

Abstract
Because interactions between activated CD4+ T cells and antigen-presenting cells (APCs) are crucial for optimal APC function, defective CD4+ T-cell activation may contribute to APC dysregulation in HIV infection. Here, we show that CD4+ T cells exposed during stimulation to noninfectious HIV having functional envelope glycoproteins failed to provide activation signals to autologous dendritic cells (DCs). Consequently, important DC functions, including production of immunoregulatory cytokines (interleukin-12 p40 and interleukin-10) and up-regulation of costimulatory molecules (CD86, CD40, CD83), as well as the capacity to stimulate naive allogeneic T cells, were all adversely affected. The blunted up-regulation of CD154 in CD4+ T cells that were activated in the presence of noninfectious viruses is likely to be the major underlying mechanism for these defects. Addition of recombinant trimeric CD154 could restore production of cytokines by DCs cocultured with HIV-exposed T cells. Moreover, the functional defects mediated by coculture with HIV-exposed T cells were similar to those following antibody blockade of CD40-CD154 interactions. HIV-mediated blunted CD154 expression may thus play an important role in the suppression of cell-mediated immunity seen in HIV infection.
Introduction
HIV infection is associated with a gradual loss of immune competence, leading to an increased susceptibility to infection and cancer. Although HIV infection is associated with abnormalities in most compartments of the immune system, defects in cell-mediated immunity appear to be of the greatest clinical importance. Impaired antigen-presenting cell (APC) function is thought to be a critical component of HIV-associated immunodeficiency (reviewed in Chougnet1 and Donaghy et al2), although the extent of this impairment remains controversial.3,4 However, the mechanism(s) underlying these defects have not been clearly delineated. In particular, it is not well established whether these defects are directly due to infection of APCs by HIV, or exposure of APCs to HIV gp120, as suggested by recent papers,5-8 or are a consequence of dysregulation of CD4+ T cells, because interactions between activated CD4+ T cells and APCs are crucial for optimal APC function.
Among the various receptor-ligand pairs important for CD4+ T cell-APC communication, CD40-CD154 interactions are crucial. CD40, a member of the tumor necrosis factor (TNF)-receptor superfamily, is constitutively expressed on the surface of APCs, including B cells, macrophages, and dendritic cells (DCs) (reviewed in van Kooten and Banchereau9). CD40 ligand or CD154, a member of the TNF superfamily, undergoes tightly regulated inducible expression on the surface of CD4+ T cells as a result of signaling via the T-cell receptor (TCR).9 CD40-CD154 interactions are critical for the induction and regulation of cell-mediated immunity, as demonstrated in individuals presenting with genetic CD154 deficiency (the X-linked hyper-IgM syndrome) along with a variety of mouse models. In particular, engagement of CD40 on DCs and macrophages induces profound phenotypic changes and the production of immune regulatory mediators, as well as promotes APC survival (reviewed in Chougnet1). Although CD40 engagement is one of several pathways of maturation/activation of DCs (reviewed in Chougnet1 and Guermonprez et al10), its unique role is underscored by the inability of TNF-α to rescue DC function in CD40-/- mice.11 Of note, patients with X-linked hyper-IgM syndrome also present with a severe impairment of DC maturation,12 along with a range of opportunistic infections similar to that seen with AIDS.13
The capacity of CD4+ T cells to up-regulate CD154 following stimulation through the TCR becomes progressively impaired in HIV infection, in parallel with overall immune suppression.14-16 Defective CD40-CD154 interactions could thus play a crucial role in the APC abnormalities described for HIV-infected donors, which include decreased levels of CD80 and CD86 expression by lymph node DCs,17 defective allostimulatory capacity of circulating DCs,18 and decreased numbers of circulating DCs.19-21 Consistent with this hypothesis, we and others have shown that providing exogenous recombinant CD154 increases interleukin 12 (IL-12) production by peripheral mononuclear cells (PBMCs) from HIV-infected donors.22-24 In addition, our recent data indicate that levels of IL-12 p70 production correlate with CD154 expression.16 Importantly, the CD154 dysregulation described in HIV-infected donors can also be modeled in vitro, following engagement of the CD4 receptor by noninfectious viruses having functional envelope glycoproteins or by native HIV gp120 itself prior to T-cell activation, as we and others have previously shown.16,25 Our previous data also suggest that HIV-mediated blunted CD154 up-regulation results from defects in the signaling cascades upstream of CD154 promoter activation16 and particularly in defects in the early events that follow TCR engagement (C.C., unpublished data, April 2004), which is in agreement with previous studies.26-28
We therefore hypothesize that engagement of the CD4 receptor by HIV gp120 leads to decreased CD154 expression on TCR-activated CD4+ T cells. This in turn will impair the maturation/activation of APCs, with consequences for immune responses dependent on optimal APC function. HIV-mediated blunted CD154 expression on T cells may thus contribute importantly to the suppression of cell-mediated immunity seen in HIV infection.
Materials and methods
Cell preparation
Monocytic and nonmonocytic populations were separated by elutriation, starting from leukophoresis products of blood bank donors who were not infected with HIV and were frozen until use. Approval for the use of leukapheresis products was obtained from the Institutional Review Board of the National Institutes of Health Blood Bank. DCs were then differentiated in vitro by culture of monocytes in the presence of granulocyte macrophage colony-stimulating-factor (GM-CSF) and IL-4 (both 103 U/mL every 2nd day, for 7 days; PeproTech, Rocky Hill, NJ), leading to a population of greater than 80% pure immature DCs (defined as CD1a+). Autologous CD4+ T cells were purified from the nonmonocytic population by negative selection (CD4-negative selection kit; Dynal, Lake Success, NY). Purified CD4+ T cells were greater than 95% pure, as assessed by flow cytometry analysis. Cytokines exhibited undetectable levels of endotoxin, as tested by the limulus amebocyte lysate (LAL) assay (< 0.1 ng/μg). Other reagents (culture medium, serum, etc) were purchased with the specification of undetectable endotoxin levels.
T-cell stimulation
Six days after initiation of DC differentiation, purified autologous CD4+ T cells were stimulated for 24 hours with anti-CD3/CD28 beads (Dynal; 2.5 μL/106 T cells) in the presence or absence of Aldrithiol-2-inactivated HIV-1MN (AT-2 HIV). AT-2 HIV was prepared following well-established protocols to inactivate the infectivity of retroviruses, with the lack of residual infectivity after AT-2 treatment confirmed by in vitro culture, as previously described.29-31 AT-2 HIV was used at the concentration of 1 μg HIV p24gag equivalent/mL in most experiments.29 The molecular weight of p24 is approximately one-fifth that of gp120, and the average p24/gp120 molar ratio on HIV-1 virions is approximately 50:1 to 100:1.32 Therefore, the mass difference will be 10-fold less gp120 than p24; that is, a gp120 concentration of approximately 1 nM was used in most experiments.
