


Abstract
Evidence is provided that some condensed linker histone-containing chromatin structures are highly flexible in solutions containing 2 mM Mg2+. Chromatin assembled in vitro ± histone H5 on a 6.3 kb linear DNA fragment in 90 mM NaCl using the polyglutamic acid method sedimented fairly homogeneously. The H5-containing sample had s20, w values that were 58–69% greater than the sample lacking H5. Chromatin assembled on linear pUC19 plasmid DNA was treated with T4 DNA ligase in solutions containing 2 mM Mg2+ over a range of DNA concentrations. It was found that the intramolecular DNA ends of the chromatin could be joined together more efficiently than the intramolecular ends of the naked DNA at the higher DNA concentrations. This result could not be attributed to the effective reduction in DNA length by nucleosome formation. The chromatin structures formed did not have naked DNA tails extending from the ends as assessed by exonuclease III digestion. Chromatin assembled on DNA shortened by up to 420 bp gave very similar results, suggesting that the structure was a flexible one, rather than a rigid one having DNA ends that were fortuitously juxtaposed.
INTRODUCTION
Gene regulation probably requires DNA looping between transcription promoters and distal regulatory elements where regulatory proteins bind (1–3). Because the intervening DNA exists in the form of chromatin, it seems important to know the inherent flexibility of chromatin. For example, in the idealized solenoid model (4,5), where nucleosomes of successive coils contact each other, the fiber would likely not be flexible enough to loop unless it were significantly distorted or unfolded. For some other models, considerable variability of the chromatin higher-order structure is allowed (6–9). In this case, some of the possible structures are expected to be fairly flexible, even without linker histone removal and/or core histone hyperacetylation (10). In the former case, mechanisms that disrupt the ‘30 nm fiber’ would be of primary interest. Thus, the extent to which the structure of chromatin is dynamic influences how one thinks about DNA looping and mechanisms of gene regulation.
It has been shown recently, by using FLP recombination, that DNA integrated into genomic DNA in cultured cells can loop efficiently, although with slow kinetics requiring a time course of several days (11). However, the DNA was integrated into active chromatin regions permissive for expression of the puromycin resistance gene. The flexibility of inactive chromatin has not been addressed.
In this work, the flexibility of chromatin was assessed by the ligation of linear 2–3 kb DNA fragments into circles after their assembly into chromatin in vitro. The polyglutamic acid system (12) can be used in order to assemble chromatin on DNA restriction fragments several thousand base pairs in length in vitro. Arrays of physiologically spaced nucleosomes are generated on some genomic DNA sequences by this procedure (13–15). The resultant chromatin is soluble in 90 mM NaCl, and makes a good substrate for T4 DNA ligase. The ligation products can be readily identified by gel electrophoresis.
Mg2+ concentrations of 5–10 mM are optimum for ligation of DNA with T4 DNA ligase. However, Mg2+ concentrations >2 mM cause aggregation and precipitation of chromatin (16). Under these conditions, the circularization of DNA fragments that have been assembled into chromatin does not occur efficiently. However, it was found that at 2 mM Mg2+, there is still sufficient enzyme activity for the ligation of DNA or chromatin fragments possessing GC-rich sticky ends such as AvaI, where the 5′ overhang is 5′-CCGG-3′ for pUC19. These conditions allowed the efficiency of chromatin circularization to be studied.
In this study, it was found that chromatin assembled in vitro on linearized pUC19 plasmid DNA is remarkably flexible even when it is compacted by linker histone and assayed in the presence of 2 mM Mg2+. In concentrated solutions intramolecular chromatin ends find each other more efficiently than the ends of naked DNA molecules.
MATERIALS AND METHODS
DNA constructs
Supercoiled plasmids were prepared by standard protocols using rich media (TB), and were purified by banding in CsCl/ethidium bromide or by using Qiagen Maxiprep Kits. Plasmid pUC19 with a 420 bp deletion (pUCΔ420) was prepared by cutting pUC19 with SspI (cutting at nucleotide number 2501) and SfoI (cutting at nucleotide number 235) and re-circularization. Plasmid pUC19 with a 140 bp deletion (pUCΔ140) was prepared by cutting with TfiI (cutting at nucleotide numbers 641 and 781) and re-circularization. Deletions were verified by restriction mapping. All of the constructs used for chromatin assembly plus ligation contained the same AvaI cohesive ends and >175 bp of flanking sequences. The linear construct (6.26 kb) used for chromatin assembly and analytical sedimentation was prepared by AvaI digestion of a construct consisting of a 1901 bp NdeI fragment from the 3′-end of the chicken ovalbumin gene inserted into the EcoRI site of pBR322. AvaI cut this construct once at nucleotide number 1425 of pBR322. Nucleosome ladders resulting from chromatin assembly of this construct have been described previously (13). The 615 bp DNA used to assess the efficiency of short naked DNA circularization at increasing DNA concentrations was prepared by Sau3AI digestion of pUC19 DNA and gel purification.
