


Abstract
Sequence tagged microsatellite profiling (STMP) enables the rapid development of large numbers of co-dominant DNA markers, known as sequence tagged microsatellites (STMs). Each STM is amplified by PCR using a single primer specific to the conserved DNA sequence flanking the microsatellite repeat in combination with a universal primer that anchors to the 5′-ends of the microsatellites. It is also possible to convert STMs into conventional microsatellite, or simple sequence repeat (SSR), markers that are amplified using a pair of primers flanking the repeat sequence. Here, we describe a modification of the STMP procedure to significantly improve the capacity to convert STMs into conventional SSRs and, therefore, facilitate the development of highly specific DNA markers for purposes such as marker-assisted breeding. The usefulness of this technique was demonstrated in bread wheat.
INTRODUCTION
Microsatellites, or simple sequence repeats (SSRs), are molecular markers based on tandem repeats of short (1–6 bp) DNA sequences that are abundant and widely dispersed throughout most eukaryotic genomes (1). High polymorphism, ease of scoring and amenability to automation make microsatellites suitable for a large number of genetic studies including DNA fingerprinting (2), paternity testing (3), genetic mapping (4) and the analysis of genetic diversity (5).
Sequence tagged microsatellite profiling (STMP) (6) enables the cost efficient and rapid development of large numbers of SSR markers from genomic or cDNA. It differs from other published methods for developing SSRs by eliminating the need to screen a DNA library for SSR-containing clones, and providing a 25-fold increase in sequencing throughput. STMP permits the construction of a comprehensive profile of the abundance and frequency of SSRs present within a pool of amplified restriction fragments, where each SSR is defined by a 22 bp nucleotide sequence tag. The conversion of sequence tags into PCR primers allows the amplification of corresponding SSRs, called sequence tagged microsatellites (STMs), from the pool of amplified restriction fragments or directly from genomic DNA. STM amplification is performed using a sequence tag primer in combination with a universal primer that anchors to the 5′-end of the targeted microsatellite repeat. Thus STMP reduces the cost of developing a SSR marker to the synthesis of a single primer specific to the conserved DNA sequence flanking a target SSR. Sequence tag primers can also be used to amplify the corresponding restriction fragments from the pool of amplicons used to construct a sequence tag profile, thereby enabling the full characterisation of a SSR locus by DNA sequence analysis, and design of a pair of primers flanking the microsatellite repeat. The latter procedure is especially useful as it allows the conversion of STMs into more specific PCR markers, if required, by providing a pair of conventional sequence-specific primers for SSR amplification.
In our experience, the isolation of restriction fragments containing target SSRs from the pool of amplicons used to construct a sequence tag profile can be difficult when STMP is applied to large and complex genomes. This difficulty impedes the full characterisation of a SSR locus and the development of a pair of primers specific to the conserved DNA sequence flanking the microsatellite repeat. In this paper, we describe a modification of the STMP procedure to facilitate the isolation of restriction fragments corresponding to sequence tags from the pool of amplicons used to construct a sequence tag profile. This procedure significantly improves the identification of amplified target restriction fragments by reducing the complexity of the amplicon pool, thereby facilitating the conversion of STMs into conventional SSR markers. The procedure is easy to perform and requires only the preparation of a pool of SSR-enriched restriction fragments from the same DNA template that was used to construct the sequence tag profile.