To control for possible confounding effects of nonviral, host cell proteins present even in the highly purified virus preparations tested, we used microvesicles, which were prepared from uninfected cultures of the CEM X 174 cell line using the same procedures as those used to prepare AT-2 HIV.33 Microvesicles were added at a concentration that provided an equal amount of total protein as AT-2 HIV.
After 24 hours, stimulating beads were removed by magnetic separation, the CD4+ T cells were washed 3 times in RPMI to remove unbound viruses or nonviral cell proteins, and live cells were counted. In parallel, some CD4+ T cells were cultured for 24 hours without stimulation. To determine the efficiency of the washing procedures to remove AT-2 HIV bound to the surface of activated T cells, we treated AT-2 HIV-exposed cells with trypsin. CD4+ T cells obtained from 3 individual subjects prepared as described (ie, activated for 24 hours with anti-CD3/CD28 in the presence of 1 μg equivp24 AT-2 HIV, then washed 3 times) were treated or not with trypsin for 5 minutes at room temperature, followed by washes, following the standard procedure to remove bound viruses.34 Cells were then lysed, and cell-associated p24 was determined in trypsin-treated versus untreated cells by enzyme-linked immunosorbent assay (ELISA; Coulter, Miami, FL). Cell-associated p24 was reduced in trypsin-treated cells by 13.2% ± 1.0%, indicating that about 0.13 μg equivp24 AT-2 HIV may still remain on the surface of T cells after the washing steps.
To evaluate whether engagement of CD4 by other ligands during activation resulted in effects similar to those observed with AT-2 HIV, CD4+ T cells were incubated for 1 hour at 4°C with Ab against domain 1 of the CD4 molecule (clone QS4120) or an isotype-matched control Ab (mouse IgG1); both Ab (Ancell, Bayport, MN) were used at 10 μg/mL. As a control, CD4+ T cells were also left untreated. After several washes to remove unbound Ab, cells were stimulated with anti-CD3/CD28 beads, as described for HIV-exposed T cells.
DC/T-cell cocultures
CD4+ T cells that had been previously stimulated with anti-CD3/CD28 beads or had been left unstimulated, as described in “T-cell stimulation,” were mixed with autologous DCs at a ratio of 5:1 (7.5 × 105 T cells and 1.5 × 105 DCs/well). DCs were also cocultured with unstimulated autologous CD4+ T cells, at the same ratio. Cocultures were carried on for 24 hours. As a control, DCs were also cultured alone. In some experiments, blocking anti-CD154 Ab (clone M91, 10 μg/mL; Immunex, Seattle, WA) or recombinant trimeric CD154 (0.5-5 μg/mL; Amgen) were added to the DC/T-cell cocultures. These reagents exhibited undetectable levels of endotoxin (< 0.1 ng/μg). Of note, we have previously used the appropriate isotype-matched control Abs for the CD154 Abs from Immunex.22,35 In particular, human PBMCs stimulated in the presence of such control Abs exhibited normal production of IL-12.22 Similarly, production of IL-12 p40 by mouse DCs stimulated in a very similar experimental system as the one used herein (ie, in vitro coculture of mouse DCs with autologous activated T cells) was not inhibited by the control Ab.35 On the basis of consistent results for these controls in past experiments, the isotype-matched control Ab for the anti-CD154 Ab was not included again in the present study.
Determination of DC maturation
After culture, DCs were washed with fluorescence-activated cell sorting (FACS) buffer (PBS, 10% FCS, 0.01% sodium azide) and incubated with human IgG (20 μg/mL; Sigma, St Louis, MO) for 10 minutes at 4°C, to block Fc receptors. They were then stained with labeled Abs that recognize CD40, CD83, CD86, CD1a, or with isotype-matched control Abs (Ab from BD PharMingen, Mountain View, CA or eBiosciences, San Diego, CA), for 30 minutes at 4°C. The cells were then washed twice before being fixed in FACS buffer containing 4% paraformaldehyde. Surface expression was analyzed using a FACSCalibur and the CellQuest software (Becton Dickinson, Franklin Lakes, NJ). A minimum of 20 000 CD1a+ cells was analyzed. Mean fluorescence index (MFI) for each parameter was determined on gated CD1a+ cells.
Cytokine production
Production of IL-12 p40 (R&D Systems, Minneapolis, MN; sensitivity, 4 pg/mL), IL-10 (PharMingen; sensitivity, 10 pg/mL), or interferon γ (IFN-γ; PharMingen; sensitivity, 10 pg/mL) was measured by ELISA. Undetectable values were assigned an arbitrary value of half the sensitivity limit, that is 2, 5, and 5 pg/mL for IL-12 p40, IL-10, and IFN-γ, respectively.
Allostimulatory capacity of DCs
DCs were cocultured with autologous T cells stimulated with anti-CD3/CD28 beads in the presence or absence of AT-2 HIV, as described in “T-cell stimulation.” After 24 hours, the number of DCs in each preparation was calculated by counting large cells, and equal numbers of DCs were used to stimulate allogeneic naive CD4+ T cells. To do that, cells (DCs and T cells) that had been cocultured were irradiated (30 Gy [3000 rad]) and used to stimulate allogeneic naive T cells. As a control, DCs that had been cultured alone were also irradiated and similarly used. Allogeneic naive CD4+ T cells had been purified from the T cells of another HIV-uninfected donor by sorting on size and 4-color staining (CD8-CD19-CD45RA+ CD62L+). Allogeneic T cells were mixed with DCs at several ratios of DCs to allogeneic T cells (6 × 104 allogeneic T cells/well; triplicate wells/condition). Baseline activation of allogeneic T cells was determined by culturing them alone. After 4 days, supernatants were harvested for measurement of cytokine production (IFN-γ and IL-10), and cells were pulsed overnight with 3H Thymidine (1 μCi [0.037 MBq]/well) to determine cell proliferation. Proliferation results are expressed as simulation indexes, defined as the ratio between the average cpm in wells containing DCs and T cells and the average cpm in wells containing T cells alone.
Statistical analysis
Paired t tests were used to determine the difference between in vitro conditions. Dose-dependence was tested by ANOVA tests. P values less than .05 were considered as significant.