In vitro chromatin assembly
Histones, both core and H5, were prepared from chicken erythrocyte nuclei as described previously (12). In these methods of histone preparation nuclei are first washed several times with buffer containing 0.35 M, or greater, concentrations of NaCl, essentially removing all soluble non-histone chromosomal proteins. No high mobility group (HMG) proteins or other non-histone proteins were detected by SDS–PAGE for overloaded samples. Nucleosome formation on linear DNA molecules and nucleosome alignment using histone H5 were performed just as described previously for supercoiled DNA (12), except where indicated otherwise. Basically, core histones were deposited on DNA by salt gradient dialysis using 0.9 µg of core histones per µg of DNA at a DNA concentration of 200 µg/ml. After dialysis to 10 mM Tris–HCl pH 8.0, 1 mM Na2EDTA, the core histone-reconstituted DNA was adjusted to achieve final concentrations of 100 µg/ml DNA, 0.15 M NaCl, 2 mg/ml sodium polyglutamate, 0.5 µg of histone H5 per µg of DNA and incubated for 12 h at 37°C. At this ratio of H5 to DNA, approximately one H5 molecule per nucleosome is associated with the chromatin (17).
Ligation reactions
Aliquots of chromatin or DNA (4 µl) (at 100 µg DNA/ml) in 0.15 M NaCl, 10 mM Tris–HCl, pH 8.0 2 mg/ml polyglutamic acid were added to appropriate volumes of pre-mixed reagents to achieve final solutions containing 2 mM MgCl2, 1 mM ATP, 10 mM DTT, 100 µg/ml BSA. Final solution volumes were 400, 50, 25, 12.5 or 6.2 µl for final DNA concentrations of 1, 8, 16, 32 or 64 µg/ml, respectively. Six (Weiss) Units of T4 DNA Ligase (New England BioLabs) were added to the three more dilute DNA solutions and 3 (Weiss) Units were added to the two more concentrated solutions. Samples were incubated overnight at room temperature, although the reactions appeared to be 90% complete after 1 h. Reactions were then stopped by adjusting the samples to 10 mM EDTA, 0.5% SDS and samples were phenol extracted, ethanol precipitated and dissolved in sample loading buffer for gel electrophoresis. The initial plasmid was ∼25% nicked. Some additional nicking arose from the incubations in Mg 2+-containing buffers due to trace levels of nuclease contamination in the restriction enzyme buffer and in the core histone prep.
DNA topoisomer analysis and gel electrophoresis
The mean number of nucleosomes on the circularized DNA fragments was measured accurately by their linking number change (ΔL), relative to naked DNA. A value of ΔL = –1.0 per nucleosome was used (18,19). Supercoiled (Form I) pUC19 was determined to have a ΔL of –13.5, corresponding to one supercoil per ∼200 bp, by topoisomer counting on two-dimensional gels (20). Deletion mutants of pUC19 were also determined to contain one supercoil per ∼200 bp. These supercoiled plasmids provided convenient markers for measuring the number of nucleosomes contained on the corresponding circularized chromatin fragments by electrophoresis on 2% agarose gels containing 0.5× TBE and 0.2 µg/ml chloroquine sulfate, where all of the topoisomers generated from the chromatin samples corresponded to negatively supercoiled DNA. It was found that small 2% agarose gels readily separated the linear and circular DNA forms in the size range of 2–3 kb in 4 h at 100 V. However, linear DNA fragments are resolved poorly on these gels; DNA fragments greater than ∼5 kb compress into a single band. Gels were photographed under UV illumination using a yellow filter after ethidium bromide staining using Polaroid Polapan 55PN film. The percentage of monomer circles was determined from scanned (AGFA ARCUS II high resolution scanner) Polaroid gel photographic negatives using IPLab Gel (Signal Analytics Corp.) volume analysis software on a Power Mac G4.
