


Abstract
Genetic evidence suggests that the Bacillus subtilis dnaX gene only encodes for the τ subunit of both DNA polymerases III (Pol IIIs). The B.subtilis full-length protein and their mutant derivatives τ(373– 563) (lacking the N-terminal, domains I–III or amino acid residues 1–372) and τ(1–372) (lacking the C-terminal region or amino acids 373–563) have been purified. The τ protein forms tetramers, τ(373– 563) forms dimers, whereas τ(1–372), depending on the ionic strength, forms trimers or tetramers in solution. In the absence of single-stranded (ss) DNA and a nucleotide cofactor, τ interacts with the SPP1 hexameric replicative G40P DNA helicase in solution or with G40P-ATP bound to ssDNA, with a 1:1 stoichiometry. G40P(109–442), lacking the N-terminal amino acid residues 1–108, interacts with the C-terminal moiety of τ. The data indicate that the interaction of G40P with the τ subunit of Pol III, is relevant for the loading of the Pol IIIs into the SPP1 G38P-promoted open complex.
INTRODUCTION
Initiation of DNA replication requires the coordination of multiple genes products. Studies in different systems have shown three common steps: (i) a replicon-specific initiation protein (e.g. DnaA) recognises and binds to a specific cis-acting replication sequence (e.g. oriC), forming a nucleoprotein complex; (ii) a recruitment of the replication machinery; and (iii) a replication complex remodelling takes place (1–3). In the case of Escherichia coli, DnaA-ATP upon binding to its cognate sites, at oriC, catalyses unwinding of the adjacent AT-rich sequences and promotes open complex formation (2,3). The complex formed by the replication fork DNA helicase (DnaB-ATP) and its loader (DnaC-ATP) interacts with the ATP-activated DnaA–oriC open complex. Here, DnaB-ATP of the ATP-DnaB–DnaC-ATP complex interacting with DnaA-ATP as well as DnaC-ATP interacting with the single-stranded (ss) DNA region of the open complex (2–7). After loading, a remodelling of the replication proteins implies the release of DnaC-ADP from the complex with the subsequent activation of DnaB-ATP (7). DnaB bound at the open complex directly interacts with the DnaG primase (8). DnaB and DnaG, at the open complex, stimulate the loading of the DNA polymerase III (Pol III) holoenzyme and therefore complete the formation of the replication forks (8–10).
Escherichia coli Pol III has three functional subassemblies: (i) the polymerase core, composed of α, which has the polymerase activity, ε which provides the 3′–5′ exonuclease activity, and θ with no defined function; (ii) the β subunit or β-sliding clamp (11); and (iii) the clamp loader formed by τ2γ2δδ′χψ (11–13). In vitro the γ3δδ′ complex is involved in β-sliding clamp loading, but lacks the ability to contact with the polymerase core (14,15). The τ protein that interacts with the α subunit plays an active role in the β-sliding clamp loading onto both the leading and lagging strand ‘polymerases’, and in the interaction of the replicative DNA helicase with the Pol III enzyme, making this protein a key component for proper formation of the replisome activity (16–18). In E.coli, the dnaX gene encodes both τ (643 amino acids) and γ (431 amino acids) products. A ribosomal frameshift signal gives rise to the γ subunit, which lacks the C-terminal 212 amino acid residues when compared with τ (reviewed in 2).
Previous studies with proteolytic fragments of the E.coli τ protein, generated by limited proteolysis, indicate the presence of five discrete domains (I–V) (17–19). Domain I contains the two Walker motives, domain III contains the structural element required for δδ′ and χψ interaction and the oligomerisation domain, and domains IV and V contain the DnaB and the α subunit interacting region, respectively (17–19). The E.coli τ protein shares a high degree of identity with the τ proteins of Gram-positive bacteria at domains I (∼56%) and II (∼45%), but there is a low degree of identity at domain III (∼14%), and no significant level of identity could be detected between the E.coli τ protein at domains IV and V and equivalent regions of the τ protein of Gram-positive bacteria (20,21).
The initiation of DNA replication in Gram-positive bacteria is poorly understood. In Bacillus subtilis, the products DnaA, DnaC, DnaD, DnaB, DnaI, DnaG and both Pol IIIs are essential for initiation of DNA replication (22–24). DnaA, DnaC, DnaG and DnaI have their counterpart in E.coli DnaA, DnaB, DnaG and DnaC, respectively, whereas the presence of proteins homologues to DnaB and DnaD is not obvious in the E.coli genome (22). Gram-positive bacteria contain two Pol III enzymes; one of them has the DnaE (homologous to E.coli α subunit, DnaE/α), and the other has the PolC polymerising subunit (20,24). Furthermore, from genome sequence, the genes encoding for β, τ, δ and δ′ can be easily predicted, but the presence of ε, θ, γ, χ and ψ subunits is not obvious in Gram-positive bacteria (20,23). The B.subtilis dnaX gene codes for a 62.5 kDa protein termed τ (20,21). The B.subtilis τ protein shares a high degree of identity with equivalent proteins of different Gram-positive bacteria at domain I (75–82%), II (47–52%), III (34–38%) and the C-terminal region (44–47%, equivalent to domains IV and V of E.coli τ). Bacillus subtilis and Streptococcus pyogenes τ protein interacts with δ, δ′, PolC and DnaE/α subunits (20,23). In S.pyogenes the Pol III PolC2βτ4δδ′ complex extends DNA with high processivity, whereas the DnaE2βτ4δδ′ complex does it with low processivity (20). Unless otherwise stated, the indicated genes and products in this work are from B.subtilis origin.
The initiation of DNA replication of the B.subtilis bacteriophage SPP1 is a simple system. It requires the phage-encoded G38P (replisome organiser), G39P (helicase loader), G40P (hexameric replicative DNA helicase) and G36P (SSB) products and the host-encoded DnaG (DNA primase) and both Pol IIIs (25,26). In the absence of ATP, G38P specifically interacts with its cognate sites and locally denatures the AT-rich region at the SPP1 replication origin (oriL) to form an open complex (27). The G39P–G40P-ATP complex is recruited to the G38P bound oriL. Then G38P–G39P heterodimers leave the origin with the subsequent G40P-ATP activation (27). Hexameric G40P also interacts with monomeric DnaG primase (28). How B.subtilis Pol III(s) are loaded at oriL remains an open question. In this work we provide evidence that one τ tetramer interacts with one G40P hexamer. We have shown that the C-terminal moiety of τ interacts with the C-terminal region of G40P.