Results
HIV-exposed activated T cells fail to provide optimal maturation signals to DCs
We have previously reported that exposure of CD4+ T cells to AT-2 HIV affects the level of activation of these cells.16 To assess the functional consequences of this effect, we determined what type of signal HIV-exposed CD4+ T cells give to autologous DCs, using an in vitro system of DC/T-cell cocultures. In this experimental system, CD4+ T cells were either stimulated with anti-CD3/CD28 beads alone (“untreated”) or in the presence of AT-2 HIV (“AT-2 HIV-exposed”) or control microvesicles (“vesicle-exposed”). As a control, unstimulated CD4+ T cells (“unstimulated”) were also used. After 24 hours, T cells were extensively washed and cocultured with autologous DCs for an additional 24-hour period. To assess the functional consequences of AT-2 HIV exposure of T cells, we determined the level of activation of DCs, by measuring the expression of 3 classic markers of DC activation/maturation (CD86, CD40, and CD83), as well as the production of immunoregulatory cytokines (IL-12 p40 and IL-10) by DCs. As shown in Figure 1A-B, AT-2 HIV-exposed T cells failed to provide optimal maturation signal to DCs, as evidenced by a marked inhibition in the expression of the 3 measured markers of DC activation, compared with DCs cocultured with untreated T cells (all P < .02, paired t test). Furthermore, DCs cocultured with AT-2 HIV-exposed T cells produced approximately 2-fold less IL-10 and IL-12 p40 than DCs cocultured with untreated T cells (both P < .05, paired t test; Figure 1C). In contrast, exposure of T cells to control microvesicles during activation induced only minimum inhibition, as compared with untreated T cells (all P > .18, paired t test; Figure 1A-C). As a negative control, unstimulated T cells were mixed with DCs at a similar ratio and, as expected, induced minimum DC activation (Figure 1A-C).
Figure 1.
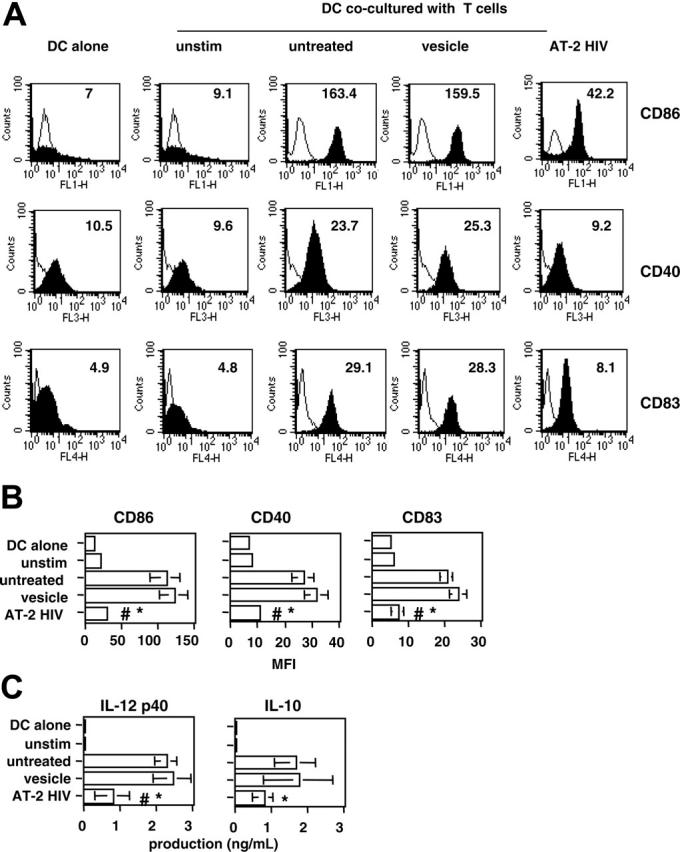
Decreased maturation of DCs cocultured with T cells activated in presence of AT-2 HIV. Purified CD4+ T cells were left unstimulated (unstim) or stimulated with anti-CD3/CD28 beads, either without treatment (untreated) or in the presence of control microvesicles (vesicle) or AT-2 HIVMN (AT-2 HIV). After 24 hours, T cells were mixed with autologous monocyte-derived DCs at a ratio of 5:1. Similar percentages of dead T cells (∼ 10%) were observed in the different cultures before coculture (results not shown). (A) Inhibition of DC activation marker expression in one representative subject. Expression markers were determined on DCs, gated on forward-scatter and CD1a expression. Open histograms represent the staining by the isotype-matched control Ab. Closed histograms represent the staining by the Ab of interest. Numbers in the graph represent the MFI for each condition. (B) Inhibition of DC activation marker expression in 7 subjects. Results are shown as the mean (± SE) MFI obtained in 7 individual subjects. (C) Inhibition of production of immunoregulatory cytokines. Levels of IL-12 p40 and IL-10 in 24-hour coculture supernatants were determined by ELISA. Results are shown as the mean (± SE) levels measured in 7 individual subjects. (B-C) *Significant inhibition (P < .05, paired t test) between untreated and AT-2 HIV exposed; #significant inhibition (P < .05, paired t test) between vesicle- and AT-2 HIV-exposed.
We then determined whether the effect of exposure to AT-2 HIV was dose dependent, because our earlier results have shown a dose-dependent inhibitory effect of AT-2 HIV exposure on CD154 up-regulation.16 We therefore exposed the T cells to graded concentrations of AT-2 HIV (from 0.125-1 μg equivp24/106 cells) during the activation phase. Exposure to lower concentrations of AT-2 HIV also affected the activation signal those T cells provided to DCs, because CD86 expression was inhibited by an average of 51% and 22% when DCs had been cultured with T cells exposed to 0.5 and 0.25 μg equivp24, respectively. A lower dose (0.125 μg equivp24) had no effect on CD86 expression by DCs (Figure 2A). In general, the effect of AT-2 HIV exposure was dose dependent (all P ≤ .05, ANOVA; Figure 2B). Interestingly, p40 production was the function the most sensitive to AT-2 HIV exposure, because exposure to 0.25 μg equivp24 only was still able to significantly inhibit it (P < .05).
Figure 2.

Dose dependence of the effect of exposure to AT-2 HIV. DCs were cocultured with autologous CD4+ T cells as described in Figure 1. T cells had been activated in the presence of either control microvesicles (vesicles) or graded doses of AT-2 HIV (0.125-1 μg equivp24/106 cells). (A) Inhibition of CD86 expression in one representative subject. CD86 was determined on DCs, gated on forward-scatter and CD1a expression. Numbers in the graph represent the MFI for each condition. (B) Summary of the inhibition observed in 4 subjects. Results are shown as the mean ± SE of the results obtained in 4 individual subjects. Numbers in the graph represent the P value for each comparison (ANOVA).