MATERIALS AND METHODS
Enrichment procedure
A pool of amplified PstI–MseI restriction fragments was prepared for STMP analysis as described by Hayden and Sharp (6). Enrichment of the amplicon pool for DNA fragments containing SSRs was performed in a 100 µl reaction mixture containing 0.2 mM dNTP, 1× PCR buffer, 1.5 mM MgCl2, 40 pmol biotinylated PCT6 primer mix, 10 pmol PstI adapter primer (CTC GGA AGC CTC AGT C-OH), and the preamplified PstI–MseI fragments as DNA template. The PCT6 primer mix consisted of equimolar amounts of two HPLC-purified synthetic oligomers (biotin–KKV RVR VCT CTC TCT CTC T-OH and biotin–KKR VRV RTC TCT CTC TCT C-OH, where K was G and T, R was A and G, and V was A, C and G). The reaction mixture was heated at 94°C for 5 min and 2 U Taq DNA polymerase was added. Incubation was continued at 55°C for 10 min, then at 72°C for 10 min. The reaction was terminated by adding 100 µl of ice-chilled 20 mM EDTA, pH 8.0. Biotinylated DNA molecules were removed from the reaction mixture by affinity capture using streptavidin-coated magnetic beads (Merck) as follows: 0.5 mg of prewashed beads suspended in 200 µl of 10× SSC (1.5 M NaCl, 0.15 M trisodium citrate, pH 7.0) was added to the reaction mix, followed by incubation at room temperature for 1 h with gentle agitation. The beads were washed twice with 200 µl of 10× SSC to remove unbound DNA fragments, and resuspended in 100 µl of 10× SSC. The bead suspension was transferred to a new tube, incubated at 80°C for 10 min, and the supernatant was removed. Single-stranded PstI–MseI fragments containing SSR sequences were released from the beads by incubating with 100 µl of alkaline buffer (10 mM NaOH, 1 mM EDTA, 10 mM Tris–HCl, pH 8.0). The eluted DNA fragments were ethanol precipitated and resuspended in 30 µl of reaction mix containing 0.2 mM dNTP, 1× PCR buffer, 1.5 mM MgCl2, 5 pmol each MseI adapter primer (GAG CAA GGC TCT CAC A-OH) and PstI adapter primer and 1 U Taq DNA polymerase. PCR amplification was performed for 25 cycles (60 s at 92°C, 60 s at 56°C and 60 s at 72°C). The reaction products were diluted 1:20 with TE buffer.
Amplification of PstI–MseI fragments containing target SSRs
The amplification of PstI–MseI fragments containing target SSRs was performed in a 20 µl reaction mixture containing 0.2 mM dNTP, 1× PCR buffer, 1.5 mM MgCl2, 5 pmol each of sequence tag (Table 1) and MseI adapter primers, 2 µl of diluted SSR-enriched PstI–MseI fragments as template DNA and 1 U Taq DNA polymerase. PCR amplification was performed for 25 or 30 cycles (60 s at 92°C, 60 s at 56°C and 60 s at 72°C) and the reaction products were separated on a 6% sequencing gel and visualised by silver staining. To confirm the amplification of target restriction fragments, putative target bands were excised from the polyacrylamide gel (7), reamplified under identical conditions and tested for the presence of a SSR. This test was performed using the PCR conditions described above with 5 pmol each of appropriate sequence tag and PCT6 primers and 1:50 diluted reamplified restriction fragment as template DNA. The amplification of a STM fragment of expected size confirmed the successful isolation of a target PstI–MseI fragment.
Table 1. Sequence tag primers used to amplify restriction fragments containing target SSRs from the pools of PstI–MseI amplicons.
| Codea | Primer sequence (5′→3′) | Size of isolated putative PstI–MseI fragment (bp) | Observed size of STMb (bp) | Expected size of target STM (bp) |
|---|---|---|---|---|
| STM 43 | CTGCAGCAGCGAACAGGTG | 360 | 190 | 190 |
| STM 114 | CTGCAGAAAACAGCGGGAATG | 290 | 100 | 100 |
| 270 | 75 | 75 | ||
| STM 271 | CTGCAGAGACCATTGCGTTCAC | 325 | 110 | 110 |
| STM 297 | CTGCAGCAAAAGACTTCAGTGC | 215 | 110 | 110 |
| STM 314 | CTGCAGGGGCTAGATGTTTTTC | 315 | 185 | 185 |
| STM 380 | CTGCAGGTGCGAGTATTCAATG | 260 | 110 | 110 |
| 220 | 110 | 110 | ||
| 210 | 105 | c | ||
| STM 409 | CTGCAGGGACACCTTTACA | 310 | 75 | 75 |
| STM 471 | CTGCAGAAAGTAAACAACCATC | 310 | 120 | 120 |
| STM 639 | CTGCAGATTTTTCCAACTTCAG | 255 | 155 | 155 |
| STM 773 | CTGCAGCTGAGGACACTCTTCA | 255 | 225 | 225 |
| 230 | 190 | 190 | ||
| 220 | 180 | 180 |
aThe full set of primer sequences used in the present study are available from the authors upon request.
bSize of STM fragment amplified from the isolated putative target restriction fragment.
cAmplification of an STM fragment of the observed size was not expected.