MATERIALS AND METHODS
Strains and plasmids
Escherichia coli XL1 Blue (25) and BL21DE3 pLysS (29) were used for cloning and protein overexpression. Bacillus subtilis YB886 wild-type (wt) and its isogenic derivatives BG201 (dnaX51) and BG661 (dnaX51 ΔrecA6) were used for complementation assays (30; this work). The E.coli pT712 (GIBCO-BRL), pQE9 (Qiagen), and pET3a (Stratagene) and E.coli–B.subtilis pHP13 (31) plasmids have been previously described.
Plasmid constructions
The dnaX gene, which encodes for a 563 amino acid protein, was PCR amplified from the B.subtilis chromosomal DNA and then a hexa-histidine sequence at the 3′ end of the dnaX gene was introduced (Fig. 1). The product was cloned into pT712 to generate pCB506 or pHP13 to generate pCB508. The dnaX gene, from codon 1 to 372, was PCR amplified, and cloned into pQE9, which introduced a hexa-histidine sequence at the 5′ end of the gene, to generate pCB529. The dnaX gene, from codon 373–563, was PCR amplified, and a hexa-histidine sequence at the 3′ end of the gene was introduced. The product was cloned into pET3a to generate pCB509. The nucleotide sequence of the clones was determined.
Figure 1.

Scheme of the proteins used in this study. The hexa-histidine sequence is denoted by a filled bar. (A) τ amino acid sequence is represented by a grey bar. The τ(1–372) protein contains amino acid residues 1–372, and τ(373–563) contains amino acid residues 373–563 of the τ protein. (B) G40P amino acid sequence is represented by an open bar. G40P (109–442) contains amino acid residues 109–442 of G40P.
DNA complementation analysis
The nucleotide sequence of YB886 dnaX, used as template, differs at position 1598 with the one deposited (NCBI-GB NC 000964). A cytosine was found in YB886 instead of an adenine, with a subsequent change of amino acid 533 (Ala to Glu). A Glu at position 533 is conserved in τ proteins from other Gram-positive bacteria (e.g. Lactococcus lactis). The nucleotide sequence of the thermosensitive (ts) dnaX51 gene was also determined. A thymine was found at position 1393 instead of a cytosine, implying the substitution of the conserved Pro465 for Ser465 in the dnaX51ts gene.
Bacillus subtilis wt and dnaX51ts ΔrecA6 strain, which is thermosensitive for dnaX and defective in homologous DNA recombination, were transformed with the low-copy plasmids pCB508-borne dnaX gene and pHP13 (vector) (five copies per cell), and the cells were plated at permissive (30°C) and non-permissive (55°C) temperatures.
Enzymes and reagents
DNA-modifying enzymes were from New England Biolabs and MBI Fermentas. [γ-32P]ATP was from Amersham Biosciences. Sepharose G50 and G25, Superose 6 and 12, Superdex 75 and Q Sepharose were from Amersham Pharmacia Biotech. The Ni-NTA agarose was from Qiagen, and IPTG and rifampicin were from Calbiochem.
Protein purification
G40P and G40P(109–442), lacking amino acid residues 1–108 (Fig. 1), were overproduced and purified as previously described (32; P. Mesa, unpublished results). BL21DE3 [pLysS] bearing pCB506 or pCB509 and XL1 Blue bearing pCB529 were grown as previously described (32). Cells were harvested, resuspended in buffer A (50 mM phosphate pH 7.5, 5% glycerol) containing 100 mM NaCl and 5 mM imidazole, lysed by ultrasonication, and the cell debris removed by centrifugation.
The soluble fraction of τ, τ(373–563) or τ(1–372) was loaded onto a Ni resin, washed at low imidazole concentration and the τ and τ(373–563) proteins eluted at 100 mM imidazole, whereas τ(1–372) eluted at 50 mM imidazole. The imidazole eluted τ or τ(1–372) was loaded onto a Q Sepharose column equilibrated with buffer A containing 30 mM NaCl, and the pure proteins eluted at 180 or 200 mM NaCl, respectively. The imidazole eluted τ(373–563) protein was loaded onto a Q Sepharose column equilibrated with buffer A containing 10 mM NaCl. The τ(373–563) protein, present in the flow-through fraction, was free of contaminating proteins. The pure proteins were dialysed against buffer B (50 mM phosphate pH 7.5, 50% glycerol, 100 mM NaCl, 0.1 mM DTT) and stored at –20°C. Polyclonal antibodies against purified τ protein were raised.
The protein concentration was determined by UV absorbance, using the corresponding molar extinction coefficients at 280 nm. The molar extinction coefficient for G40P, G40P(109–442), τ, τ(1–372) and τ(373–563) were calculated to be 29 400, 22 900, 31 630, 17 570 and 13 940 M–1 cm–1. G40P, G40P(109–442) are expressed as moles of hexamers, τ and τ(1–372) as moles of tetramers, whereas τ(373–563) is expressed as moles of dimer.
Molecular mass determination
The native molecular mass of τ and τ(1–372) was determined by gel-filtration FPLC using a Superose 12 column, whereas for τ(373–563) a Superdex 75 column was used. A standard curve of Kav against log10 of molecular mass was determined as recommended by the supplier. The standard proteins were: thyroglobulin, 669 kDa; ferritin, 440 kDa; catalase, 232 kDa; aldolase, 158 kDa; bovine serum albumin (BSA), 67 kDa; ovoalbumin, 43 kDa; chymotrysinogen A, 25 kDa; and RNase, 13 kDa.
The stoichiometry of G40P and τ interaction was determined using a Superose 6 column. Chromatography was carried out on buffer C (50 mM Tris pH 7.5, 5% glycerol, 5 mM MgCl2, 1 mM DTT) containing 90 mM NaCl or 600 mM NaCl in the presence or absence of 0.1 mM ATP. The flow rate was 0.2 ml/min and the A260 was measured. Fractions (0.6 ml) were collected and then analysed by SDS–polyacrylamide gel electrophoresis (PAGE).
Electrophoretic mobility shift assay (EMSA)
The sequence of the oligonucleotides that were used in the binding studies were: 50-nt (5′-GTACGGACGTCCAGCTGAGAAGAGGATCCCCGGGTACCAGCTCGAATTC-3′) and 18-nt (5′-GCATATTACCATTCCGCC-3′). The oligonucleotides shorter than 18-nt were made by subsequent removal from the 3′ end of the 18-nt segment; hence all oligonucleotides smaller than 18-nt share the same 5′ end and extend for the indicated length towards the 3′ end (e.g. the sequence of 6-nt is 5′-GCATAT-3′).