To evaluate whether engagement of CD4 by other ligands during activation resulted in effects similar to those observed with AT-2 HIV, we used an anti-CD4 Ab. As shown in Table 1, CD4+ T cells activated in the presence of anti-CD4 Ab provided defective activation signal to DCs, as evidenced by decreased IL-12 p40 production and CD40 expression. Moreover, this effect was very similar to that of AT-2 HIV-exposed T cells (Figure 1). These data are in agreement with our previous results, which have shown that AT-2 HIV is inhibiting T-cell activation principally through interactions between the CD4 receptor and HIV gp120.16
Table 1.
Decreased maturation of DC co-cultured with T cells activated in presence of anti-CD4 Ab
| IL-12 p40 production | CD40 expression | |
|---|---|---|
| Donor 1 | ||
| DCs alone | < 4 | 10 |
| DCs + T-cell stimulated | 1628 | 49.1 |
| DCs + T-cell stimulated + anti-CD4 Ab | 1000 | 27.4 |
| DCs + T-cell stimulated + control Ab | 2008 | 44.8 |
| Donor 2 | ||
| DCs alone | 13 | 9.7 |
| DCs + T-cell stimulated | 2453 | 49.6 |
| DCs + T-cell stimulated + anti-CD4 Ab | 809 | 2.2 |
| DCs + T-cell stimulated + control Ab | 1824 | 52.4 |
CD4+ T cells purified from 2 individuals were incubated with Ab against the domain 1 of the CD4 molecule or control Ab (both Ab at 10 μg/mL) or left untreated. After 1 hour, cells were washed and stimulated with anti-CD3/CD28 beads. After 24 hours, T cells were mixed with autologous monocyte-derived DCs at a ratio of 5:1, as described in the legend of Figure 1. IL-12 p40 production (expressed in pg/mL) and CD40 expression on DCs (expressed in MFI) were analyzed as described in Figure 1.
DCs cultured with stimulated HIV-exposed T cells have decreased allostimulatory capacity
A major functional hallmark of DCs is their capacity to stimulate naive T cells (reviewed in Guermonprez et al10). We therefore determined whether DCs cultured with AT-2 HIV-exposed T cells had defective allostimulatory properties compared with DCs cultured with untreated T cells. Allogeneic naive CD4+ T cells were mixed with DCs at different ratios of DCs to T cells. As shown in Figure 3, DCs cultured with AT-2 HIV-exposed T cells did not provide an optimal activation signal to naive T cells, as evidenced by the 2-fold reduction of the stimulation index. However, the capacity of such DCs to stimulate T cells remained higher than that of unstimulated DCs (Figure 3A).
Figure 3.

Decreased allostimulation capacity of DCs cocultured with HIV-exposed stimulated T cells. DCs cocultured with autologous T cells as described in Figure 1 were irradiated (30 Gy [3000 rad]) and used to stimulate allogeneic naive CD4+ T cells, at the indicated ratios of DCs to T cells. As a control, allogeneic T cells were cultured alone. After 4 days, proliferation and production of cytokines was measured. (A) Proliferation. Results are expressed as simulation index (average radioactivity in wells containing DCs and T cells/average radioactivity in wells containing control T cells). Average cpm measured in wells containing only T cells was 471. Results are from 1 representative individual of 2 individual subjects tested. (B) Cytokine production. Results are expressed as mean production of IL-10 or IFN-γ (average of 3 wells containing DCs and T cells).
Interestingly, the type of cytokines produced by these T cells was also different, depending on the DCs used for stimulation. As shown in Figure 3B, IFN-γ production by allogeneic T cells stimulated by DCs cocultured with AT-2 HIV-exposed T cells was markedly lower than that by T cells stimulated by DCs cocultured with untreated T cells (2113 pg/mL versus 239 pg/mL). In contrast, IL-10 production by the former T cells was higher than that by the latter T cells (30 pg/mL versus < 10 pg/mL).
Taken together, the data presented in Figures 1, 2, 3 suggest that exposure of CD4+ T cells to HIV during activation broadly decreased the capacity of these T cells to provide activation signals to DCs. Consequently, important DC functions, namely production of immunoregulatory cytokines, up-regulation of crucial costimulatory molecules, and stimulation of naive cells, were all adversely affected.
Impaired CD154 induction on HIV-exposed CD4+ T cells is likely to be the origin of defective DC maturation
Our previous results have demonstrated that CD154 up-regulation is impaired on AT-2 HIV-exposed CD4+ T cells.16 As CD40-CD154 interactions are crucial for DC activation (reviewed in Chougnet1), defective CD154 expression by AT-2 HIV-exposed T cells could explain their poor capacity to activate DCs. To determine whether this is the case, we first compared the effect of a blocking anti-CD154 Ab with that mediated by AT-2 HIV-exposed T cells. Three different culture conditions were established: DCs were cultured with either (1) stimulated T cells (“untreated”), (2) T cells stimulated in the presence of AT-2 HIV (“AT-2 HIV exposed”), and (3) untreated T cells, and the cocultures were performed in the presence of blocking anti-CD154 Ab. Functional inhibition in AT-2 HIV-exposed or anti-CD154-treated T cells was defined by comparison with untreated cells. As expected on the basis of our previous results,35 anti-CD154 Ab resulted in incomplete DC maturation and activation, as evidenced by a 60% to 75% decreased induction of CD86, CD83, and CD40, as well as decreased IL-12 p40 production (Figure 4A). IL-12 p40 production, and expression of CD40 and CD83 were significantly decreased in anti-CD154-treated cultures (condition 3) compared with control cultures (condition 1) (all P < .02). The same trend was observed for CD86 expression; however, the difference did not reach statistical difference (P = .10). Importantly, coculture with AT-2 HIV-exposed T cells (condition 2) had the same effect on DC activation as blocking CD40-CD154 interactions (condition 3) (Figure 4A, all P > .19, paired t test).
Figure 4.

Defective DC maturation is likely due to impaired CD154 induction on AT-2 HIV-exposed CD4+ T cells. (A) Exposure to AT-2 HIV and neutralizing anti-CD154 Ab mediates similar defective DC maturation. Three different culture conditions were established: DCs were cultured with either untreated T cells, AT-2 HIV exposed T cells, or untreated T cells, and the cocultures were performed in the presence of blocking anti-CD154 Ab (10 μg/mL). Functional inhibition in AT-2 HIV exposed T cells or untreated T cells was defined by comparison with untreated T cells. Results are shown as the mean percentage of inhibition ± SE for each parameter (3 donors). (B) Decreased IL-12 p40 induction because of exposure of T cells to AT-2 HIV is corrected by exogenous rCD154. DCs were cocultured with AT-2 HIV-exposed T cells and rCD154 (+ +), AT-2 HIV-exposed T cells (+ -), untreated T cells and rCD154 (- +), and untreated T cells (- -). Results are shown as the mean ± SE IL-12 p40 production (4 donors). *Significant difference (P < .03, paired t test); NS indicates a nonsignificant difference (P > .5).