Conversion of STMs into conventional SSR markers
A second (reverse) primer specific to the conserved region flanking a target microsatellite repeat was designed from the DNA sequence of the isolated PstI–MseI fragment using NetPrimer (Premier Biosoft International) and the parameters: primer length 18–27 nt, 3′ end stability –5.5 to –9.0 kcal/mol, oligomer Tm 55–65°C, GC content 40–70% and primer rating >80. The amplification of target SSRs was performed in a 20 µl reaction mixture containing 0.2 mM dNTP, 1× PCR buffer, 1.5 mM MgCl2, 1 U Taq DNA polymerase, 5 pmol each of appropriate sequence tag (forward) and reverse primers (Table 2) and 100 ng genomic DNA from Triticum aestivum cv. Halberd. A touchdown PCR profile of 32 cycles was used for amplification: 60 s at 92°C, 60 s at 62°C and 30 s at 72°C, where the annealing temperature was reduced by 1°C per cycle for the first seven cycles. Microsatellite fragments were separated on 6% sequencing gels and visualised by silver staining.
Table 2. Primer sequences of conventional SSR markers developed from isolated PstI–MseI fragments.
| Code | Isolated PstI–MseI fragment | SSR structure | Primer sequences | SSR fragment size (bp) | Chromosomal location |
|---|---|---|---|---|---|
| sun24 | STM 43 (360 bp) | (CT)8 | Fwd: CTGCAGCAGCGAACAGGTG | 237 | 4B |
| Rev: CCTGCCTGCCTGTTTTGTCTC | |||||
| sun25 | STM 114 (290 bp) | (CT)21 | Fwd: CTGCAGAAAACAGCGGGAATG | 192, 172 | 7B, 7B |
| STM 114 (270 bp) | (CT)10 | Rev: GCGAAGACGACACAGCAGCAG | |||
| sun26 | STM 271 (325 bp) | (CT)17 | Fwd: CTGCAGAGACCATTGCGTTCAC | 155 | 7A |
| Rev: GCAAGGAGCTGAACAAGCCACA | |||||
| sun27 | STM 297 (215 bp) | (CT)8 | Fwd: CTGCAGAGACCATTGCGTTCAC | 156 | 1D |
| Rev: GTTTGTTTTCACCTGCACTATAGAATC | |||||
| sun28 | STM 314 (315 bp) | (CT)16 | Fwd: CTGCAGGGGCTAGATGTTTTTC | 217 | 7A |
| Rev: GACATACTCTATCACTTCACTCTCA | |||||
| sun29 | STM 380 (260 bp) | (CT)6 | Fwd: CTGCAGGTGCGAGTATTCAATG | 138 | 5D |
| Rev: CCACCCATCCATCCATCCTAC | |||||
| sun30 | STM 380 (210 bp) | (CT)7 | Fwd: CTGCAGGTGCGAGTATTCAATG | 119 | 5A |
| Rev: CCGTCCATCCTACCATCTATCTG | |||||
| sun31 | STM 380 (220 bp) | (CT)10 | Fwd: CTGCAGGTGCGAGTATTCAATG | 134 | 5B |
| Rev: CCATCCATCCATCCTACTCCTAC | |||||
| sun32 | STM 409 (310 bp) | (CT)21 | Fwd: CTGCAGGGACACCTTTACA | 174 | 3B |
| Rev: GCCTTCTTCCGTCACCTCAC | |||||
| sun33 | STM 471 (310 bp) | (CT)17 | Fwd: CTGCAGAAAGTAAACAACCATC | 167 | 2D |
| Rev: CGGAGCGTCCCTAATGTGAGAT | |||||
| sun34 | STM 639 (255 bp) | (CT)9 | Fwd: CTGCAGATTTTTCCAACTTCAG | 178 | 3A |
| Rev: CTGTACCATATTCTTAGGTCCTTGG | |||||
| sun35 | STM 773 (255 bp) | (CT)23 | Fwd: CTGCAGCTGAGGACACTCTTCA | 185 | 2B, 2A, 2D |
| STM 773 (230 bp) | (CT)12 | Rev: AAACGCCCCAACCACCTCTCTC | |||
| STM 773 (220 bp) | (CT)7 | ||||
| sun36 | STM 773 (255 bp) | (CT)23 | Fwd: ATGGTTTGTTGTGTTGTGTGTAGG | 230, 210, 205 | 2B |
| Rev: AAACGCCCCAACCACCTCTCTC |
Chromosomal assignment of conventional SSRs
The chromosomal origin of the developed conventional SSRs was determined through PCR amplification using the reaction conditions described above and DNA from the set of 21 nullisomic-tetrasomic lines of T.aestivum cv. Chinese Spring (8).