ExoIII/S1 reactions
ExoIII digestions of chromatin or DNA were performed in 0.5 mM MgCl2, 10 mM Tris–HCl, pH 8.0, 0.2 mM EGTA (21). DNA or chromatin samples were incubated with 100 U ExoIII/µg DNA for the stated time at room temperature. Samples were then phenol extracted, ethanol precipitated and dissolved in de-ionized water. An equal volume of 2× filter sterilized S1 buffer (1× buffer was 0.25 M NaCl, 0.5 mM ZnSO4, 30 mM sodium acetate, pH 4.6) was added, and samples were incubated for 30 min at 37°C with 20 U S1 nuclease/ml (21). Samples were then neutralized with 1 M Tris–HCl pH 10.0 and EDTA was added to 10 mM. Samples were ethanol precipitated and dissolved in 10 mM Tris–HCl pH 8.0 for subsequent restriction enzyme digestion (under conditions recommended by the manufacturer) or gel analysis.
Analytical ultracentrifugation
Sedimentation was performed in a Beckman XL-A analytical ultracentrifuge at 20 000 r.p.m. at 20°C using 12 mm double sector cells and a four-hole analytical rotor. Sample concentrations were ∼0.5 A260 U/ml. Scans were taken at 265 nm. The data were collected in digital form using the continuous scan mode set at 0.001 cm radial increments. Sedimentation boundaries were analyzed according to van Holde and Weischet (22). The plateau region of the slowly sedimenting polyglutamic acid present in the samples contributed ∼0.002 A260 units to the solvent baseline for the sedimenting chromatin, and this small value was subtracted out to correct the sedimenting chromatin boundary analysis for the additional small boundary that was not due to chromatin. This analysis removes the contribution of diffusion to the boundary shape and yields the integral distribution of sedimentation coefficients present in the sample.
RESULTS
Histone H5 condenses chromatin under assembly conditions
Using the standard chromatin assembly protocol described in Materials and Methods, histone H5 was incubated with one portion of a core histone-reconstituted linear DNA (6 kb plasmid plus insert) in the presence of polyglutamic acid in 0.090 M NaCl for 12 h at 37°C. Another portion of the same sample was incubated under the same conditions without histone H5. The NaCl concentration was lowered from the standard 0.15 M in order to eliminate aggregation of the H5-containing sample. The resulting chromatin ± H5 was analyzed by analytical ultracentrifugation. It can be seen in Figure 1 that, in the absence of H5, the chromatin sedimented homogeneously with s20, w values in the range of 36–43 S. In the presence of H5, the chromatin sedimented significantly faster, and still fairly homogeneously, with s20, w values in the range of 57–71 S. Thus, H5 complexation, which leads to a mass increase of only ∼10% caused a 58–69% increase in the sedimentation coefficient due predominantly to a reduction in the frictional coefficient. This indicates that a significant degree of compaction of the chromatin was induced by histone H5 in our in vitro system, mimicking what has been found for native chromatin (23). Approximating the sedimenting macromolecules as prolate ellipsoids of equivalent volumes, the ellipsoid representing the H5-containing chromatin would have a >10-fold reduced axial ratio (24). In the standard 150 mM NaCl chromatin assembly buffer or in the 2 mM Mg 2+-containing ligation buffer, even greater compaction of the chromatin occurred, but some aggregation was apparent, consistent with the properties of native chromatin (25). These results suggest that the in vitro assembled chromatin was capable of forming physiological levels of compaction.
Figure 1.

Sedimentation analysis of chromatin assembled in vitro on a 6.3 kb linear DNA fragment in the absence or presence of linker histone H5. The NaCl concentration was 90 mM. Sedimentation boundaries were analyzed according to van Holde and Weischet (22). The point with boundary fraction 0.05 was omitted from the analysis because of the larger error imparted to this point by subtracting out the small constant plateau value resulting from the slow sedimenting polyglutamic acid present in the samples (see Materials and Methods). The y-axis gives the fraction of the sample that has an s20, w less than or equal to the value indicated on the x-axis.
Chromatin circularizes more readily than naked DNA at high DNA concentrations
The circularization of naked DNA is well understood. The ring closure probability is defined as the ratio of the equilibrium constant for cyclization of a linear DNA fragment with cohesive ends to the equilibrium constant for bimolecular joining (26). It can be understood in terms of the effective concentrations of the various types of DNA ends in solution. In dilute solutions cyclization is highly preferred because intramolecular ends are tethered together and the ends from different DNA molecules are far apart. As the DNA concentration is increased bimolecular and higher-order joining of the DNA molecules become preferred. DNA fragments with lengths between ∼300 and several thousand bp readily circularize, and are well represented theoretically as flexible chains (11,27,28).
