that two types of half-duplexes are in equilibrium in solution: one is a parallel duplex between CGAA parts and the other is an anti-parallel duplex between AGCT parts. Once a parallel half duplex is formed, the conformation with two extended arms could be preferred due to base–base interactions. In crystallization, the parallel duplexes easily associate to form a long column, with the anti-parallel duplex associated in a head-to-tail manner. In addition, these columns are laterally bound through stacking interactions between the parallel duplexes. It will be interesting to clarify which structure occurs when these arms are missing. The crystal structure of the tetramer d(CGAA) will give an answer to this question.
Another question is whether the parallel duplex structure is involved in some biological function. As the major part of living systems is established by using the Watson–Crick pairing between complementary strands, it may be difficult to find such a parallel duplex (P-DNA) in the stable structure of a double helix. Once Z-DNA was discovered, it took 20 years to find Z-DNA-binding proteins (6). For the parallel duplex, too, it is expected that when DNA is unwound to perform a function in a single-stranded state and slightly acidic conditions, P-DNA will occur as a local structure, as reported by Tchurikov and co-workers (17–19). As single-strand DNAs from DNA viruses or phages can adopt many types of structures (like RNA), sometimes these could have a specific function. A typical example is a DNA enzyme derived by in vitro selection (53). In practice, functional DNAs are being developed for several purposes (54), because DNA is more stable thermally and chemically than RNA. The present finding of P-DNA will be a useful structural guide for designing such functional DNAs. Furthermore, the structural features found in the present investigation may be useful for controlling DNA function by manipulating the pH.
DATA BANK ACCESSION CODES
The atomic coordinates have been deposited in the Protein Data Bank with the ID 1ixj.
Acknowledgments
ACKNOWLEDGEMENTS
We thank N. Sakabe, M. Suzuki, N. Igarashi and A. Nakagawa for facilities and help during data collection, and T. Simonson for proof-reading of the original manuscript. This work was supported in part by a grant for the RFTF (97L00503) from the Japanese Society for the Promotion of Science, by the Structural Biology Sakabe Project and by Grants-in-Aid for Scientific Research (nos 12480177 and 14035217) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
PDB no. 1ixj
REFERENCES
- 1.Watson J.D. and Crick,F.H. (1953) Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid. Nature, 171, 737–738. [DOI] [PubMed] [Google Scholar]
- 2.Fuller W., Wilkins,M.H.F., Wilson,H.R. and Hamilton,L.D. (1965) The molecular configuration of deoxyribonucleic acid. IV. X-ray diffraction study of the A form. J. Mol. Biol., 12, 60–80. [DOI] [PubMed] [Google Scholar]
- 3.Leslie A.G.W., Arnott,S., Chandrasekaran,R. and Ratliff,R.L. (1980) Polymorphism of DNA double helices. J. Mol. Biol., 143, 49–72. [DOI] [PubMed] [Google Scholar]
- 4.Wang A.H.-J., Quigley,G.J., Kolpak,F.J., Crawford,J.L., van Boom,J.H., van der Marel,G. and Rich,A. (1979) Molecular structure of a left-handed double helix DNA fragment at atomic resolution. Nature, 282, 680–686. [DOI] [PubMed] [Google Scholar]