RESULTS AND DISCUSSION
Rationalisation of method
In the original STMP procedure (6), the amplification of a target restriction fragment was achieved using a sequence tag primer in combination with a primer specific to the MseI adapter sequence. However, it was observed during the application of STMP to bread wheat, an allohexaploid with an extremely large (1C = 17.3 pg) genome consisting of more than 80% repetitive DNA (8), that the specific amplification of target restriction fragments was often difficult to achieve. Rather, target DNA fragments were typically observed as the more strongly amplified fragment among the multiple (non-target) amplicons that resulted from the use of only a single primer determining PCR specificity (6; M.J.Hayden, unpublished data). This result was not unexpected since the probability of achieving specific amplification of a target fragment is reduced with increased complexity of the amplicon pool from which the target is to be amplified. To achieve more specific amplification of restriction fragments corresponding to sequence tags, the STMP procedure was modified to include a step to specifically enrich the PstI– MseI amplicon pool for DNA fragments containing SSRs. This enrichment step significantly reduced the complexity of the amplicon pool and thereby promoted more specific amplification of a target restriction fragment.
To enrich the PstI–MseI amplicon pool for DNA fragments containing SSRs, the preamplified restriction fragments were hybridised with a biotinylated oligonucleotide repeat probe that was identical in sequence to the 5′-anchored SSR primer used to construct the sequence tag profile (Fig. 1). The biotinylated DNA fragments were then isolated by affinity capture using streptavidin-coated magnetic beads and washed under stringent (80°C) conditions. The captured single-stranded PstI–MseI fragments were eluted from the beads under alkaline conditions and reamplified by PCR. The resulting SSR-enriched PstI–MseI amplicon pool served as template DNA for subsequent amplification of target restriction fragments. To enhance the efficiency of the enrichment procedure, Taq DNA polymerase was used to extend biotinylated SSR probes that were hybridised to single-stranded PstI–MseI fragments to form a complement DNA strand. By forming a partially double-stranded DNA molecule the possibility of cross-hybridisation with non-target PstI–MseI fragments during affinity capture was minimised. Similarly, the extension by DNA polymerase of a non-biotinylated primer hybridised to the PstI adapter sequence of non-target DNA molecules also reduced cross-hybridisation.
Figure 1.
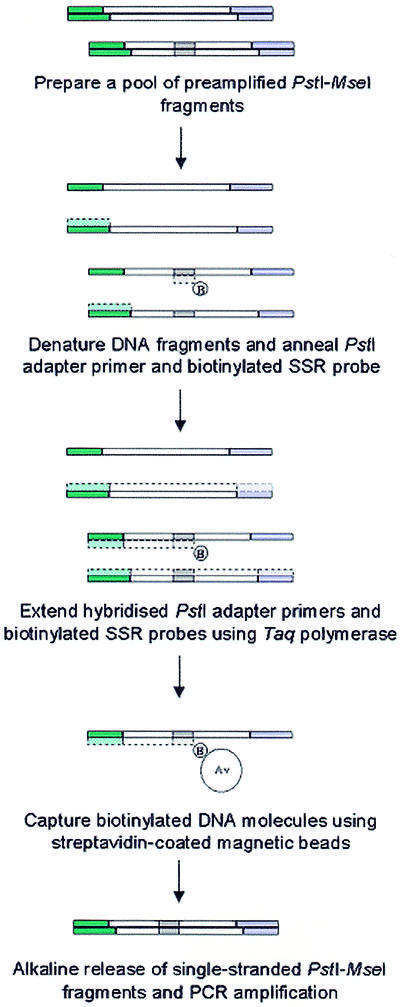
Schematic diagram of the SSR enrichment procedure.