Whereas sufficiently long chromatin fibers should be flexible enough to circularize, chromatin fibers with DNA lengths in the range of 2–3 kb might be expected to be too stiff to circularize effectively, based upon the traditional solenoid model (4,5). However, end-to-end ligation should still occur at higher DNA concentrations as long as the ends are available for ligation. Thus, ligation products were analyzed over a range of DNA concentrations for pUC19 plasmid (2.7 kb), cut with AvaI restriction enzyme, before and after its assembly into chromatin. Surprisingly, the chromatin readily formed monomer circles. Moreover, at high DNA concentrations the chromatin circularized to a much greater extent than the corresponding naked DNA, as shown in Figure 2.
Figure 2.

DNA products obtained from a linear 2.7 kb pUC19 DNA fragment treated with T4 DNA ligase in the form of naked DNA or chromatin at increasing DNA concentrations. The 2% agarose gels contained 0.2 µg/ml chloroquine sulfate in order to resolve the topoisomers resulting from the closed circular DNA forms. Monomer circle and linear DNA forms are indicated. (A) Lanes labeled 1–64 µg DNA/ml show the ligation products resulting from the naked DNA. M denotes linear λHindIII DNA size markers; the 2.0 and 2.3 kb bands are resolved, whereas the 6.6, 9.4 and 23 kb bands run together. The lane labeled U denotes the unligated 2.7 kb DNA. (B) Lanes labeled 1–64 µg DNA/ml show the DNA products obtained from the H5+ chromatin. One chromatin sample lacking histone H5 (H5–) ligated at 16 µg DNA/ml is shown. The unligated DNA starting material used in this experiment is shown in A, lane U. Additionally, a core histone reconstituted DNA sample at a concentration of 16 µg/ml that was incubated overnight under ligation conditions, but without the T4 DNA ligase, ran indistinguishably from the starting DNA (not shown). M denotes linear unresolved 4.4–23 kb λHindIII DNA markers; I denotes the (Form I) supercoiled form of pUC19 having one supercoil per ∼200 bp. (C) Quantitation of the percentage monomer circles formed for DNA or chromatin ligated over a range of DNA concentrations. The single black triangle at 16 µg/ml denotes the sample lacking histone H5.
Figure 2A shows that, as the naked DNA concentration increased from 1 to 64 µg/ml, end-to-end ligation competed more effectively with circularization, and the percentage monomer circles formed decreased substantially, as expected. The results for the chromatin are shown in Figure 2B. The similar level of DNA supercoiling from the chromatin circles and the Form I plasmid marker shows that the chromatin contained approximately one nucleosome per 200 bp. In contrast with the naked DNA, the percent monomer circles for the H5-containing chromatin was high and remained nearly constant. Results are quantitated in Figure 2C. Whereas the percent monomer circles for naked DNA decreased from 95% at 1 µg/ml to 12% at 64 µg/ml, the percent monomer circles for the chromatin remained in excess of 70%. Based upon the mechanism of naked DNA cyclization (29), these results suggest that, at the higher DNA concentrations, the ends of the same molecules collide with each other more frequently for the chromatin than they do for the naked DNA.
This experiment also shows that the presence of histone H5 did not alter the supercoiling (ΔL) of the chromatin circles (compare the 16 µg/ml samples ± H5). Thus, the presence of linker histones does not appear to increase the DNA wrapping of the histone core of the nucleosome or to impart any additional writhe to the pUC19 chromatin. However, the possibility remains that fragments of eukaryotic DNA that form more ordered nucleosome arrangements than that of pUC19 plasmid DNA might form different higher order structures that behave somewhat differently.
In the absence of linker histone it was found that >60% of the DNA formed monomer circles even at saturating levels of core histones (1.2 µg core histones per µg of DNA) for which the mean ΔL value corresponded to one supercoil per 150 bp (data not included).
The more efficient circularization of chromatin versus naked DNA cannot be attributed simply to the effective reduction in DNA length by nucleosome formation
The ring closure probability, or j factor, for naked DNA circularization increases ∼3-fold as the DNA length is decreased from 2.7 kb to 600 bp, where it is maximal (29). Because nucleosome formation (the 1st-order chromatin structure) reduces the effective DNA length by ∼4.6-fold for each ∼150 bp packaged into a nucleosome, this effective length reduction could conceivably contribute to the apparently greater circle-forming ability of chromatin relative to the naked DNA. Thus the DNA concentration dependence of ligation was repeated for a 615 bp (2686 bp/4.4) naked DNA fragment over an equivalent molar DNA concentration range as in the previous experiment. Figure 3 shows that, at the highest equivalent concentration, considerable end-to-end ligation still occurred. Quantitation of the results indicates <20% monomer circles (CC + NC) were formed compared with >70% for the chromatin (Fig. 2C). Thus, the 2.7 kb chromatin circularizes more readily than naked DNA of length equivalent to that of the bead-on-a-string structure of the 2.7 kb chromatin.