- 5.Drew H., Takano,T., Tanaka,S., Itakura,K. and Dickerson,R.E. (1980) High-salt d(CpGpCpG): a left-handed Z′ DNA double helix. Nature, 286, 567–573. [DOI] [PubMed] [Google Scholar]
- 6.Brown B.A. 2nd and Rich,A. (2001) The left-handed double helical nucleic acids. Acta Biochim. Pol., 48, 295–312. [PubMed] [Google Scholar]
- 7.Cruse W.B., Egert,E., Kennard,O., Sala,G.B., Salisbury,S.A. and Viswamitra,M.A. (1983) Self base pairing in a complementary deoxydinucleoside monophosphate duplex: crystal and molecular structure of deoxycytidylyl-(3′–5′)-deoxyguanosine. Biochemistry, 12, 1833–1839. [DOI] [PubMed] [Google Scholar]
- 8.Westhof E. and Sundaralingam,M. (1980) X-ray-structure of a cytidylyl-3′,5′-adenosine-proflavine complex: a self-paired parallel-chain double helical dimer with an intercalated acridine dye. Proc. Natl Acad. Sci. USA, 77, 1852–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson H., van der Marel,G.A., van Boom,J.H. and Wang,A.H.-J. (1992) Unusual DNA conformation at low pH revealed by NMR: parallel-stranded DNA duplex with homo base pairs. Biochemistry, 31, 10510–10517. [DOI] [PubMed] [Google Scholar]
- 10.Robinson H. and Wang,A.H.-J. (1993) 5′-CGA sequence is a strong motif for homo base-paired parallel-stranded DNA duplex as revealed by NMR analysis. Proc. Natl Acad. Sci. USA, 90, 5224–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y. and Patel,D.J. (1994) Solution structure of the d(T-C-G-A) duplex at acidic pH. A parallel-stranded helix containing C+.C,G.G and A.A pairs. J. Mol. Biol., 242, 508–526. [PubMed] [Google Scholar]
- 12.Parvathy V.R., Bhaumik,S.R., Chary,K.V., Govil,G., Liu,K., Howard,F.B. and Miles,H.T. (2002) NMR structure of a parallel-stranded DNA duplex at atomic resolution. Nucleic Acids Res., 30, 1500–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soyfer V.N. and Potaman,V.N. (1996) Triple-helical Nucleic Acids. Springer-Verlag, New York.
- 14.Keniry M.A. (2000) Quadruplex structures in nucleic acids. Biopolymers, 56, 123–146. [DOI] [PubMed] [Google Scholar]
- 15.Chen L., Cai,L., Zhang,X. and Rich,A. (1994) Crystal structure of a four-stranded intercalated DNA: d(C4). Biochemistry, 33, 13540–13546. [DOI] [PubMed] [Google Scholar]
- 16.Snoussi K., Nonin-Lecomte,S. and Leroy,J.L. (2001) The RNA i-motif. J. Mol. Biol., 309, 139–153. [DOI] [PubMed] [Google Scholar]
- 17.Tchurikov N.A., Chernov,B.K., Golova,Y.B. and Nechipurenko,Y.D. (1989) Parallel DNA: generation of a duplex between two Drosophila sequences in vitro. FEBS Lett., 257, 415–418. [DOI] [PubMed] [Google Scholar]
- 18.Tchurikov N.A., Shchyolkina,A.K., Borissova,O.F. and Chernov,B.K. (1992) Southern molecular hybridization experiments with parallel complementary DNA probes. FEBS Lett., 297, 233–236. [DOI] [PubMed] [Google Scholar]
- 19.Borisova O.F., Shchyolkina,A.K., Chernov,B.K. and Tchurikov,N.A. (1993) Relative stability of AT and GC pairs in parallel DNA duplex formed by a natural sequence. FEBS Lett., 322, 304–306. [DOI] [PubMed] [Google Scholar]
- 20.Tchurikov N.A., Chistyakova,L.G., Zavilgelsky,G.B., Manukhov,I.V., Chernov,B.K. and Golova,Y.B. (2000) Gene-specific silencing by expression of parallel complementary RNA in Escherichia coli. J. Biol. Chem., 275, 26523–26529. [DOI] [PubMed] [Google Scholar]