The DNA binding activity of G40P or G40P(109–442) in the presence of different concentrations of τ and their mutants derivatives was studied. Oligonucleotides of different length were used to study the stoichiometry of the ternary complex. Proteins were pre-incubated in buffer C containing 50 mM NaCl, 4 mM AMP-PNP or 5 mM ATP for 5 min. Then [γ32P]ssDNA (1 nM) was added (final volume 20 µl) and incubated for 10 min at 37°C. Glutaraldehyde was used only when indicated to a final concentration of 0.012% and incubated for 5 min at room temperature. Loading buffer was added and samples were loaded and separated in a 4% non-denaturing (nd) PAGE in 25 mM Tris, 250 mM glycine supplemented with 5 mM MgCl2, 30 µM AMP-PNP or 50 µM ATP. Gels with AMP-PNP were run at room temperature for 1 h at 150 V. Gels with ATP were run at 4°C for 1 h at 200 V. Gels were dried and exposed to autoradiography.
The half-life of the G40P–ssDNA complex was measured as previously described (33).
ATPase activity measurement
The ATPase activity of G40P or τ protein was determined by measuring the amount of phosphate set free upon hydrolysis as previously described (27). Reaction mixtures containing different proteins concentrations were incubated in buffer C containing 50 mM NaCl. ATP was added to a final concentration of 0.6 mM (specific activity 0.015 µCi/nmol) to measure the G40P ATPase activity. A specific activity of 0.1 µCi/nmol was used to measure the τ ATPase activity.
DNA unwinding assay
A constant amount of G40P was pre-incubated with τ and mutants derivatives at different concentrations for 5 min at room temperature, then the helicase substrate (0.5 nM, viral M13 mp18 annealed to a 50-nt [γ 32P]ssDNA containing a 3′ tail of 22 nt) was added and the mixture incubated for 30 min at 37°C in buffer C containing 50 mM NaCl and 2 mM ATP in a 20 µl reaction as described (27). The reaction was stopped and the samples were loaded onto a 8% ndPAGE in TBE (90 mM Tris, 90 mM boric acid, 2.5 mM EDTA) (27). Gels were run at room temperature for 1 h at 180 V. The dried gels were exposed to autoradiography.
RESULTS AND DISCUSSION
The hexa-histidine tagged τ is an active protein
The B.subtilis dnaX gene encodes only for the 62.5 kDa τ protein, because by western blot analysis one single product of 62.5 kDa was observed (data not shown). This is consistent with the observation that the S.pyogenes dnaX gene encodes only for the τ protein (20). Furthermore, the C-terminal region of the τ protein is essential for viability because in the dnaX51ts strain replacement of Pro465 for Ser renders the cell thermosensitive for growth (this work).
We have fused a sequence coding hexa-histidine tag at the 3′ end of the dnaX gene (dnaXh) and performed complementation studies to learn whether the tag affects the activity of the fused τ protein in vivo. These studies have been carried out either under conditions in which homologous recombination is blocked by the presence of a recA null allele (dnaX51 ΔrecA6) or in the recombination proficient (dnaX51ts) background. The pCB508-borne dnaXh and the pHP13 vector, as a control, were transferred into B.subtilis dnaX51ts ΔrecA6, and the transformants grown at 30°C. Cells bearing the plasmid were grown on LB plates at 30 or 55°C (non-permissive temperature). The dnaX51 ΔrecA6 cells bearing pCB508 were able to grow at both 30 and 55°C, whereas B.subtilis dnaX51ts ΔrecA6 bearing the pHP13 vector only grew at 30°C. Furthermore, B. subtilis dnaX51 cells bearing pCB508 grew at both temperatures, whereas those cells transformed with pHP13 only grew at 30°C. Altogether, these results suggest that the τ protein containing a hexa-histidine at the C-terminal part of the protein, is active in vivo. Similar results have been reported when the gfp gene was fused to the N-terminal or C-terminal part of the τ protein (22,34).
Purification and native state of τ, τ(1–372) and τ(373–563) mutant proteins
Escherichia coli τ protein could be cleaved, by using limited proteolysis, into different fragments representing major protein domains (17). Based on these data and amino acid sequence comparison, we have PCR amplified and cloned the coding sequence of τ variants lacking, either the N-terminal τ(373–563), where domains I–III or amino acids 1–372 are deleted, or the C-terminal moiety τ(1–372), where the C-terminal domain or amino acids 373–563 are deleted (see Fig. 1A). The 63 727 Da τ, 22 117 Da τ(373–563) and 42 849 Da τ(1–372) protein were purified to >99% as judged by SDS–PAGE (data not shown).
The mass of the purified τ protein was analysed by MALDI-TOF spectrometry under conditions were only monomers are detected and the result was a protein with a mass of 63 727 Da. These data were consistent with the predicted molecular mass of wt τ (62 550 Da) plus the hexa-histidine tail. The native mass of τ protein was then determined by gel-filtration chromatography using FPLC and a Superose 12 column. The elution profile of τ protein at low or high ionic strength (90–600 mM NaCl) compared to the standard proteins allowed us to estimate the Mr of the τ protein to be ∼250 000 Da (Fig. 2A). If we assumed that τ is spherical in shape it is likely that the protein is a tetramer in solution. Light-scattering analysis, under similar experimental conditions, showed a population of homogeneous molecules with a radio that corresponded to a tetramer (A. A. Antson and M. I. Martínez-Jiménez, unpublished results). Moreover, preliminary electron microscopy studies suggest that the τ protein is a tetramer (D. Lanzarot, R. Nuñez and M. I. Martínez-Jiménez, unpublished results). It is likely that the results from gel-filtration chromatography are not an artefact and the shape of the τ protein does not grossly affect its mass. Furthermore, the τ subunits of both E.coli and S.pyogenes are tetramers in solution (20,35).
Figure 2.

Molecular mass determination of τ, τ(1–372) and τ(373–563) measured by gel-filtration chromatography. The log of the molecular mass against Kav is represented. Closed circles represent the standard protein markers (see Materials and Methods). (A) The elution of τ and τ(1–372) from a Superose 12 column are shown. The elution of τ at both high and low ionic strength concentrations is identical and denoted with an open arrow (a). The elution of τ(1–372) at 90 mM (b) and at 600 mM NaCl (c) is denoted with filled arrows. (B) The elution of τ(373–563) from a Superdex 75 column. The elution of τ(373–563) at both high and low ionic strength concentrations is identical and denoted by an open arrow (d).