Figure 3.
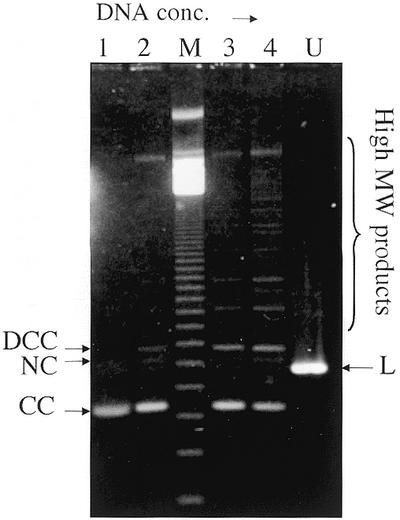
DNA products obtained from a linear 615 bp pUC19 DNA fragment treated with T4 DNA ligase in the form of naked DNA at increasing DNA concentrations. The 1.3% agarose gel in 0.5× TAE buffer contained 1 µg/ml ethidium bromide and was run at 80 V. Lanes 1–4 show the ligation products resulting from the ligation of DNA at 0.4, 4, 8 and 16 µg/ml, respectively. These concentrations are approximately equivalent to those used in Figure 2 on a molar basis. DCC, NC, L and CC denote the dimer closed circle, nicked circle, linear and monomer closed circle form of the DNA fragment, respectively. The lane labeled U contained unligated DNA; lane M contained a 123 bp linear DNA ladder. The bracket on the right indicates high MW DNA ligation products.
The chromatin structures formed do not have naked DNA tails extending from the ends
Conceivably the efficient circularization of chromatin could be a consequence of the presence of naked DNA tails extending from the ends of the chromatin structures formed. This possibility was assessed by ExoIII digestion of chromatin lacking H5, followed by S1 trimming of the purified DNA in Figure 4. It was verified from the degree of DNA supercoiling (not shown) that the reconstituted DNA contained approximately one nucleosome per 200 bp, as in the circularization experiments described above (Fig. 2). Figure 4A shows that the nucleosome-containing DNA was quite resistant to ExoIII, whereas the naked DNA was shortened by more than 1400 bp (2686–1230 bp) from the two ends. In Figure 4B and C the nuclease-treated DNAs were further digested with a restriction enzyme to better assess the ExoIII activity of each chromatin end relative to naked DNA. The results indicate that ∼90% of each end is resistant to ExoIII digestion. These experiments rule out the presence of naked DNA tails on >10% of the chromatin. Including histone H5 makes the chromatin even more resistant to ExoIII digestion.
Figure 4.

Assessment of the extent to which the chromatin samples have naked DNA protruding from their ends. Chromatin (H5–) or naked DNA samples were treated with exonuclease III for the times indicated under identical conditions; then the purified DNA samples were treated with S1 nuclease. Lanes M contained a 123 bp DNA ladder. (A) The mobilities of 2686, 1230 and 246 bp DNA fragments are indicated. The small apparent increase in size of the band labeled 2686 bp resulted from a slight frowning of the gel. (B) DNA from the same sample as used in (A) was further digested with AlwNI, located 805 bp from the left end. The mobilities of the 805, 1881 (the remaining portion of the 2686 bp original fragment) and 123 bp fragments are indicated. (C) DNA from the same sample as used in (A) was further digested with XmnI, located 804 bp from the right end. The mobilities of the 804, 1882 (the remaining portion of the original 2686 bp fragment) and 123 bp fragments are indicated.
Chromatin assembled on DNA shortened by up to 420 bp gives very similar results
It is conceivable that the high efficiency of the condensed H5-containing chromatin arises not so much from its flexibility, but from the fortuitous juxtaposition of ends in a relatively rigid condensed structure. If this were the case, then a DNA fragment two to three nucleosomes shorter than AvaI-cut pUC19 should probably condense in a different way, where the ends might not be in close proximity. To test this hypothesis a 420 bp deletion was made in pUC19 several hundred base pairs away from the AvaI site, and the experiments were repeated using this DNA linearized by AvaI. Figure 5 shows that results obtained for the deletion construct were very similar to those obtained for the full length DNA. Approximately 75% monomer circles were formed in the H5-containing chromatin. The chromatin in this experiment contained a slightly higher nucleosome density than the experiment shown in Figure 2B, as assessed from the slightly higher supercoiling obtained from the circularized chromatin samples compared with the Form I plasmid. Thus, the high percentage of monomer circles formed is not due to a deficiency of histones relative to the intact pUC19 chromatin. Additionally, similar results were obtained with a 140 bp deletion at a different position (not shown). These experiments suggest that the DNA ends were not fortuitously juxtaposed in all three constructs, and they support the idea that the chromatin structures formed were highly flexible.