- 21.Hirao I., Naraoka,T., Kanamori,S., Nakamura,M. and Miura,K. (1988) Synthetic oligodeoxyribonucleotides showing abnormal mobilities on polyacrylamide gel electrophoresis. Biochem. Int., 16, 157–162. [PubMed] [Google Scholar]
- 22.Hirao I., Nishimura,Y., Naraoka,T., Watanabe,K., Arata,Y. and Miura,K. (1989) Extraordinary stable structure of short single-stranded DNA fragments containing a specific base sequence: d(GCGAAAGC). Nucleic Acids Res., 17, 2223–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirao I., Nishimura,Y., Tagawa,Y., Watanabe,K. and Miura,K. (1992) Extraordinarily stable mini-hairpins: electrophoretical and thermal properties of the various sequence variants of d(GCGAAAGC) and their effect on DNA sequencing. Nucleic Acids Res., 20, 3891–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirao I., Ishida,M., Watanabe,K. and Miura,K. (1990) Unique hairpin structures occurring at the replication origin of phage G4 DNA. Biochim. Biophys. Acta, 1087, 199–204. [DOI] [PubMed] [Google Scholar]
- 25.Shepard W., Cruse,W.B., Fourme,R., de la Fortelle,E. and Prangé,T. (1998) A zipper-like duplex in DNA: the crystal structure of d(GCGAAAGCT) at 2.1 Å resolution. Structure, 6, 849–861. [DOI] [PubMed] [Google Scholar]
- 26.Sunami T., Kondo,J., Chatake,T., Hirao,I., Watanabe,K., Miura,K. and Takénaka,A. (2001) X-ray analyses of d(GCGAAAGC) and d(GXGAAAGCT), where X=2′-deoxy-5-iodocytidine. Nucleic Acids Symp. Ser., [Suppl 1], 191–192. [DOI] [PubMed] [Google Scholar]
- 27.Berger I., Kang,C., Sinha,N., Wolters,M. and Rich,A. (1996) A highly efficient 24-condition matrix for the crystallization of nucleic acid fragments. Acta Crystallogr., D52, 465–468. [DOI] [PubMed] [Google Scholar]
- 28.Leslie A.G.W. (1992) Molecular data processing. In Moras,D., Podjarny,A.D. and Thierry,J.-C. (eds), Crystallographic Computing 5, From Chemistry to Biology. Oxford University Press, Oxford, UK, pp. 50–61.
- 29.Steller I., Bolotovsky,R. and Rossmann,M.G. (1997) An algorithm for automatic indexing of oscillation images using Fourier analysis. J. Appl. Crystallogr., 30, 1036–1040. [Google Scholar]
- 30.Rossmann M.G. and van Beek,C.G. (1999) Data processing. Acta Crystallogr., D55, 1631–1640. [DOI] [PubMed] [Google Scholar]
- 31.Powell H.R. (1999) The Rossmann Fourier autoindexing algorithm in MOSFLM. Acta Crystallogr., D55, 1690–1695. [DOI] [PubMed] [Google Scholar]
- 32.Collaborative Computational Project Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr., D50, 760–763. [DOI] [PubMed] [Google Scholar]
- 33.Terwilliger T.C. and Berendzen,J. (1999) Automated structure solution for MIR and MAD. Acta Crystallogr., D55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brünger A.T., Adams,P.D., Clore,G.M., DeLano,W.L., Gros,P., Grosse-Kunstleve,R.W., Jiang,J.-S., Kuszewski,J., Nilges,M., Pannu,N.S., Read,R.J., Rice,L.M., Simonson,T. and Warren,G.L. (1998) Crystallography & NMR System: a new software suite for macromolecular structure determination. Acta Crystallogr., D54, 905–921. [DOI] [PubMed] [Google Scholar]
- 35.Bansal M., Battacharyya,D. and Ravi,B. (1995) NUPARAM and NUCGEN: software for analysis and generation of sequence dependent nucleic acid structures. Comput. Appl. Biosci., 11, 281–287. [DOI] [PubMed] [Google Scholar]
- 36.Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr., A47, 110–119. [DOI] [PubMed] [Google Scholar]