Analysis of the native state of τ(1–372) protein by gel-filtration chromatography, using a Superose 12 column, showed that in the presence of 90 mM NaCl the estimated Mr was ∼170 000 Da, whereas in the presence of 600 mM NaCl the Mr was ∼130 000 Da (Fig. 2A). It is likely, therefore, that τ(1–372) is a tetramer or trimer depending on the ionic strength. Thus, the interactions between τ(1–372) protomers are weaker than those of the full-length protein, and therefore the τ(1–372) tetramer exists in a dynamic equilibrium with trimers. It is likely that the C-terminal region of the protein contributes to the stability of the tetramer, although it is not required for tetramerisation per se. This is consistent with the observation that the oligomerisation domain of E.coli τ lay in domain III (18,36), however, the C-terminal domain of B.subtilis τ seems to stabilise the tetrameric form of the τ protein.
Gel-filtration chromatography using a Superdex 75 column allowed us to determine the native mass of τ(373–563). The elution profile of τ(373–563) at low or high ionic strength (90– 600 mM NaCl) compared to that of the standard proteins corresponded to a protein with an Mr of ∼44 000 Da, which corresponds to twice the mass of the τ(373–563) protomer (Fig. 2B). This is consistent with the observation that τ(373–563) protein, in the presence of a protein cross-linker, migrates as a dimer in SDS–PAGE (data not shown). In contrast, an E.coli τ mutant protein containing domains IV and V is a monomer in solution (37). It is likely, therefore, that B.subtilis τ has two discrete domains for protein oligomerisation, one in the N-terminal and another in the C-terminal of the protein.
G40P interacts with τ in the absence of ATP
In E.coli, an interaction between domain IV of the τ protein and the replicative helicase DnaB has been described (18). To address whether the SPP1 replicative DNA helicase, G40P, interacts with the τ protein gel-filtration chromatographic studies were performed using a Superose 6 column. The elution profile was obtained and fractions containing proteins [18–26 (10.8–15.6 ml)] were analysed by SDS–PAGE (Fig. 3). The τ protein was identified in fractions 23–25, with a peak at fraction 24, corresponding to an Mr of ∼250 000 (Fig. 3A), whereas the replicative G40P DNA helicase, in the absence of a nucleotide, eluted mainly with a peak at fraction 23 corresponding to an Mr of ∼300 000 (27,32,38) (Fig. 3B). This is consistent with the observation that τ is a tetramer in solution (see Fig. 2) and that in the absence of ATP the ring-shaped hexameric G40P is slightly unstable (27,32,38). In order to know whether G40P and τ interact, G40P (0.4 µM as hexamer) was incubated in the presence of an excess of τ (1.6 µM as tetramer) in buffer C containing 90 mM NaCl for 10 min at 20°C, then the reaction was loaded onto a Superose 6 column, and the native mass of the mixed proteins was estimated. The G40P and τ proteins shifted from a peak at fraction 23 and 24, respectively, to a peak of both proteins at fraction 22, which corresponded to an Mr of 550 000–500 000 (Fig. 3C). Molecular mass complexes >550 kDa were not observed. As expected, all G40Ps shift to a higher molecular mass and the excess of free τ moves as a 250 kDa product. Identical results were observed when 0.1 mM ATP is present in the incubation and the chromatography running buffer (data not shown). It is likely, therefore, that hexameric G40P and tetrameric τ interact, and that the interaction between both proteins, which shows a 1:1 stoichiometry, is independent of the presence of ATP or DNA in the reaction mixture.
Figure 3.
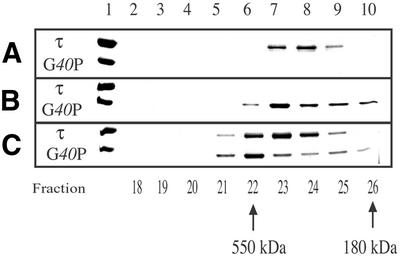
τ interacts with G40P with a 1:1 stoichiometry. Gel-filtration chromatographic analysis of τ (A), G40P (B) and both proteins (C) using a Superose 6 column. The proteins from fractions 18 to 26 were separated by SDS–PAGE as described in Materials and Methods. Fractions numbers are indicated below. The G40P and τ proteins were loaded as markers (lane 1). The estimated mass of spherical proteins, in fractions 22 and 26, are indicated.
G40P interacts with τ and τ(373–563)
The G40P helicase upon binding to a 50-nt-long ssDNA segment gives rise to two discrete complexes (termed I and II) (32). It was suggested that, under these conditions, there is one G40P molecule bound in complex I and two G40P molecules bound to DNA in complex II (32) (Fig. 4A). To study whether the interaction of G40P with the τ protein has any effect on the DNA binding affinity of G40P, EMSA was used (Fig. 4A). The full-length τ protein was not able to form a stable complex with a [γ32P]50-nt or a [γ32P]200-nt ssDNA segment, even in the presence of a large protein excesses (500 molecules per DNA molecule) or in the presence of 0.012% glutaraldehyde (data not shown).
Figure 4.

τ and τ mutants interact with G40P or G40P(109–442) bound to ssDNA in the presence of 4 mM AMP-PNP. (A) G40P (doubling from 1.25 to 20 nM) was incubated with [γ32P]50-nt ssDNA (1 nM) (lanes 2–6). A constant amount of G40P (5 nM) (lanes 7–18) was pre-incubated with doubling concentrations of τ or τ(1–372) (10–80 nM, lanes 7–10 or lanes 11–14, respectively) or with τ(373–563) (40–320 nM, lanes 15–18), and then incubated with [γ32P]50-nt ssDNA (1 nM). (B) The protein concentrations used are the same as that in (A). G40P(109–442) was incubated with [γ32P]50-nt ssDNA (1 nM) (lanes 2–6). A constant amount of G40P(109–442) (lanes 7–18) was pre-incubated with increasing concentrations of τ or τ(1–372) (lanes 7–10 or lanes 11–14, respectively) or τ(373–563) (lanes 15–18), and then incubated with [γ32P]50-nt ssDNA (1 nM). Glutaraldehyde (0.012% final concentration) was added to the reaction mixture before loading into the 4% ndPAGE. The assays were performed as described in Material and Methods. In lane 1, no protein was added. The three type of complexes obtained are denoted by I–III, and FD denotes the free ssDNA. –, the absence of the indicated protein; +, the presence of the indicated protein.