The fact that AT-2 HIV exposure and anti-CD154 Ab had similar consequences only suggests a causal relationship between HIV-mediated blunted CD154 expression and nonoptimal DC function. Therefore, to further evaluate such a potential causative link, we determined whether addition of recombinant trimeric CD154 (rCD154) could reverse the functional defects exhibited by DCs cocultured with AT-2 HIV-exposed T cells. Four different culture conditions were established (conditions 1 to 4, from top to bottom in Figure 4B): DCs were cultured with (1) AT-2 HIV-exposed T cells and rCD154, (2) AT-2 HIV-exposed T cells, (3) untreated T cells and rCD154, and (4) untreated T cells. As shown in Figure 4B, providing exogenous rCD154 restored similar levels of IL-12 p40 production by DCs cultured with AT-2 HIV-exposed T cells as that of DCs cultured with untreated T cells (condition 1 versus condition 3, P = .32, paired t-test). Providing exogenous rCD154 also corrected IL-10 production by DCs (condition 1, 5.4 ± 1.3 ng/mL; condition 3, 5.1 ± 0.6 ng/mL; P = .77). Of note, addition of rCD154 induced higher IL-12 p40 and IL-10 production in DCs cocultured with untreated T cells (condition 3 versus condition 4; both P < .05). Such increases in cytokine production by rCD154-treated DCs could be because the amount of CD154 expressed by untreated T cells (condition 4) was not sufficient to induce maximum DC activation, which was then induced by providing additional exogenous rCD154 (condition 3). Interestingly, addition of lower amounts of exogenous rCD154 (as low as 1 μg/mL) also restored IL-10 and IL-12 p40 production in 2 individual subjects (Table 2).
Table 2.
Effect of graded doses of exogenous rCD154 on production of immune regulatory cytokines
| IL-12 p40 | IL-10 | |
|---|---|---|
| Donor 1 | ||
| DCs alone | < 4 | < 10 |
| DCs + untreated T cells | 4008 | 5260 |
| DCs + AT-2 HIV exposed T cells | 1755 | 2172 |
| DCs + AT-2 HIV exposed T cells + 1 μg/mL rCD154 | 4725 | 6458 |
| DCs + AT-2 HIV exposed T cells + 2.5 μg/mL rCD154 | 5118 | 6942 |
| DCs + AT-2 HIV exposed T cells + 5 μg/mL rCD154 | 5154 | 7091 |
| Donor 2 | ||
| DCs alone | < 4 | < 10 |
| DCs + untreated T cells | 3013 | 539 |
| DCs + AT-2 HIV exposed T cells | 1400 | 93 |
| DCs + AT-2 HIV exposed T cells + 0.5 μg/mL rCD154 | 2246 | 633 |
| DCs + AT-2 HIV exposed T cells + 1 μg/mL rCD154 | 4050 | 1141 |
| DCs + AT-2 HIV exposed T cells + 2.5 μg/mL rCD154 | 4018 | 2098 |
| DCs + AT-2 HIV exposed T cells + 5 μg/mL rCD154 | 4461 | 3644 |
Purified CD4+ T cells from 2 individuals were stimulated with anti-CD3/CD28 beads, in the presence of AT-2 HIV (AT-2—exposed T cells) or in the absence of AT-2 HIV (untreated), as described in Figure 1. After several washes, T cells were cultured with autologous DCs in the absence or presence of graded doses of rCD154 (0.5-5 μg/mL). Production of IL-12 p40 and IL-10 (expressed in pg/mL) was analyzed as described in the legend of Figure 1.
Taken together, the data presented in Figure 4 strongly suggest that the blunted CD154 up-regulation on CD4+ T cells that follows CD4 engagement by HIV represents a major mechanism underlying the failure of such cells to provide optimal maturation/activation signals to DCs.
Discussion
A variety of functional defects have been reported in APCs isolated from HIV-infected subjects. However, the mechanism(s) underlying these defects have not been clearly delineated. In particular, it is not well established whether these defects are directly due to infection of APCs by HIV, or exposure of APCs to HIV gp120, as suggested by recent papers,5-8 or are a consequence of dysregulation of CD4+ T cells. In the present study, we focused on the latter mechanism. We used a model in which DCs are cultured with T cells that had been exposed to noninfectious HIV, to exclude any possible confounding effects due to direct infection by HIV. Our results clearly show that HIV-exposed T cells provide a suboptimal activation/maturation signal to DCs, as evidenced by low levels of expression of the costimulatory molecules CD40 and CD86, as well as of the classical maturation marker, CD83. Such exposure also profoundly reduced secretion of IL-10 and IL-12 p40. Thus, this in vitro experimental system closely replicates several of the defects described in APCs from HIV-infected donors. It is known that HIV infection of DCs can directly affect their function, although results have varied, depending on the experimental system used. Recently, using measurement of intracellular cytokines, it has been shown that CD154-stimulated HIV-infected DCs did not produce IL-12 p40, in contrast to uninfected DCs,7 and failed to express DC-LAMP.6 Similarly, HIV-infected DCs were poor stimulators of allogeneic T cells.5 Our results do not exclude the possibility that direct infection plays a role in vivo; however, the percentage of HIV-infected DCs in vivo is always very low,36,37 making it highly unlikely that HIV infection of DCs could explain the extent of defective APC function in HIV-infected donors. Our results thus provide an alternative mechanism, in which defective APC function arises from CD4+ T-cell dysregulation, without a requirement of infection of the affected APCs.