Figure 5.
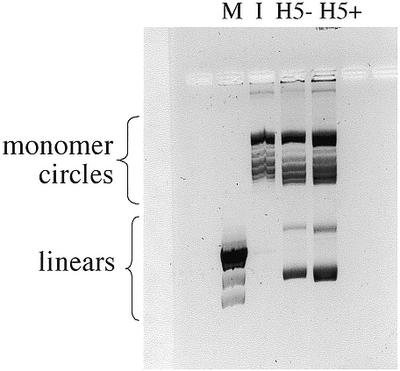
DNA ligation products obtained from chromatin assembled on a linear 2.27 kb DNA fragment (pUC19 with a 420 bp deletion) with AvaI ends. The chromatin sample in the absence (H5–) or presence (H5+) of histone H5 was incubated with T4 DNA ligase at a DNA concentration of 25 µg/ml. Lane M denotes linear λHindIII DNA markers; lane I denotes supercoiled plasmid (pUC19 with a 420 bp deletion) containing one supercoil per ∼200 bp. The gel contained 0.2 µg/ml chloroquine sulfate. Linear and monomer circle DNA forms are indicated.
DISCUSSION
The experiments reported here suggest that in vitro assembled chromatin which is condensed by linker histone and maintained in a condensed state by the presence of 2 mM Mg 2+ can, nevertheless, readily circularize. The data presented in Figure 1 show that histone H5 considerably condenses in vitro assembled chromatin in 90 mM NaCl. Similar extents of compaction by histone H5 and sedimentation coefficient heterogeneity have been obtained using the 2.5 kb chromatin assembled in vitro on the twelve 208 bp tandem repeats (208–12) of Lytechinus 5S rDNA (30). It is likely that in the presence of 2 mM Mg2+ and the NaCl concentrations used in the ligation experiments the 2–3 kb H5-containing chromatin structures were nearly maximally compacted (30). It could be ruled out that the circularization resulted from naked DNA ends extending from the chromatin in any appreciable fraction of the material (Fig. 4).
At higher DNA concentrations, the percentage of the chromatin molecules that formed monomer circles significantly exceeded that of naked DNA (Fig. 2). The preference for forming monomer circles rather than end-to-end products even exceeded that of a 4.4-fold shortened length of naked DNA with the maximum ‘j factor’ for circle formation. This 615 bp DNA should have had approximately the same contour length as the 2.7 kb chromatin viewed as a bead-on-a-string model, providing a control for the effect of the first order compaction of DNA resulting from its wrapping around histone cores. It is unlikely that a nucleosome could bridge the two DNA ends to bring them together to form a circle. In reconstitutions of ∼200 bp DNA fragments with core histones, no complexes consisting of two DNA molecules and one histone octamer are observed (31,32). Moreover, there is no evidence that the DNA ends contained within such a structure, if it existed, would be accessible to T4 DNA ligase. Thus, there is every reason to believe that the core histone reconstituted 2.7 kb linear DNA used here contained ordinary nucleosomes throughout its length. The preference of the condensed chromatin over naked DNA to form monomer circles must therefore be due to its higher order structure. It is unlikely that an inflexible higher order structure formed that had the DNA ends fortuitously juxtaposed, because two different deletions within the DNA construct resulted in essentially the same results (Fig. 5). These results support a flexible higher order structure.
The formation of monomer circles was likely slightly inhibited by two factors that were unrelated to the inherent chromatin flexibility. First, while the ionic conditions used in the ligation reactions (2 mM Mg2+, 1.5–96 mM Na+) have been determined to result predominantly in the formation of a compact 30 nm fiber state for native chromatin, they are close to conditions where the 30 nm fibers aggregate (25). Therefore, incipient aggregation was likely occurring during ligation, as was observed by analytical sedimentation under these conditions. Second, it is likely that nucleosomes formed very close to the ends on some fraction of the molecules inhibiting their ligation. Thus, in the absence of these two technical problems, the percentage monomer circle formation might have been even higher.