G40P, at a concentration that gives ∼50% of protein– ssDNA complexes (5 nM, Fig. 4A, lane 4), was pre-incubated with increasing concentrations of τ, τ(1–372) or τ(373–563) in buffer C containing 50 mM NaCl and 4 mM AMP-PNP, and then [γ32P]50-nt ssDNA (1 nM) was added. In the presence of 10 nM τ or τ(1–372) or 40 nM τ(373–563) the amount of free DNA decreased >3-fold when compared to the presence of G40P alone. Such an effect is independent of the addition of a large excess (>200-fold) of BSA protein (data not shown). It is likely that τ, τ(1–372) and τ(373–563) interact with free G40P and contribute to G40P–ssDNA complex formation by increasing the interaction of free G40P with ssDNA or stabilising the preformed complex (see below). Under our standard reaction conditions the half-life of the G40P–ssDNA complex was longer than 60 min (data not shown), thus we favour the hypothesis that τ, τ(1–372) or τ(373–563) induces a conformational change in G40P that increases its ssDNA binding efficiency.
In the presence of τ (80 nM) or τ(373–563) (320 nM) G40P forms a complex that migrates with a lower mobility than complex II, termed complex III (Fig. 4A). Although τ(1–372) did not seem to form a stable complex with G40P–ssDNA, a decrease of the free DNA was observed. From these results we could infer that G40P bound to ssDNA interacts with tetrameric τ or with lower affinity to dimeric τ(373–563). This is consistent with the observation that E.coli hexameric DnaB helicase interacts with tetrameric τ or with two or more N-Δ430τ (domains IV and V) molecules (17). We propose that the τ(1–372) protein might also induce a G40P conformational change that increases its affinity to DNA.
Complex III was only observed at a high τ/G40P ratio (>8:1) (see below), but this stoichiometry would not be expected in the replication fork. Moreover, the co-localisation of both proteins in the same DNA segment, however, cannot be ascertained (see below).
G40P(109–442) interacts with τ and τ(373–563)
The interaction of G40P(109–442), which lacks the first 108 amino acid residues, with τ, τ(1–372) or τ(373–563) (see Fig. 1) was measured by EMSA (Fig. 4B). G40P(109–442) has reduced ssDNA binding activity or a reduced rate when complexed with ssDNA (S. Ayora and P. Mesa, unpublished results). Therefore, glutaraldehyde (0.012%) was added to stabilise the G40P(109–442)–ssDNA interaction before loading the reaction into the 4% ndPAGE. In the presence of 0.012% glutaraldehyde the amount of G40P(109–442)– ssDNA complex formed is similar to G40P wt at an equivalent protein concentration (Fig. 4). However, under the experimental conditions used, the G40P(109–442)–DNA complexes were retained in the well, indicating that G40P(109–442) can not bind ssDNA as the wt protein does. Such an effect cannot be reversed by the presence of a >200-fold excess of BSA (data not shown).
G40P(109–442), at a concentration that gives ∼50% of protein–ssDNA complexes (5 nM), was pre-incubated with increasing concentration of τ, τ(1–372) or τ(373–563) in buffer C containing 50 mM NaCl and 4 mM AMP-PNP, and then [γ32P]50-nt DNA (1 nM) was added. In the presence of 10 nM τ, a complex III was observed. A lower mobility complex is also observed with τ(373–563), but here a higher protein concentration was required (data not shown).
In the presence of increasing concentrations of τ(1–372) or τ(373–563) type I and II complexes, as described for G40P–ssDNA, were detected, but large networks retained in the well were not observed (Fig. 4B). Such an effect is independent of the addition of a large excess of BSA protein (data not shown). It is likely, therefore, that τ(1–372) or τ(373–563) induce a conformational change in G40P(109– 442) that increases its ssDNA binding efficiency (see above).
τ Does not bind DNA when complexed with G40P
Escherichia coli DnaB and G40P enzymes bind and encircle an ∼20 ± 3-nt ssDNA (32,39). The 20-nt ssDNA segment is located inside the central hole of the hexameric helicases (32,38). Previously, it was shown that the DnaB binding site is bipartite with ∼10-nt ssDNA defining the strong binding site to the C-terminal part and ∼10-nt ssDNA of weak binding to the N-terminal half of the enzyme (39). Furthermore, nuclease protection studies show that G40P encircles and fully protects a 16-nt-long segment at the 5′ tail of a forked molecule (32).
G40P–ssDNA binding assays were performed with ssDNA segments of different length (18–6-nt) to estimate the minimal G40P binding site. G40P binds to ssDNA segments as short as 10-nt in length (Fig. 5A), but fails to form stable complexes with 8- and 6-nt ssDNA segments (Fig. 5B).6
Figure 5.
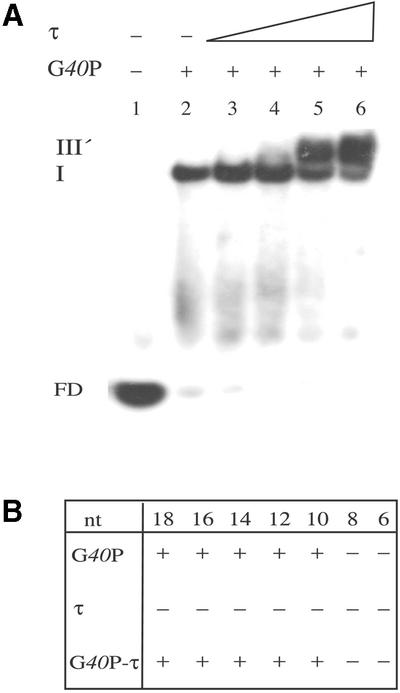
τ interacts with G40P bound to short ssDNA segments in the presence of 4 mM AMP-PNP. (A) Lane 1, no protein was added. Lane 2, G40P (5 nM). Lanes 3–6, G40P (5 nM) was pre-incubated with increasing concentrations of τ (20, 40, 80 nM), and then incubated with [γ32P]10-nt ssDNA as described in Material and Methods. Abbreviations are those of Figure 4, except that in complex III′ it is assumed to have tetrameric τ protein bound to one hexameric G40P–ssDNA complex. (B) DNA binding of G40P, τ or both proteins to ssDNA segments of different length. –, the absence of binding to the specific ssDNA segment; +, binding to the specific ssDNA segment. The length of the oligonucleotides (from 18 to 6 nt) is indicated.
Figure 6.
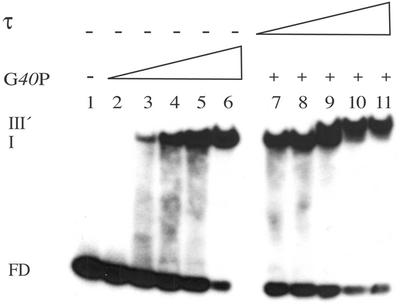
τ interacts with G40P bound to ssDNA with a 1:1 stoichiometry in the presence of 5 mM ATP. G40P (doubling concentrations from 6.25 to 100 nM, lanes 2–6) was incubated with [γ32P]18-nt ssDNA (1 nM). A constant amount of G40P (50 nM) was pre-incubated with τ (doubling from 12.5 to 200 nM, lanes 7–10), and then incubated with [γ32P]18-nt ssDNA (1 nM). The assay was performed as described in Material and Methods. The abbreviations are those of Figure 4.