The mechanism by which HIV-exposed T cells are responsible for suboptimal DC activation likely involves defective CD40-CD154 interactions. Indeed, our results show that the addition of recombinant CD154 overcame impaired DC activation. Furthermore, the effect of a blocking anti-CD154 Ab was very similar to that of HIV-exposed T cells. Blunted CD154 up-regulation on HIV-exposed T cells, as well as on CD4+ T cells isolated from HIV-infected donors, has been demonstrated by our laboratory and others.14-16,25 The foremost mechanism underlying this defect was the interaction between the major HIV surface glycoprotein, gp120, and the CD4 receptor on the surface of CD4+ T cells.16,25 HIV gp120 can be detected in tissues38,39 and circulates in the blood of HIV-infected donors on the surface of virions (both infectious and noninfectious) and as a free protein.40 Of note, only a limited fraction (< 0.1%) of circulating virions are demonstrably infectious41,42; therefore, exposure to inactivated viruses may mimic the most frequent type of CD4-HIV interactions that occur in vivo. One potential concern is the concentration of AT-2 HIV we used in this study. Klasse and Moore43 have recently raised concerns that the doses of gp120 or inactivated virions used in vitro may not reflect accurately what is present in vivo. In this paper, we report a significant effect of a concentration of AT-2 HIV that corresponds to approximately 1 nM gp120, which is higher than the reported gp120 plasma levels (reviewed in Klasse and Moore43). However, it must be noted that gp120 concentrations in tissues are essentially unknown, especially in the tissue microenvironment of interstitial lymph node spaces. However, they are predicted to be higher than in plasma, because cell-associated HIV RNA and DNA can be much higher in lymph nodes than in blood.44 Moreover, we have shown that T cells exposed to as little as 250 pM gp120 also failed to provide optimal activation signal to DCs. In particular, IL-12 p40 induction was significantly inhibited after exposure of T cells to such a concentration of gp120 (Figure 2). Interestingly, p40 production was more sensitively affected by the addition of AT-2 HIV to T-cell cultures than the other markers of DC activation were. One potential reason underlying this differential sensitivity is that p40 production also depends on the presence of IFN-γ. IFN-γ synergizes with other stimuli to induce p40, through transcriptional activation principally mediated by the IFN-γ-induced IFN consensus sequence binding protein (ICSBP).45 Stimulation of CD4+ T cells in the presence of HIV gp160 has been shown to decrease their production of IFN-γ.46 Therefore, it is possible that the addition of AT-2 HIV into T-cell cultures may also have affected the capacity of these CD4+ T cells to produce IFN-γ and not only their CD154 expression. Both mechanisms could thus have participated into the profound inhibition of IL-12 p40 production.
In this study, we only used AT-2 HIVMN, a CXCR4-using strain. However, we have previously shown that the AT-2-treated CCR5-using strain ADA inhibited CD154 induction as much as the CXCR4-using strain MN did.16 The underlying mechanism was dependent on CD4-gp120 interactions, not on interactions between gp120 and the chemokine receptors.16 In addition, our present results show that T cells exposed to anti-CD4 Ab also induced defective DC activation (Table 1). Taken together, these data further suggest that the mechanism for defective DC function described herein is not likely to be restricted to a few specific HIV strains.
In several studies, HIV-exposed DCs showed a partial activation profile, as they harbored a phenotype typical of activated DCs (up-regulation of CD80, CD86, CD40, HLA class II molecules, as well as the appearance of the maturation marker CD83), but lacked allostimulatory capacity and did not secrete IL-12, TNF-α, or chemokines.8 Therefore, these HIV-exposed DCs did not display as profound a defect as the one demonstrated in our experimental system. It must also be noted that the direct deleterious effect of AT-2 HIV on DC function remains controversial, because in some studies inactivated simian immunodeficiency virus-pulsed DCs were shown to be efficient at inducing antiviral T-cell responses47 and proved effective therapeutic vaccines in a primate model.48 Of note, it is unlikely that a direct effect of AT-2 HIV on DCs plays a significant role in our results. Indeed, using trypsin treatment to determine the amount of AT-2 HIV that may have remained bound on the surface of T cells after washing and could thus directly interact with DCs, we estimated that amount to be about 0.13 μg equivp24 AT-2 HIV, when the original viral input was 1 μg equivp24. Such a concentration was however shown to be inefficient at decreasing DC activation (Figure 2). The data from Fantuzzi et al8 and our data thus underscore the fact that gp120 and/or noninfectious viruses could play a major role in defective APC function in HIV-infected donors, either through impairment of CD4+ T-cell activation leading to subsequent defect in DC function (the present study) or via direct effects on DCs.8,49
The HIV-mediated suboptimal DC activation described herein may also have profound and durable effects by switching the profile of T-cell activation and potentially inducing regulatory T cells. It has been recently postulated that stimulation of T cells by immature DCs can induce active immunosuppression through the induction of such regulatory cells. Indeed, repetitive stimulation of T cells with human immature DCs induced IL-10-producing T-cell regulatory 1-like cells (Tr1 cells), which, in addition, showed an early up-regulation of the negative regulator cytotoxic T-lymphocyte-associated molecule 4 (CTLA-4).50 Our results are in agreement with that hypothesis, because we have shown that naive T cells, following their stimulation by DCs cultured with HIV-exposed T cells, exhibit a profile reminiscent of that of Tr1 cells, such as low proliferation and reduced production of IFN-γ but increased production of IL-10. Interestingly, similar results were recently obtained in cocultures of DCs and T cells purified from HIV-infected donors, which were shown to be suppressive of “third-party” immune responses, through an IL-10-dependent mechanism.6 Taken together, our results and those of Granelli-Piperno et al6 suggest that HIV-mediated suboptimal activation of DCs may play an important role in HIV pathogenesis, through the induction of regulatory T cells, because such cells can hamper antiviral effector immune responses (reviewed in Belkaid and Rouse51).
In the present study, we have shown that defective APC function in HIV-infected donors may occur as a consequence of primary CD4+ T-cell dysregulation, in particular as a result of defective CD154 expression. Importantly, noninfectious viruses induced this defect, underscoring the crucial role that such defective particles likely play in the pathogenesis of HIV infection. Our results thus suggest a model in which exposure of CD4+ T cells to HIV, infectious or not, starts a vicious cycle, whereby suboptimal activation of T cells leads to functional impairment of APCs, which then fail to give optimal feedback signal to T cells and may favor the emergence of regulatory T cells.
Acknowledgments
We thank Dr Larry Wahl for providing purified cell populations from HIV-uninfected blood bank donors and Julian Bess Jr for the production of the AT-2 inactivated virus preparations used in these studies. We also thank Drs. Christopher Karp, Julie Nelson, and Gene M. Shearer for their critical reading of this manuscript, and Norah Shire for her expert help in statistical analyses.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government.
Prepublished online as Blood First Edition Paper, November 3, 2005; DOI 10.1182/blood-2005-07-2731.
Supported by the National Institutes of Health (grant AI 056927; C.C.) and federal funds from the National Cancer Institute, National Institutes of Health (contract no. NO1-CO-12400; J.D.L.).
An Inside Blood analysis of this article appears at the front of this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 U.S.C. section 1734.