Mechanistic studies with naked DNA indicate that EcoRI cohesive ends associate and dissociate rapidly many times before covalent sealing by T4 DNA ligase (29). If a similar mechanism is operative here with AvaI cohesive ends, the higher preference for forming monomer circles for chromatin rather than forming end-to-end products, as with naked DNA, suggests that the chromatin has a flexing mode which allows the DNA ends to collide often. It is difficult to see how intramolecular ends could collide frequently for solenoid models (4,5) (Fig. 6A). Even if a solenoid transiently dissociates for short periods of time (25), it is hard to understand how intramolecular ends could readily associate with high preference over intermolecular ends in concentrated DNA solutions. On the other hand, the accordion-like ‘Zig-Zag’ model (6,7,33) would seem to be able to flex in a way that the DNA ends could readily collide while being largely internal to the chromatin structure (Fig. 6B).
Figure 6.

Rope chromatin models containing 12 nucleosomes. The rope represents the path of the DNA. Histones are not shown. The stars indicate the DNA ends. (A) Solenoid-like model; the rope nucleosomes are wrapped around a helical support, which is not a part of the structure. (B) Accordion-like model.
It is not clear to what extent DNA sequence affects the flexibility of chromatin. The linearized (or supercoiled) plasmid studied here does not form a highly ordered nucleosome array, in contrast with some regions of genomic DNA (15). In non-solenoid models, the higher order structure formed might be influenced by the regularity of the nucleosome arrangement. Model building studies (6) suggest that the more irregular chromatin structures should be more condensed than the regular structures. It should be possible to study the circularization of genomic DNA fragments using the methods described here.
It has been shown that significant unfolding of chromatin higher order structures, as assessed by sedimentation, at physiological NaCl concentrations occurs only when linker histone is absent and the core histones are acetylated (10). In this study, the chromatin was highly condensed, as assessed by sedimentation (Fig. 1); nevertheless it was flexible. Irrespective of which chromatin higher order structure model is correct, the results presented here strongly suggest that some condensed chromatin structures are highly flexible, even when linker histone is present and the core histones are not acetylated. Thus, DNA looping in chromatin as a means of delivering regulatory proteins to transcription promoters or histone-modifying enzymes to nucleosomes may be able to occur in initially inactive chromatin.
Acknowledgments
ACKNOWLEDGEMENT
This work was supported by US Public Health Service grant GM62857 (to A.S.).
REFERENCES
- 1.Schleif R. (1992) DNA looping. Annu. Rev. Biochem., 61, 199–223. [DOI] [PubMed] [Google Scholar]
- 2.Wijgerde M., Grosveld,F. and Fraser,P. (1995) Transcription complex stability and chromatin dynamics in vivo. Nature, 377, 209–213. [DOI] [PubMed] [Google Scholar]
- 3.Ptashne M. and Gann,A. (1997) Transcriptional activation by recruitment. Nature, 386, 569–577. [DOI] [PubMed] [Google Scholar]
- 4.Finch J.T. and Klug,A. (1976) Solenoidal model for superstructure in chromatin. Proc. Natl Acad. Sci. USA, 73, 1897–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thoma F., Koller,T.H. and Klug,A. (1979) Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. J. Cell Biol., 83, 403–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woodcock C.L., Grigoryev,S.A., Horowitz,R.A. and Whitaker,N. (1993) A chromatin folding model that incorporates linker variability generates fibers resembling the native structures. Proc. Natl Acad. Sci. USA, 90, 9021–9025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zlatanova J., Leuba,S.H., Yang,G., Bustamante,C. and van Holde,K. (1994) Linker DNA accessibility in chromatin fibers of different conformations: a reevaluation. Proc. Natl Acad. Sci. USA, 91, 5277–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woodcock C.L. and Horowitz,R.A. (1995) Chromatin organization re-evaluated. Trends Cell Biol., 5, 272–277. [DOI] [PubMed] [Google Scholar]
- 9.van Holde K.E. and Zlatanova,J. (1995) Chromatin higher order structure: chasing a mirage? J. Biol. Chem., 270, 8373–8376. [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Ramirez M., Rocchini,C. and Ausio,J. (1995) Modulation of chromatin folding by histone acetylation. J. Biol. Chem., 270, 17923–17928. [DOI] [PubMed] [Google Scholar]