In a previous section it was shown that G40P and τ interact in the absence of a nucleotide cofactor and DNA (Fig. 3). Furthermore, the presence of τ reduces the mobility of G40P–ssDNA complexes, to render a complex III of undefined stoichiometry (Fig. 4). We could hypothesise that in complex III there are one or two G40P molecules bound to ssDNA and τ protein bound to them. Alternatively, when G40P is already bound to ssDNA, a cryptic τ DNA binding motif might be exposed. To ascertain which of those hypotheses is correct, G40P DNA binding assays to a 10-nt ssDNA segment, in the presence of an increasing concentration of τ, were performed (Fig. 5B). As expected, G40P bound to a 10-nt ssDNA segment formed only one type of complex (I) (32) (Fig. 5A). When the τ protein is added to the G40P–ssDNA a low migration complex, termed III′ was formed (Fig. 5A). Complex III′ has to be formed by a tetramer of τ bound to a hexamer of G40P bound to ssDNA, because the entire 10-nt ssDNA segment was covered by G40P.
The stoichiomety of G40P-ATP and τ is 1:1
The affinity of G40P for ssDNA increased when ATP was replaced by non-hydrolysable ATP analogue AMP-PNP by ∼20-fold (32). In the presence of ATP, G40P binds and releases the ssDNA substrate, whereas in the presence of AMP-PNP, stable binding to ssDNA is mainly observed (32). In a previous section it was shown that there was a significant difference in the stoichiometry of G40P and τ depending on the condition tested. In the absence of a nucleotide cofactor both proteins interact with a 1:1 stoichiometry by gel-filtration chromatography (Fig. 3), but in the presence of AMP-PNP and ssDNA a >8-fold excess of τ is needed for τ–G40P–ssDNA complex formation by EMSA (Fig. 4A). The stoichiometry of G40P and τ, in the presence of an 18-nt ssDNA segment and 5 mM ATP, was measured by EMSA (Fig. 6). G40P, at a concentration that gives ∼50% of protein–ssDNA complexes (50 nM), was incubated with increasing concentrations of τ. In the presence of 50 nM τ, the preformed G40P–ssDNA shifts to a lower migration form, suggesting that there are one G40P hexamer and one τ tetramer. Since, in the presence of AMP-PNP, a higher ratio of τ/G40P is required for reaching an equilibrium, we might assume either that the AMP-PNP bound to τ reduces its ability to interact with the helicase or the AMP-PNP bound to the helicase reduces its ability to interact with τ. Alternatively, τ interacts with G40P at or near the ATP binding pocket and AMP-PNP reduces the τ–G40P interaction.
Here, again, in the presence of 2 or 4 τ per G40P an increment of G40P–ssDNA complex formation was observed (see above). Since, τ at a ratio of 2:1 and 4:1 increases ∼2-fold G40P-ATP–ssDNA complex formation, whereas a τ ratio of 8:1 and 16:1 increases also ∼2-fold G40P-AMP-PNP–ssDNA complex formation, we assume that τ–AMP-PNP is limited to interact with G40P or vice versa.
τ Enhances G40P ATPase and helicase activities
The ATP hydrolysis of G40P in the presence or the absence of τ and the presence of ssDNA was measured. The τ protein contains the two typical Walker domains (21), therefore, the ATPase activity of the protein was measured. We could show that τ protein has a low ATPase activity (0.02 µmol/min/mg) that is independent of ssDNA (Table 1). This is consistent with the observation that E.coli τ has a weak ATPase activity, but in this case, the ATPase activity is stimulated by DNA (40).
Table 1. Effect of τ in G40P ATPase and helicase activities.
| ssDNA | ATPasea | Helicase (%)b | |
|---|---|---|---|
| G40P | – | 1.8 | |
| + | 11.8 | 5% | |
| τ | – | 0.02 | |
| + | 0.02 | NA | |
| G40P–τ | – | 4.8 | |
| 1:1 | + | 15.8 | 6 |
| G40P–τ | – | 5.1 | |
| 1:4 | + | 16.5 | 9 |
aThe ATPase activity is expressed in µmol/min/mg.
bSuboptimal G40P unwinding activity. NA, not applied.
When the G40P ATPase activity is measured in the presence of increasing concentration of τ only a marginal increase in ATPase activity (<2-fold) was observed at a 1:1 stoichiometry to reach saturation at 1:2 or higher τ ratios (Table 1). In a previous section we have observed that the presence of τ, τ(1–372) or τ(373–563) proteins increases the affinity of G40P for ssDNA by ∼2-fold (see Fig. 4). It is likely, therefore, that the stimulation of the ATPase activity results from an increase in ssDNA binding, but an indirect effect cannot be ruled out.
A similar marginal increase (2-fold) in the G40P helicase activity is observed when τ is present in a 1:2 stoichiometry (Table 1). This is consistent with the observation that the addition of E.coli τ has no effect on the DnaB unwinding activity, but τ in the Pol III enzyme increases the DnaB unwinding activity >10-fold (17).
CONCLUSIONS
The interaction of the τ subunit and the replicative G40P DNA helicase is critical for the coupling of both polymerases (PolC and DnaE) and the G38P–G39P–G40P primosomal apparatus at the SPP1 oriL region. We expressed and purified τ and two truncated protein derivatives: τ(1–372) (contains domains I–III) and τ(373–563) (contains domains IV and V). G40P hexameric helicase seems to interact with tetrameric τ with a 1:1 stoichiometry. For the first time we provide evidence that the C-terminal domain of the replicative helicase, G40P(109–442), interacts with the dimeric τ(373–563). A weak interaction between τ(1–372) and G40P is also suggested. The tetramerisation domain of τ is mapped in the N-terminal region of the protein, but is modulated by the C-terminal domain of the protein. As shown for the E.coli clamp loader, we assume that δ and δ′ interact with the τ N-terminal moiety (reviewed in 13). The E.coli asymmetric clamp loader has each subunit of the τ/γ complex occupying a unique position (τγτ) with each τ protein interacting with one α subunit to form an asymmetric dimeric Pol III (α4εθβτ2γδδ′χψ) holoenzyme (13). Here, the asymmetric distribution of subunits within the τγτ complex may aid the two core polymerases in their distinct function of continuous leading and discontinuous lagging strand (13). In Gram-positive bacteria there are different Pol III enzymes (PolC2βτ4δδ′, DnaE2βτ4δδ′ and perhaps PolCDnaEβτ4δδ′) (20,23), but the PolC4βτ4δδ′ or DnaE4βτ4δδ′ enzymes were not observed. It is unlikely, therefore, that each τ subunit is interacting with each of the polymerising subunits of the Pol III enzymes, hence a model that accommodates the findings presented here is illustrated in Figure 7. In the τ N-terminal domain lays the primary tetramerisation site and in the C-terminal lays a dimerisation domain. The C-terminal region of hexameric G40P would interact with the dimeric τ C-terminal end, rather than with each subunit of tetrameric τ. We hypothesised that G40P binds to one or both C-terminal moiety of each dimeric τ (Fig. 7). If the replicative Pol III holoenzyme contains one PolC and one DnaE/α polymerising core, we propose that the G40P–τ complex provides an asymmetric component on the interaction of one or both asymmetric Pol IIIs. The τ C-terminal dimer would place the DnaE/α at the lagging and PolC at the leading strand. This is consistent with the observation that the S.pyogenes tetrameric τ subunit interacts with two polymerising subunits (PolC and DnaE/α) to form either the processive (PolC2βτ4δδ′) or the poorly processive (DnaE2βτ4δδ′) enzymes (20). Furthermore, G40P hexameric helicase interacts with tetrameric τ with a 1:1 stoichiometry (this work).