References
- 1.Chougnet C. Role of CD40 ligand dysregulation in HIV-associated dysfunction of antigen-presenting cells. J Leukoc Biol. 2003;74: 702-709. [DOI] [PubMed] [Google Scholar]
- 2.Donaghy H, Stebbing J, Patterson S. Antigen presentation and the role of dendritic cells in HIV. Curr Opin Infect Dis. 2004;17: 1-6. [DOI] [PubMed] [Google Scholar]
- 3.Fan Z, Huang XL, Borowski L, Mellors JW, Rinaldo CR Jr. Restoration of anti-human immunodeficiency virus type 1 (HIV-1) responses in CD8+ T cells from late-stage patients on prolonged antiretroviral therapy by stimulation in vitro with HIV-1 protein-loaded dendritic cells. J Virol. 2001;75: 4413-4419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang XL, Fan Z, Colleton BA, et al. Processing and presentation of exogenous HLA class I peptides by dendritic cells from human immunodeficiency virus type 1-infected persons. J Virol. 2005;79: 3052-3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawamura T, Gatanaga H, Borris D, Connors M, Mitsuya H, Blauvelt A. Decreased stimulation of CD4(+) T cell proliferation and IL-2 production by highly enriched populations of HIV-infected dendritic cells. J Immunol. 2003;170: 4260-4266. [DOI] [PubMed] [Google Scholar]
- 6.Granelli-Piperno A, Golebiowska A, Trumpfheller C, Siegal FP, Steinman RM. HIV-1-infected monocyte-derived dendritic cells do not undergo maturation but can elicit IL-10 production and T cell regulation. Proc Natl Acad Sci U S A. 2004; 101: 7669-7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smed-Sorensen A, Lore K, Walther-Jallow L, Andersson J, Spetz AL. HIV-1-infected dendritic cells up-regulate cell surface markers but fail to produce IL-12 p70 in response to CD40 ligand stimulation. Blood. 2004;104: 2810-2817. [DOI] [PubMed] [Google Scholar]
- 8.Fantuzzi L, Purificato C, Donato K, Belardelli F, Gessani S. Human immunodeficiency virus type 1 gp120 induces abnormal maturation and functional alterations of dendritic cells: a novel mechanism for AIDS pathogenesis. J Virol. 2004;78: 9763-9772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Kooten C, Banchereau J. CD40-CD40 ligand. J Leukoc Biol. 2000;67: 2-17. [DOI] [PubMed] [Google Scholar]
- 10.Guermonprez P, Valladeau J, Zitvogel L, Thery C, Amigorena S. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002;20: 621-667. [DOI] [PubMed] [Google Scholar]
- 11.Miga A, Masters S, Durell B, et al. Dendritic cell longevity and T cell persistence is controlled by CD154-CD40 interactions. Eur J Immunol. 2001; 31: 959-965. [DOI] [PubMed] [Google Scholar]
- 12.Fontana S, Moratto D, Mangal S, et al. Functional defects of dendritic cells in patients with CD40 deficiency. Blood. 2003;102: 4099-4106. [DOI] [PubMed] [Google Scholar]
- 13.Levy J, Espanol-Boren T, Thomas C, et al. Clinical spectrum of X-linked hyper-IgM syndrome. J Pediatr. 1997;131: 47-54. [DOI] [PubMed] [Google Scholar]
- 14.Vanham G, Penne L, Devalck J, et al. Decreased CD40 ligand induction in CD4 T cells and dysregulated IL-12 production during HIV infection. Clin Exp Immunol. 1999;117: 335-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subauste CS, Wessendarp M, Portilllo JA, et al. Pathogen-specific induction of CD154 is impaired in CD4+ T cells from human immunodeficiency virus-infected patients. J Infect Dis. 2004;189: 61-70. [DOI] [PubMed] [Google Scholar]
- 16.Zhang R, Fichtenbaum C, Hildeman D, Lifson J, Chougnet C. CD40 Ligand dysregulation in HIV infection: HIV gp120 inhibits signaling cascades upstream of CD40 ligand transcription. J Immunol. 2004;172: 2678-2686. [DOI] [PubMed] [Google Scholar]
- 17.Lore K, Sonnerborg A, Brostrom C, et al. Accumulation of DC-SIGN+CD40+ dendritic cells with reduced CD80 and CD86 expression in lymphoid tissue during acute HIV-1 infection. AIDS. 2002; 16: 683-692. [DOI] [PubMed] [Google Scholar]
- 18.Donaghy H, Gazzard B, Gotch F, Patterson S. Dysfunction and infection of freshly isolated blood myeloid and plasmacytoid dendritic cells in patients infected with HIV-1. Blood. 2003;101: 4505-4511. [DOI] [PubMed] [Google Scholar]
- 19.Pacanowski J, Kahi S, Baillet M, et al. Reduced blood CD123+ (lymphoid) and CD11c+ (myeloid) dendritic cell numbers in primary HIV-1 infection. Blood. 2001;98: 3016-3021. [DOI] [PubMed] [Google Scholar]
- 20.Donaghy H, Pozniak A, Gazzard B, et al. Loss of blood CD11c(+) myeloid and CD11c(-) plasmacytoid dendritic cells in patients with HIV-1 infection correlates with HIV-1 RNA virus load. Blood. 2001;98: 2574-2576. [DOI] [PubMed] [Google Scholar]
- 21.Chehimi J, Campbell D, Azzoni L, et al. Persistent decreases in blood plasmacytoid dendritic cell number and function despite effective highly active antiretroviral therapy and increased blood myeloid dendritic cells in HIV-infected individuals. J Immunol. 2002;168: 4796-4801. [DOI] [PubMed] [Google Scholar]
- 22.Chougnet C, Thomas E, Landay AL, et al. CD40 Ligand and IFN-γ synergistically restore IL-12 production in HIV-infected patients. Eur J Immunol. 1998;28: 646-656. [DOI] [PubMed] [Google Scholar]
- 23.Chougnet C, Kovacs A, Baker R, et al. Influence of human immunodeficiency virus-infected maternal environment on development of infant interleukin-12 production. J Infec Dis. 2000;181: 1590-1597. [DOI] [PubMed] [Google Scholar]
- 24.Subauste C, Wessendarp M, Smulian A, Frame P. Role of CD40 ligand signaling in defective type 1 cytokine response in human immunodeficiency virus infection. J Infect Dis. 2001;183: 1722-1731. [DOI] [PubMed] [Google Scholar]
- 25.Chirmule N, McCloskey T, Hu R, Kalyanaraman V, Pahwa S. HIV gp120 inhibits T cell activation by interfering with expression of costimulatory molecules CD40 ligand and CD80 (B7-1). J Immunol. 1995;155: 917-924. [PubMed] [Google Scholar]
- 26.Goldman F, Crabtree J, Hollenback C, Koretzky G. Sequestration of p56(lck) by gp120, a model for TCR desensitization. J Immunol. 1997;158: 2017-2024. [PubMed] [Google Scholar]