- 37.Kraulis P.J. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- 38.Sayle R.A. and Milner-White,E.J. (1995) RASMOL: biomolecular graphics for all. Trends Biochem. Sci., 20, 374–376. [DOI] [PubMed] [Google Scholar]
- 39.Matthews B.W. (1968) Solvent content of protein crystals. J. Mol. Biol., 33, 491–497. [DOI] [PubMed] [Google Scholar]
- 40.Takénaka A., Matsumoto,O., Chen,Y., Hasegawa,S., Chatake,T., Tsunoda,M., Ohta,T., Komatsu,Y., Koizumi,M. and Ohtsuka,E. (1995) Structural composition of hammerhead ribozymes. J. Biochem., 117, 850–855. [DOI] [PubMed] [Google Scholar]
- 41.Hossain T.M., Kondo,J., Ueno,Y., Matsuda,A. and and Takénaka,A. (2002) X-Ray analysis of d(CGCGAATTXGCG)2 containing a 2′-deoxy-N4-methoxycytosine residue at X: a characteristic pattern of sugar puckers in the crystalline state of the Dickerson–Drew type DNA dodecamers. Biophys. Chem., 95, 69–77. [DOI] [PubMed] [Google Scholar]
- 42.Langridge R. and Rich,A. (1963) Molecular structure of helical polycytidylic acid. Nature, 198, 725–728. [DOI] [PubMed] [Google Scholar]
- 43.Shibata M., Takénaka,A. and Sasada,Y. (1985) Structure of 9-(2-hydroxyethyl)guanine. Acta Crystallogr., C41, 1501–1503. [Google Scholar]
- 44.Takénaka A. and Sasada,Y. (1982) Elementary patterns in protein-nucleic acid interactions. III. Crystal structure of adenine:phthalic acid (3:1) complex hexahydrate. Bull. Chem. Soc. Jpn, 55, 680–686. [Google Scholar]
- 45.Suck D., Manor,P.C. and Saenger,W. (1976) The structure of a trinucleoside diphosphate: Adenylyl-(3′,5′)-adenylyl-(3′,5′)-adenosine hexahydrate. Acta Crystallogr., B32, 1727–1737. [Google Scholar]
- 46.Arora S.K. and Larson,S.B. (1987) Structure of arprinocid [9-(2-chloro-6-fluorobenzyl)adenine], a coccidiostat. Acta Crystallogr., C43, 912–914. [Google Scholar]
- 47.Ohki M., Takénaka,A., Shimanouchi,H. and Sasada,Y. (1977) 3-(9-Adenyl)propionyltryptamine monohydrate. Acta Crystallogr., B33, 2954–2956. [Google Scholar]
- 48.Cai L., Chen,L., Raghavan,S., Ratliff,R., Moyzis,R. and Rich,A. (1998) Intercalated cytosine motif and novel adenine clusters in the crystal structure of the Tetrahymena telomere. Nucleic Acids Res., 26, 4696–4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saenger W. (1984) Principles of Nucleic Acid Structure. Springer-Verlag, New York.
- 50.Sunami T., Chatake,T., Hirao,I., Yokoyama,S., Watanabe,K., Miura,K. and Takénaka,T. (1998) X-Ray analysis of a deoxy-oligonucleotide containing a stable hairpin structure. The Annual Meeting 1998 of CrSJ, Abstract, 118. Yokohama, Nov., CrSJ, Tokyo.
- 51.Albert A. (1973) Ionization constants of pyrimidines and purines. In Zorbach,W.W. and Tipson,S. (eds), Synthetic Procedures in Nucleic Acid Chemistry. John Wiley & Sons, Vol. 2, pp. 1–46.
- 52.Edwards E.L., Patrick,M.H., Ratliff,R.L. and Gray,D.M. (1990) A.T and C.C+ base pairs can form simultaneously in a novel multistranded DNA complex. Biochemistry, 29, 828–836. [DOI] [PubMed] [Google Scholar]
- 53.Breaker R.R. and Joyce,G.F. (1994) A DNA enzyme that cleaves RNA. Chem. Biol., 1, 223–229. [DOI] [PubMed] [Google Scholar]
- 54.Sioud M. (2001) Nucleic acid enzymes as a novel generation of anti-gene agents. Curr. Mol. Med., 1, 575–588. [DOI] [PubMed] [Google Scholar]
- 55.Brünger A.T. (1992) Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature, 355, 472–475. [DOI] [PubMed] [Google Scholar]