Evaluation of method
To determine the capacity of the enrichment procedure to improve the isolation of restriction fragments containing target SSRs, 46 sequence tag primers were employed in PCR to amplify corresponding restriction fragments from both SSR-enriched and untreated PstI–MseI amplicon pools. PCR was performed for both 25 and 30 cycles to determine the optimal conditions for the amplification of each target fragment. The identification of putative target PstI–MseI fragments was significantly improved for 28 (61%) of the sequence tag primers tested. Improved identification in reactions using the SSR-enriched amplicon pool as template DNA was generally observed as an increase in yield (2- to 50-fold) of the target fragment(s) relative to non-specific (background) amplicons (Fig. 2A) and the amplification of fewer non-target fragments (Fig. 2B). The failure to achieve improved identification of target fragments for the remaining 18 (39%) sequence tag primers arose for two reasons. In 11 (24%) instances, putative target restriction fragments were easily identified in reactions using the non-enriched pool of amplicons as template DNA due to the amplification of a single (or few) fragments. Hence, further improvement for the identification of target fragments was limited (Fig. 2C). Alternatively, multiple amplicons of about equal intensity were observed in reactions using either type of template DNA, amongst which no putative target fragment(s) could be identified (data not shown). In these instances, a second round of PCR using a nested primer specific to the target restriction fragment would be needed to sufficiently reduce the complexity of the amplification pattern to permit the identification of a putative target amplicon. An appropriate nested primer could be designed from the DNA sequence of the STM corresponding to a target SSR.
Figure 2.

Amplification of PstI–MseI fragments containing SSRs corresponding to sequence tags. Each PCR was performed in replicate using six independently prepared unenriched (UE) and SSR-enriched (E) amplicon pools as template DNA. The sequence tag primers used in each reaction are: (A) STM 43, (B) STM 409 and (C) STM 639. Putative target fragments are indicated by an arrow. A 25 bp DNA ladder (Gibco BRL) was used as a size marker (M).
The successful isolation of PstI–MseI fragments corresponding to sequence tags was confirmed by PCR amplification of a STM fragment of expected size from putative target restriction fragments that were excised from polyacrylamide gels. Each PCR was performed using an appropriate sequence tag primer and the 5′-anchored SSR primer used to construct the sequence tag profile. In all instances where a single putative target restriction fragment was isolated, the amplified STM fragment was of expected size. Where more than one putative target fragment was isolated but only one corresponding PstI–MseI fragment was expected, additional SSRs were uncovered (e.g. STM 380, Table 1). These additional SSRs probably corresponded to closely related or homologous loci, an issue that could be resolved by DNA sequence analysis. The usefulness of the PCR test to validate putative target restriction fragments was also demonstrated by the finding that 11% (7/60) of the isolated fragments did not contain SSRs. Overall, PstI–MseI fragments containing target SSRs were successfully isolated for 34 of the 39 sequence tag primers for which a putative target restriction fragment was identified.
To demonstrate the conversion of STMs into conventional SSR markers, a subset of the isolated PstI–MseI fragments were sequenced and a second (reverse) primer specific to the conserved region flanking each target microsatellite was designed (Table 2). In all instances where a single PstI–MseI fragment was isolated, specific amplification of the target SSR was achieved (Fig. 3). Where multiple target fragments were isolated, the ability to achieve locus-specific SSR amplification depended on the presence of nucleotide sequence differences between related PstI–MseI fragments. In the case of STM 114, the DNA sequences flanking the target SSRs were identical in the isolated PstI–MseI fragments, and primers for single locus amplification could not be designed. Rather, the primer designed resulted in the amplification of both target loci (see sun25, Fig. 3). In the case of STM 380, nucleotide sequence variation in the regions flanking the target SSRs permitted the design of primers for the specific amplification of each SSR (Table 2 and Fig. 3). A similar situation was also possible for STM 773, except that single locus amplification could only be achieved for the SSR present in the 255 bp PstI–MseI fragment. However, a primer could be designed to permit the amplification of all three target SSRs isolated for STM 773 (see sun35, Fig. 3). Overall, pairs of flanking primers permitting the specific amplification of all target SSRs were developed, demonstrating a high conversion rate of STMs into conventional SSR markers. The assignment of each SSR to a wheat chromosome by aneuploid analysis confirmed that the developed markers were locus-specific and suitable for use in marker-assisted breeding.
Figure 3.