- 27.Gratton S, Julius M, Sekaly R. Lck-independent inhibition of T cell antigen response by the HIV gp120. J Immunol. 1998;161: 3551-3556. [PubMed] [Google Scholar]
- 28.Marschner S, Hunig T, Cambier J, Finkel T. Ligation of human CD4 interferes with antigen-induced activation of primary T cells. Immunol Lett. 2002;82: 131-139. [DOI] [PubMed] [Google Scholar]
- 29.Rossio J, Esser M, Suryanarayana K, et al. Inactivation of human immunodeficiency virus type 1 infectivity with preservation of conformational and functional integrity of virion surface proteins. J Virol. 1998;72: 7992-8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chertova E, Crise B, Morcock D, Bess JJ, Henderson L, Lifson J. Sites, mechanism of action and lack of reversibility of primate lentivirus inactivation by preferential covalent modification of virion internal proteins. Curr Mol Med. 2003;3: 265-272. [DOI] [PubMed] [Google Scholar]
- 31.Morcock DR, Thomas JA, Gagliardi TD, et al. Elimination of retroviral infectivity by N-ethylmaleimide with preservation of functional envelope glycoproteins. J Virol. 2005;79: 1533-1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chertova E, Bess JJ, Crise B, et al. Envelope glycoprotein incorporation, not shedding of surface envelope glycoprotein (gp120/SU), is the primary determinant of SU content of purified human immunodeficiency virus type 1 and simian immunodeficiency virus. J Virol. 2002;76: 5315-5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bess JJ, Gorelick R, Bosche W, Henderson L, Arthur L. Microvesicles are a source of contaminating cellular proteins found in purified HIV-1 preparations. Virology. 1997;230: 134-144. [DOI] [PubMed] [Google Scholar]
- 34.Spenlehauer C, Kirn A, Aubertin AM, Moog C. Antibody-mediated neutralization of primary human immunodeficiency virus type 1 isolates: investigation of the mechanism of inhibition. J Virol. 2001;75: 2235-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chougnet C, Freitag C, Schito M, Thomas E, Sher A, Shearer G. In vivo CD40-CD154 (CD40 ligand) interaction induces integrated HIV expression by APC in an HIV-1-transgenic mouse model. J Immunol. 2001;166: 3210-3217. [DOI] [PubMed] [Google Scholar]
- 36.Cameron PU, Forsum U, Teppler H, Granelli PA, Steinman RM. During HIV-1 infection most blood dendritic cells are not productively infected and can induce allogeneic CD4+ T cells clonal expansion. Clin Exp Immunol. 1992;88: 226-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McIlroy D, Autran B, Cheynier R, et al. Infection frequency of dendritic cells and CD4+ T lymphocytes in spleens of human immunodeficiency virus-positive patients. J Virol. 1995;69: 4737-4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pantaleo G, Graziosi C, Demarest J, et al. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature. 1993;362: 355-358. [DOI] [PubMed] [Google Scholar]
- 39.Jones M, Bell J, Nath A. Immunolocalization of HIV envelope gp120 in HIV encephalitis with dementia. AIDS. 2000;14: 2709-2713. [DOI] [PubMed] [Google Scholar]
- 40.Oh S-K, Cruikshank WW, Raina J, et al. Identification of HIV-1 envelope glycoprotein in the serum of AIDS and ARC patients. J Acq Imm Deficiency Syndr. 1992;5: 251-256. [PubMed] [Google Scholar]
- 41.Piatak MJ, Saag M, Yang L, et al. High levels of HIV-1 in plasma during all stages of infection determined by competitive PCR. Science. 1993; 259: 1749-1754. [DOI] [PubMed] [Google Scholar]
- 42.Dimitrov D, Willey R, Sato H, Chang L, Blumenthal R, Martin M. Quantitation of human immunodeficiency virus type 1 infection kinetics. J Virol. 1993;67: 2182-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klasse PJ, Moore JP. Is there enough gp120 in the body fluids of HIV-1-infected individuals to have biologically significant effects? Virology. 2004;323: 1-8. [DOI] [PubMed] [Google Scholar]
- 44.Yerly S, Rutschmann OT, Opravil M, Marchal F, Hirschel B, Perrin L. Cell-associated HIV-1 RNA in blood as indicator of virus load in lymph nodes. The Swiss HIV Cohort Study. J Infect Dis. 1999; 180: 850-853. [DOI] [PubMed] [Google Scholar]
- 45.Wang IM, Contursi C, Masumi A, Ma X, Trinchieri G, Ozato K. An IFN-gamma-inducible transcription factor, IFN consensus sequence binding protein (ICSBP), stimulates IL-12 p40 expression in macrophages. J Immunol. 2000;165: 271-279. [DOI] [PubMed] [Google Scholar]
- 46.Hu R, Oyaizu N, Kalyanaraman VS, Pahwa S. HIV-1 gp160 as a modifier of Th1 and Th2 cytokine response: gp160 suppresses interferon-gamma and interleukin-2 production concomitantly with enhanced interleukin-4 production in vitro. Clin Immunol Immunopathol. 1994;73: 245-251. [DOI] [PubMed] [Google Scholar]
- 47.Frank I, Santos JJ, Mehlhop E, et al. Presentation of exogenous whole inactivated simian immunodeficiency virus by mature dendritic cells induces CD4+ and CD8+ T-cell responses. J Acquir Immune Defic Syndr. 2003;34: 7-19. [DOI] [PubMed] [Google Scholar]
- 48.Lu W, Arraes LC, Ferreira WT, Andrieu JM. Therapeutic dendritic-cell vaccine for chronic HIV-1 infection. Nat Med. 2004;10: 1359-1365. [DOI] [PubMed] [Google Scholar]
- 49.Williams MA, Trout R, Spector SA. HIV-1 gp120 modulates the immunological function and expression of accessory and co-stimulatory molecules of monocyte-derived dendritic cells. J Hematother Stem Cell Res. 2002;11: 829-847. [DOI] [PubMed] [Google Scholar]
- 50.Jonuleit H, Schmitt E, Schuler G, Knop J, Enk A. Induction of interleukin 10-producing, nonproliferating CD4(+) T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J Exp Med. 2000;192: 1213-1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Belkaid Y, Rouse BT. Natural regulatory T cells in infectious disease. Nat Immunol. 2005;6: 353-360. [DOI] [PubMed] [Google Scholar]