Figure 7.

The proposed τ model in the replisome. The tetrameric τ N-terminal moiety is depicted as large, dark-grey-filled balls, the dimeric C-terminal moiety as small, light-grey-filled balls and hexameric G40P as a ring with a central hole. The symmetric τ C-terminal moiety interacts with G40P and might interact with PolC and DnaE/α. We assume that G40P bound to the lagging strand DNA introduces an asymmetric component in the B.subtilis replisome.
Acknowledgments
ACKNOWLEDGEMENTS
We are very grateful to S. Ayora for her interest in this project. This research was partially supported by Grants BMC2000-0548 and SAF2001-4339 from MCyT-DGI and QLK2-CT-2000-00634 from the European Union to J.C.A.
REFERENCES
- 1.Jacob F., Brenner,S. and Cuzin,F. (1963) On the regulation of DNA replication in bacteria. Cold Spring Harbor Symp. Quant. Biol., 28, 329–348. [Google Scholar]
- 2.Kornberg A. and Baker,T.A. (1992) DNA Replication, 2nd Edn. W.H.Freeman and Co., New York.
- 3.Messer W. and Weigel C. (1996) Initiation of chromosome replication. In Neidhardt,F.C (ed.), Escherichia coli and Salmonella: Cellular and Molecular Biology, 2nd Edn. ASM Press, Washington, DC, Vol. II, pp. 1579–1601.
- 4.Wahle E., Lasken,R.S. and Kornberg,A. (1989) The dnaB–dnaC replication protein complex of Escherichia coli. II. Role of the complex in mobilizing dnaB functions. J. Biol. Chem., 264, 2469–2475. [PubMed] [Google Scholar]
- 5.Marszalek J. and Kaguni,J.M. (1994) DnaA protein directs the binding of DnaB protein in initiation of DNA replication in Escherichia coli. J. Biol. Chem., 269, 4883–4890. [PubMed] [Google Scholar]
- 6.Learn B.A., Um,S.J., Huang,L. and McMacken,R. (1997) Cryptic single-stranded DNA binding activities of the phage Lambda P and Escherichia coli DnaC replication initiation proteins facilitate the transfer of E. coli DnaB helicase onto DNA. Proc. Natl Acad. Sci. USA, 94, 1154–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker T.A. and Bell,S.P. (1998) Polymerases and the replisome: machines within machines. Cell, 92, 295–305. [DOI] [PubMed] [Google Scholar]
- 8.Chang P. and Marians,K.J. (2000) Identification of a region of Escherichia coli DnaB required for functional interaction with DnaG at the replication fork. J. Biol. Chem., 275, 26187–26195. [DOI] [PubMed] [Google Scholar]
- 9.Kim S., Dallmann,H.G., McHenrik,C.S. and Marians,K.J. (1996) Coupling of replicative polymerase and helicase: a τ–DnaB interaction mediates rapid replication fork movement. Cell, 84, 643–650. [DOI] [PubMed] [Google Scholar]
- 10.Zechner E.L., Wu,C.A. and Marians,K.J. (1992) Coordinated leading- and lagging-strand synthesis at the Escherichia coli DNA replication fork. II. Frequency of primer synthesis and efficiency of primer utilization control Okazaki fragment size. J. Biol. Chem., 267, 4045–4053. [PubMed] [Google Scholar]
- 11.Kong X.P., Onrust,R., O’Donnell,M. and Kuriyan,J. (1992) Three-dimensional structure of the subunit of E. coli DNA polymerase III holoenzyme: a sliding DNA clamp. Cell, 69, 425–437. [DOI] [PubMed] [Google Scholar]
- 12.Dallmann H.G. and McHenry,C.S. (1995). DnaX complex of Escherichia coli DNA polymerase III holoenzyme. J. Biol. Chem., 270, 29563–29569. [PubMed] [Google Scholar]
- 13.O’Donnell M., Jeruzalmi,D. and Kuriyan,J. (2001) Clamp loader structure predicts the architecture of DNA polymerase III holoenzyme and RFC. Curr. Biol., 11, 935–946. [DOI] [PubMed] [Google Scholar]
- 14.Kim D.R. and McHenry,C.S. (1996) In vivo assembly of overproduced DNA polymerase III. Overproduction, purification and characterization of the alpha, alpha-epsilon and alpha-epsilon-theta subunits. J. Biol. Chem., 271, 20681–2089. [DOI] [PubMed] [Google Scholar]
- 15.Jeruzalmi D., O’Donnell,M. and Kuriyan,J. (2001) Crystal structure of the processivity clamp loader gamma (gamma) complex of E. coli DNA polymerase III. Cell, 106, 429–441. [DOI] [PubMed] [Google Scholar]
- 16.Glover B.P. and McHenry,C.S. (2001) The DNA polymerase III holoenzyme: an asymmetric dimeric replicative complex with leading and lagging strand polymerises. Cell, 105, 925–934. [DOI] [PubMed] [Google Scholar]
- 17.Gao D. and McHenry,C.S. (2001) τ Binds and organizes Escherichia coli replication proteins through distinct domains. Partial proteolysis of terminally tagged τ to determine candidate domains and to assign domain V as the α binding domain. J. Biol. Chem., 276, 4433–4440. [DOI] [PubMed] [Google Scholar]
- 18.Gao D. and McHenry,C.S. (2001) τ Binds and organizes Escherichia coli replication proteins through distinct domains. Domain IV, located within the unique C terminus of τ, binds the replication fork helicase, DnaB. J. Biol. Chem., 276, 4441–4446. [DOI] [PubMed] [Google Scholar]
- 19.Gao D. and McHenry,C.S. (2001) τ Binds and organizes Escherichia coli replication proteins through distinct domains. Domain III, shared by γ and τ, binds δδ′ and χψ. J. Biol. Chem., 276, 4447–4453. [DOI] [PubMed] [Google Scholar]