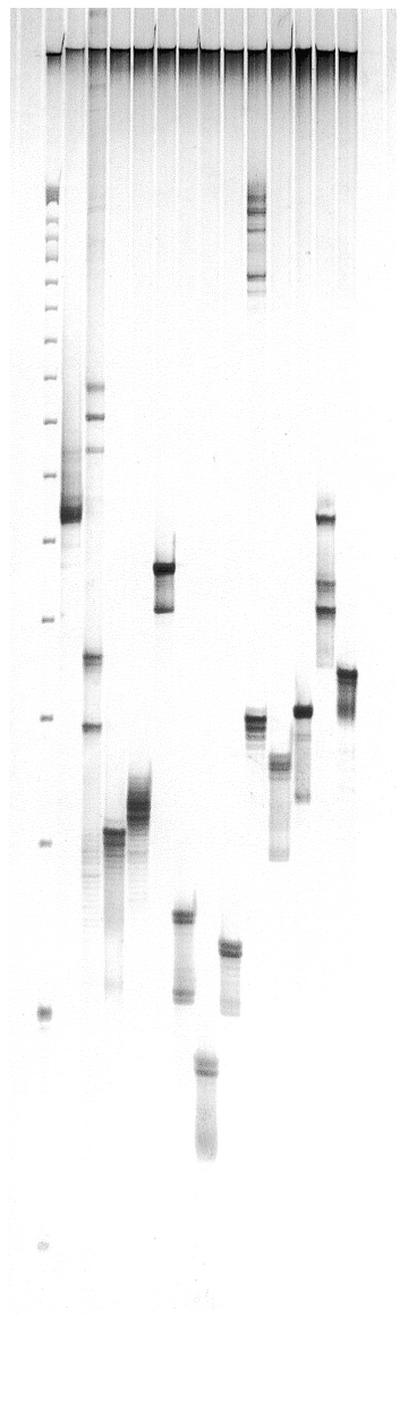
Amplification of target SSRs present in isolated PstI–MseI fragments using primer pairs specific to the conserved DNA sequences flanking the microsatellite repeats. The primer pairs used in each reaction are from left to right: sun24, sun25, sun26, sun27, sun28, sun29, sun30, sun31, sun32, sun33, sun34, sun35 and sun36. A 25 bp DNA ladder (Gibco BRL) was used as a size marker.
CONCLUSION
A rate limiting step of STMP, particularly in the analysis of large and complex genomes, is the conversion of STMs into conventional SSR markers that are amplified by PCR using a pair of primers specific to the conserved DNA sequence flanking the microsatellite repeat. This procedure is especially useful for developing locus-specific DNA markers, which are highly desirable for purposes such as the analysis of genetic diversity and marker-assisted breeding. Therefore, we have modified the STMP procedure to facilitate the isolation of restriction fragments containing target STMs from the pool of amplicons used to construct a sequence tag profile, from which a pair of primers specific to the conserved DNA sequence flanking the SSR can be designed. This method is easy to perform and requires only the preparation of a pool of SSR-enriched restriction fragments from the same DNA template that was used to construct the sequence tag profile.
REFERENCES
- 1.Weber J.L. and May,P.E. (1989) Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am. J. Hum. Genet., 44, 388–396. [PMC free article] [PubMed] [Google Scholar]
- 2.Charters Y.M., Robertson,A., Wilkison,M.J. and Ramsay,G. (1996) PCR analysis of oilseed rape cultivars (Brassica napus L. ssp. oleifera) using 5′-anchored simple sequence repeats. Theor. Appl. Genet., 92, 442–447. [DOI] [PubMed] [Google Scholar]
- 3.Amos B., Schlotterer,C. and Tautz,D. (1993) Social structure of pilot whales revealed by analytical DNA profiling. Science, 260, 670–672. [DOI] [PubMed] [Google Scholar]
- 4.Roder M.S., Korzun,V., Wendehake,K., Plaschke,J., Tixier,M.-H., Leroy,P. and Ganal,M.W. (1998) A microsatellite map of wheat. Genetics, 149, 2007–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Plaschke J., Ganal,M.W. and Roder,M.S. (1995) Detection of genetic diversity in closely related bread wheat using microsatellite markers. Theor. Appl. Genet., 91, 1001–1007. [DOI] [PubMed] [Google Scholar]
- 6.Hayden M.J. and Sharp,P.J. (2001) Sequence tagged microsatellite profiling (STMP): a rapid technique for developing SSR markers. Nucleic Acids Res., 29, e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 8.Sears E.R. and Sears,L.M.S. (1978) The telocentric chromosomes of common wheat. In Ramanujam,S. (ed.), Proceedings of the 5th International Wheat Genetics Symposium. Indian Society of Genetics and Plant Breeding, New Delhi, India, pp. 389–407.
- 9.Bennet M.D. and Smith,J.B. (1976) Nuclear DNA amounts in angiosperms. Philos. Trans. R. Soc. Lond. B Biol. Sci., 274, 227–274. [DOI] [PubMed] [Google Scholar]