- 11.Ringrose L., Chabanis,S., Angrand,P., Woodroofe,C. and Stewart,A.F. (1999) Quantitative comparison of DNA looping in vitro and in vivo: chromatin increases effective DNA flexibility at short distances. EMBO J., 18, 6630–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeong S., Lauderdale,J.D. and Stein,A. (1991) Chromatin assembly on plasmid DNA in vitro: apparent spreading of nucleosome alignment from one region of pBR327 by histone H5. J. Mol. Biol., 222, 1131–1147. [DOI] [PubMed] [Google Scholar]
- 13.Lauderdale J.D. and Stein,A. (1992) Introns of the chicken ovalbumin gene promote nucleosome alignment in vitro. Nucleic Acids Res., 20, 6589–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu K. and Stein,A. (1997) DNA sequence encodes information for nucleosome array formation. J. Mol. Biol., 270, 559–573. [DOI] [PubMed] [Google Scholar]
- 15.Stein A. and Bina,M. (1999) A signal encoded in vertebrate DNA that influences nucleosome positioning and alignment. Nucleic Acids Res., 27, 848–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwarz P.M. and Hansen,J.C. (1994) Formation and stability of higher order chromatin structures. J. Biol. Chem., 269, 16284–16289. [PubMed] [Google Scholar]
- 17.Stein A. and Bina,M. (1984) A model chromatin assembly system: factors affecting nucleosome spacing. J. Mol. Biol., 178, 341–363. [DOI] [PubMed] [Google Scholar]
- 18.Germond J.E., Hirt,B., Oudet,P., Gross-Bellard,M. and Chambon,P. (1975) Folding of the DNA double helix in chromatin-like structures from simian virus 40. Proc. Natl Acad. Sci. USA, 72, 1843–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simpson R.T., Thoma,F. and Brubaker,J.M. (1985) Chromatin reconstituted from tandemly repeated cloned fragments and core histones: A model system for study of higher order structure. Cell, 42, 799–808. [DOI] [PubMed] [Google Scholar]
- 20.Peck L.J. and Wang,J.C. (1983) Energetics of B to Z transition in DNA. Proc. Natl Acad. Sci. USA, 80, 6206–6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strauss F. and Prunell,A. (1982) Nucleosome spacing in rat liver chromatin. A study with exonuclease III. Nucleic Acids Res., 10, 2275–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Holde K.E. and Weischet,W.O. (1978) Boundary analysis of sedimentation-velocity experiments with monodisperse and paucidisperse solutes. Biopolymers, 17, 1387–1403. [Google Scholar]
- 23.Butler P.J.G. and Thomas,J.O. (1980) Changes in chromatin folding in solution. J. Mol. Biol., 140, 505–529. [DOI] [PubMed] [Google Scholar]
- 24.Cantor C.R. and Schimmel,P.R. (1980) Biophysical Chemistry, part II. W.H.Freeman and Co., San Francisco, pp. 560–561.
- 25.Widom J. (1986) Physicochemical studies of the folding of the 100Å nucleosome filament into the 300Å filament. Cation dependence. J. Mol. Biol., 190, 411–424. [DOI] [PubMed] [Google Scholar]
- 26.Jacobson H. and Stockmayer,W.H. (1950) Intramolecular reaction in polycondensations, I: the theory of linear systems. J. Chem. Phys., 18, 1600–1606. [Google Scholar]
- 27.Yamakawa H. and Stockmayer,W.H. (1972) Statistical mechanics of wormlike chains. II. Excluded volume effects. J. Chem. Phys., 57, 2843–2854. [Google Scholar]
- 28.Shimada J. and Yamakawa,H. (1984) Ring closure probabilities for twisted wormlike chains: application to DNA. Macromolecules, 17, 689–698. [Google Scholar]
- 29.Shore D., Langowski,J. and Baldwin,R.L. (1981) DNA flexibility studied by covalent closure of short fragments into circles. Proc. Natl Acad. Sci. USA, 78, 4833–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carruthers L.M., Bednar,J., Woodcock,C.L. and Hansen,J.C. (1998) Linker histones stabilize the intrinsic salt-dependent folding of nucleosomal arrays: Mechanistic ramifications for higher-order chromatin folding. Biochemistry, 37, 14776–14787. [DOI] [PubMed] [Google Scholar]
- 31.Lorch Y., La Pointe,J.W. and Kornberg,R.D. (1987) Nucleosomes inhibit the initiation of transcription but allow chain elongation with the displacement of histones. Cell, 49, 203–210. [DOI] [PubMed] [Google Scholar]
- 32.Stein A. (1979) DNA folding by histones: the kinetics of chromatin core particle assembly and the interaction of nucleosomes with histones. J. Mol. Biol., 130, 103–134. [DOI] [PubMed] [Google Scholar]
- 33.Bednar J. Horowitz,R.A., Grigoryev,S.A., Carruthers,L.M., Hansen,J.C., Koster,A.J. and Woodcock,C.L. (1998) Nucleosomes, linker DNA and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc. Natl Acad. Sci. USA, 95, 14173–14178. [DOI] [PMC free article] [PubMed] [Google Scholar]