- 20.Bruck I. and O’Donnell,M. (2000) The DNA replication machine of a Gram-positive organism. J. Biol. Chem., 275, 28971–28983. [DOI] [PubMed] [Google Scholar]
- 21.Alonso J.C., Shirahige,K. and Ogasawara,N. (1990) Molecular cloning, genetic characterization and DNA sequence analysis of the recM region of Bacillus subtilis. Nucleic Acids Res., 18, 6771–6777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imai Y., Ogasawara,N., Ishigo-oka,D., Kadoya,R., Daito,T. and Moriya,S. (2000) Subcellular localization of DNA-initiation proteins of Bacillus subtilis: evidence that chromosome replication begins at either edge of the nucleoids. Mol. Microbiol., 36, 1037–1048. [DOI] [PubMed] [Google Scholar]
- 23.Noirot-Gros M.-F., Dervyn,E., Wu,L.J., Mervelet,P., Errington,J., Ehrlich,S.D. and Noirot,P. (2002) An expanded view of bacterial DNA replication. Proc. Natl Acad. Sci. USA, 99, 8342–8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dervyn E., Suski,C., Daniel,R., Bruand,C., Chapuis,J., Errington,J., Jannière,L. and Ehrlich,S.D. (2001) Two essential DNA polymerases at the bacterial replication fork. Science, 294, 1716–1719. [DOI] [PubMed] [Google Scholar]
- 25.Pedre X., Weise,F., Chai,S., Luder,G. and Alonso,J.C. (1994) Analysis of cis and trans acting elements required for the initiation of DNA replication in the Bacillus subtilis bacteriophage SPP1. J. Mol. Biol., 236, 1324–1340. [DOI] [PubMed] [Google Scholar]
- 26.Missich R., Weise,F., Chai,S., Lurz,R., Pedre,X. and Alonso,J.C. (1997) The replisome organizer (G38P) of Bacillus subtilis bacteriophage SPP1 forms specialized nucleoprotein complexes with two discrete distant regions of the SPP1 genome. J. Mol. Biol., 270, 50–64. [DOI] [PubMed] [Google Scholar]
- 27.Ayora S., Stasiak,A. and Alonso,J.C. (1999) The Bacillus subtilis bacteriophage SPP1 G39P delivers and activates the G40P DNA helicase upon interacting with the G38P-bound replication origin. J. Mol. Biol., 288, 71–85. [DOI] [PubMed] [Google Scholar]
- 28.Ayora S., Langer,U. and Alonso,J.C. (1998) Bacillus subtilis DnaG primase stabilises the bacteriophage SPP1 G40P helicase–ssDNA complex. FEBS Lett., 439, 59–62. [DOI] [PubMed] [Google Scholar]
- 29.Studier F.W. (1991). Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J. Mol. Biol., 219, 37–44. [DOI] [PubMed] [Google Scholar]
- 30.Struck J.C.R., Alonso,J.C., Toschka,H.Y. and Erdmann,V.A. (1990) The Bacillus subtilis small cytoplasmic RNA gene and dnaX map near the chromosomal replication region. Mol. Gen. Genet., 222, 470–472. [DOI] [PubMed] [Google Scholar]
- 31.Haima P., Bron,S. and Venema,G. (1987) The effect of restriction on shotgun cloning and plasmid stability in Bacillus subtilis. Mol. Gen. Genet., 209, 335–342. [DOI] [PubMed] [Google Scholar]
- 32.Ayora S., Weise,F.,Mesa,P., Stasiak,A. and Alonso,J.C. (2002) Bacillus subtilis bacteriophage SPP1 hexameric DNA helicase, G40P, interacts with forked DNA Nucleic Acids Res., 30, 2280–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rojo F. and Alonso,J.C. (1994) A novel site-specific recombinase encoded by the Streptococcus pyogenes plasmid pSM19035. J. Mol. Biol., 238, 159–172. [DOI] [PubMed] [Google Scholar]
- 34.Lemon K.P. and Grossman,A.D. (1998) Localization of bacterial DNA polymerase: evidence for a factory model of replication. Science, 282, 1516–1519. [DOI] [PubMed] [Google Scholar]
- 35.Dallmann H.G. and McHenry,C.S. (1995) DnaX complex of Escherichia coli DNA polymerase III holoenzyme. Physical characterization of the DnaX subunits and complexes. J. Biol. Chem., 270, 29563–29569. [PubMed] [Google Scholar]
- 36.Glover B.P., Pritchard,A.E. and McHenry,C.S. (2001) τ binds and organizes Escherichia coli replication proteins through distinct domains: domain III, shared by γ and τ, oligomerizes DnaX. J. Biol. Chem., 276, 35842–35846. [DOI] [PubMed] [Google Scholar]
- 37.Dallmann H.G., Kim,S., Pritchard,A.E., Marians,K.J. and McHenry,C.S. (2000) Characterization of the unique C terminus of the Escherichia coli τ DnaX protein. Monomeric C-τ binds α and DnaB and can partially replace τ in reconstituted replication forks. J. Biol. Chem., 275, 15512–15519. [DOI] [PubMed] [Google Scholar]
- 38.Bárcena M., Martin,C.S., Weise,F., Ayora,S., Alonso,J.C. and Carazo,J.M. (1998) Polymorphic quaternary organization of the Bacillus subtilis bacteriophage SPP1 replicative helicase (G40P). J. Mol. Biol., 283, 809–819. [DOI] [PubMed] [Google Scholar]
- 39.Jezewska M.J., Rajendran,S. and Bujalowski,W. (1998) Functional and structural heterogeneity of the DNA binding of the E. coli primary replicative helicase DnaB protein. J. Biol. Chem., 273, 9058–9069. [DOI] [PubMed] [Google Scholar]
- 40.Lee S.H. and Walker,J.R. (1987) Escherichia coli DnaX product, the τ subunit of DNA polymerase III, is a multifunctional protein with single-stranded DNA-dependant ATPase activity. Proc. Natl Acad. Sci. USA, 84, 2713–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]